
Abstract
Background
Klebsiella pneumoniae strains have been divided into two major categories: classical K. pneumoniae, which are frequently multidrug-resistant and cause hospital-acquired infections in patients with impaired defenses, and hypervirulent K. pneumoniae, which cause severe community-acquired and disseminated infections in normal hosts. Both types of infections may lead to bacteremia and are associated with significant morbidity and mortality. The relative burden of these two types of K. pneumoniae among bloodstream isolates within the United States is not well understood.
Methods
We evaluated consecutive K. pneumoniae isolates cultured from the blood of hospitalized patients at Northwestern Memorial Hospital (NMH) in Chicago, Illinois between April 2015 and April 2017. Bloodstream isolates underwent whole genome sequencing, and sequence types (STs), capsule loci (KLs), virulence genes, and antimicrobial resistance genes were identified in the genomes using the bioinformatic tools Kleborate and Kaptive. Patient demographic, comorbidity, and infection information, as well as the phenotypic antimicrobial resistance of the isolates were extracted from the electronic health record. Candidate hypervirulent isolates were tested in a murine model of pneumonia, and their plasmids were characterized using long-read sequencing. We also extracted STs, KLs, and virulence and antimicrobial resistance genes from the genomes of bloodstream isolates submitted from 33 United States institutions between 2007 and 2021 to the National Center for Biotechnology Information (NCBI) database.
Results
Consecutive K. pneumoniae bloodstream isolates (n = 104, one per patient) from NMH consisted of 75 distinct STs and 51 unique capsule loci. The majority of these isolates (n = 58, 55.8%) were susceptible to all tested antibiotics except ampicillin, but 17 (16.3%) were multidrug-resistant. A total of 32 (30.8%) of these isolates were STs of known high-risk clones, including ST258 and ST45. In particular, 18 (17.3%) were resistant to ceftriaxone (of which 17 harbored extended-spectrum beta-lactamase genes) and 9 (8.7%) were resistant to meropenem (all of which harbored a carbapenemase genes). Four (3.8%) of the 104 isolates were hypervirulent K. pneumoniae, as evidenced by hypermucoviscous phenotypes, high levels of virulence in a murine model of pneumonia, and the presence of large plasmids similar to characterized hypervirulence plasmids. These isolates were cultured from patients who had not recently traveled to Asia. Two of these hypervirulent isolates belonged to the well characterized ST23 lineage and one to the re-emerging ST66 lineage. Of particular concern, two of these isolates contained plasmids with tra conjugation loci suggesting the potential for transmission. We also analyzed 963 publicly available genomes of K. pneumoniae bloodstream isolates from locations within the United States. Of these, 465 (48.3%) and 760 (78.9%) contained extended-spectrum beta-lactamase genes or carbapenemase genes, respectively, suggesting a bias towards submission of antibiotic-resistant isolates. The known multidrug-resistant high-risk clones ST258 and ST307 were the predominant sequence types. A total of 32 (3.3%) of these isolates contained aerobactin biosynthesis genes and 26 (2.7%) contained at least two genetic features of hvKP strains, suggesting elevated levels of virulence. We identified 6 (0.6%) isolates that were STs associated with hvKP: ST23 (n = 4), ST380 (n = 1), and ST65 (n = 1).
Conclusions
Examination of consecutive isolates from a single center demonstrated that multidrug-resistant high-risk clones are indeed common, but a small number of hypervirulent K. pneumoniae isolates were also observed in patients with no recent travel history to Asia, suggesting that these isolates are undergoing community spread in the United States. A larger collection of publicly available bloodstream isolate genomes also suggested that hypervirulent K. pneumoniae strains are present but rare in the USA; however, this collection appears to be heavily biased towards highly antibiotic-resistant isolates (and correspondingly away from hypervirulent isolates).
Similar content being viewed by others

Background
Klebsiella pneumoniae is a worldwide cause of bloodstream infections [1]. This bacterium can also cause severe secondary pneumonia in patients with respiratory virus infections such as SARS-CoV-2 [2]. In North America, this organism is the second leading cause of gram-negative bacteremia and is predominantly hospital-acquired or healthcare-associated rather than community acquired [3, 4]. Infections of the bloodstream are associated with higher mortality rates (20–33%) than those involving other sites [5].
Most K. pneumoniae infections are caused by “classical” (cKP) strains, which infect chronically ill patients residing in hospitals and long-term care facilities [6]. Some cKP strains have become increasingly resistant to antibiotics, including carbapenems [7,8,9,10,11,12]. A subgroup of cKP have been designated “high-risk clones,” lineages that are frequently multidrug-resistant (MDR) or extensively drug-resistant (XDR) and that have spread across continents to cause large numbers of infections [13, 14]. Some of the best characterized K. pneumoniae high-risk clones include ST11, ST14, ST15, ST17, ST147, ST45, ST258, ST307, and ST512 [15,16,17,18,19,20,21]. Within the United States, ST258 and ST307 are endemic high-risk clones that are usually resistant to carbapenems and extended-spectrum cephalosporins (e.g., ceftriaxione), respectively [22,23,24,25]. Genes conferring antimicrobial resistance in these strains are often carried by conjugative plasmids that allow for dissemination among cKP strains. Thus, it is not surprising that the IDSA, WHO, and the CDC have each deemed MDR K. pneumoniae as a serious public health priority in need of new therapeutic development [26,27,28].
A second type of K. pneumoniae, referred to as hypervirulent K. pneumoniae (hvKP), was first identified in Taiwan in 1986 as a common cause of pyogenic liver abscesses in young, otherwise healthy individuals living in the community [29,30,31,32,33,34,35,36]. Other than diabetes mellitus, these patients lacked risk factors commonly associated with cKP infections. While these strains are now common in Asia, little is known about their prevalence in the United States. Case reports document the presence of hvKP infections in North America, but only a few surveillance studies have been performed [35, 37,38,39]. hvKP strains frequently cause disseminated multi-site infections, express highly mucoid capsules, leading to a “hypermucoviscous” (hmv) colony phenotype, and are considerably more virulent in mouse models than cKP strains [40]. Their increased virulence is usually attributed to the presence of one of several large, non-conjugative “hypervirulence” plasmids [41,42,43]. The non-conjugative nature of these plasmids and the limitations imposed by the hyper-mucoid capsule on genetic exchange may restrict the hypervirulence phenotype to strains of a few sequence types (e.g., ST23, ST86, ST65, ST66) and capsule loci (e.g., KL1, KL2) [31, 44, 45]. Detection of hvKP is clinically important, as they may cause infections that require prolonged antibiotic therapy, tend to relapse, and frequently disseminate to remote sites [46].
Hypervirulence plasmids are defined by several pathogenic features. In general, these plasmids contain two distinct pathogenicity loci (PAL-1 and PAL-2) (Fig. 1A). PAL-1 usually contains a mucoid regulator gene (rmpA2), the aerobactin receptor gene (iutA), and aerobactin biosynthesis genes (iucABCD). PAL-2 usually consists of a distinct mucoid regulator operon (rmpADC), the salmochelin receptor gene (iroN), and salmochelin biosynthesis genes (iroBCD) (Fig. 1A). In addition, some of these plasmids also contain a variety of metal resistance genes including a tellurite resistance locus (ter), which is important for colonization of the gastrointestinal tract [47]. Considerable diversity exists among individual hypervirulence plasmids [43, 48]. For example, pK2044 and KP52.145pII, two of the most common and best characterized hypervirulence plasmids, differ in that KP52.145pII lacks ter and contains an incomplete PAL-1 without rmpA2 (Fig. 1A–C). We previously identified plasmid pTK421_2 from a K. pneumoniae bloodstream isolate from Northwestern Memorial Hospital (NMH) in Chicago, Illinois (Fig. 1D) [49]. This plasmid also lacks rmpA2 and ter but contains conjugation genes (Fig. 1D). The population diversity of hypervirulence plasmids remains poorly defined but is an area of active investigation.

Virulence loci associated with hypervirulence in K. pneumoniae. PAL-1 and PAL-2 contents and organization are depicted for pK2044, KP52.145pII, pTK421_2, and ICEKp1 (A). Genomic elements and plasmid architecture are depicted for pK2044 (B), KP52.145pII (C) and pTK421 (D) (drawings not to scale)
The hypervirulence plasmids contribute to the pathogenicity of hvKP strains in several ways. The mucoid regulators rmpA and rmpA2 increase capsule production and hypermucoviscosity, and the siderophores are thought to provide access to host-sequestered iron reserves [42]. However, the relative importance of the different accessory plasmid and chromosomal siderophores (aerobactin, salmochelin, and yersiniabactin) in hvKP virulence remain controversial [50, 51]. A 2015 report suggested that aerobactin was the only siderophore required for the hypervirulence phenotype of the ST86-K2 capsule type strain hvKP1 in both mouse intraperitoneal and pneumonia models [51]. In contrast, another report showed that only a triple siderophore mutant (disruption of genes encoding yersiniabactin, aerobactin, and salmochelin) of the ST23-K1 strain NTUH-K2044 was attenuated in the mouse intraperitoneal model [50]. In addition, strain KPPR1 (a rifampin-resistant variant of ATCC43816) is hypervirulent in a mouse model of pneumonia but is otherwise atypical in that it lacks a hypervirulence plasmid but contains a chromosomal version of PAL-2 in an integrative and conjugative element (ICEKp1, Fig. 1A) [52, 53]. Yersiniabactin production is commonly associated with hvKP lineages as part of a second ICE (ICEKp10) that includes genes for both yersinibactin and the genotoxin colibactin [54]. In a KPPR1 background, yersiniabactin promoted disease in the lung and salmochelin promoted dissemination to tissues [55]. Thus, mucoid regulators and siderophores play roles in conferring the hypervirulent phenotype, but significant strain-to-strain variability may occur.
As mentioned, hvKP strains were first identified based upon the unusual disease manifestations they caused (e.g., pyogenic liver abscesses), but considerable effort has been devoted to developing a microbiological definition that could be used by clinical laboratories to identify these strains. Three characteristics are commonly associated with hvKP: 1) hmv, 2) presence of hvKP pathogenicity loci, and 3) high levels of virulence in mouse models of infection [6, 29, 31, 40, 44, 46, 56,57,58,59,60,61]. Initially, hmv was used as a proxy for the hvKP phenotype. However, several reports have described hmv K. pneumoniae strains that lack typical hvKP pathogenicity loci and are not hypervirulent in mice [62,63,64]. As a result, genetic biomarkers have been used to define hvKP, but these definitions have varied from study to study [37, 39, 65]. Recently, Russo and colleagues systematically examined the accuracy of using rmpA, rmpA2, peg-344, iucA, and iroB, as diagnostic biomarkers for hvKP [40, 66]. In their cohort of 175 isolates, the presence of iucA, iroB or a hmv phenotype distinguished hvKP from cKP with an accuracy of 96%, 97%, and 90%, respectively.
Fortunately, hvKP strains remain susceptible to most antibiotics, but this appears to be changing. Antimicrobial-resistant hvKP infections are on the rise, likely due to the increase in hospital-acquired hvKp infections [3, 67, 68]. In a study from Asia, 13.7% of the iuc+ isolates contained carbapenemase genes, although only one also contained other hvKp genes such as rmpA or rmpA2 [69]. This convergence of hvKP and MDR-cKp can occur in one of two ways: an hvKP isolate acquires an antibiotic resistance mobile element, or an MDR-cKp isolate acquires a plasmid or integrative and conjugative element containing hvKP virulence genes. The more widespread emergence of K. pneumoniae strains that are both antibiotic-resistant and hypervirulent would pose a serious threat to the healthcare system.
In this work, we characterized 140 consecutive bloodstream isolates of Klebsiella cultured from patients at NMH in Chicago, Illinois from 2015 – 2017 and determined ST, KL type, and the presence of virulence and antimicrobial resistance genes. In addition, we evaluated publicly available genomes of bloodstream isolates collected within the United States from 2007 – 2021. We identified numerous MDR cKP high-risk clones and several isolates that contained genomic, clinical, and virulence properties consistent with hvKP.
Methods
Bacterial isolates and growth conditions
The isolates used in this study were from consecutive blood cultures collected between April 2015 and April 2017 and were designated as Klebsiella pnuemoniae sequence complex (KpSC) by the NMH Clinical Microbiology Laboratory using a VITEK-2 platform [70]. For the sub-analysis of isolates determined to be K. pneumoniae (sensu stricto) by sequencing, only the first isolate cultured from each unique infection was included. Isolates used in this study are listed in Additional file 13: Table S1. For experiments, bacteria were grown at 37 °C in lysogeny broth (LB) or on LB agar plates.
Antibiotic susceptibility testing
Antimicrobial susceptibility testing of NMH bloodstream islolates was done by the clinical microbiology laboratory using a VITEK-2 platform [70]. Minimum inhibitory concentrations for each antibiotic tested are listed in Additional file 13: Table S1. Clinical breakpoints for the designations of resistant, intermediate, or susceptible were based on guidelines of the Clinical and Laboratory Standards Institute [71,72,73]. MDR and XDR were defined as previously described [14].
National Center for Biotechnology Information (NCBI) Sequencing Reads
K. pneumoniae Illumina sequencing reads were obtained from NCBI (assessed NCBI on Dec 29th, 2021) by filtering for isolation source (blood) and geographical location (USA). Reads were downloaded from NCBI using Sratoolkit v 2.11.1 and trimmed using Trimmomatic (v0.36). de novo assembly was performed using SPAdes version 3.9.1 [74, 75]. Contigs were filtered based on coverage in trimmed reads using a custom perl script. Downloaded reads (n = 963) represent genomes deposited from 33 United States institutions between 2007 and 2021.
Whole-genome sequencing
Genomic DNA was prepared from a single colony cultured overnight at 37 °C in LB using the Maxwell 16 system (Promega Corp., Madison, WI, USA). Libraries for Illumina sequencing were prepared using either Nextera XT (Illumina, Inc., San Diego, CA) or Seqwell (Seqwell, MA, USA) library kits and sequenced on an Illumina MiSeq or NextSeq 500 instrument to generate paired-end 300 bp or 150 bp reads (Additional file 17: Table S5). Reads were trimmed using Trimmomatic (v0.36), and de novo assembly was performed using SPAdes version 3.9.1 [74, 75]. Nanopore sequencing was performed as described previously [76, 77]. Briefly, libraries were prepared from genomic DNA using the ligation sequencing kit (SQK-LSK109, Oxford Nanopore, UK). Libraries were sequenced on a MinION instrument using a FLO-MIN106 flow cell, and base calling and demultiplexing of sequence reads was performed using Guppy v3.4.5 [78]. Hybrid assembly and circularization of Nanopore and Illumina reads were performed using Flye v2.9 (Additional file 18: Table S6). Nanopore sequencing errors were corrected by aligning Illumina reads to the assembly using BWA v0.7.17 and using serial rounds of Pilon v1.23. Annotation was performed using the NCBI Prokaryotic Genome Annotation Pipeline [79].
Hypermucoviscosity testing
The string test was performed as described previously [80]. Briefly, Isolates were grown overnight at 37 °C on LB agar. A single colony was lifted with a loop to evaluate the formation of a viscous string between the loop and the colony. A positive string test was defined as a string length ≥ 5 mm. A centrifugation assay was also used to assess hmv. Centrifugation of overnight bacterial cultures (5 mL LB) was performed at 3220×g for 10 min [81]. hmv-positive isolates were identified qualitatively by the persistence of turbidity.
Tellurite resistance testing
For screening of tellurite resistance, K. pneumoniae isolates were grown overnight at 37 °C on LB agar plates supplemented with 3 μg/mL potassium tellurite (Sigma-Aldrich, USA).
Phylogenetic analysis
FastTree 2 was used to generate a maximum-likelihood phylogenetic tree based on the core genome, which was defined as sequences present in 95% of the isolates [82]. SNPs were identified by aligning raw Illumina reads to the genome of reference strain NTUH-K2044 using bwa-0.7.15. kSNPv3.0 was used for phylogenetic analysis of assembled genomes when raw sequencing reads were not available. The accession numbers of previously reported strain sequences used in this analysis (KPPR1, KP52.145, SB5881, K180005, NCTC9494, NTUH-K2044, RJF999, and ED2) are listed in Additional file 19: Table S7. The phylogenetic trees were visualized and annotated using iTOL (v4) [83].
Molecular typing and identification of virulence genes
Assembled whole genome sequences were analyzed with the bioinformatics tools Kleborate v2.1 and Kaptive v0.7.3 using default settings to evaluate multilocus sequence type, capsule locus type, antimicrobial resistance gene content, and the presence of virulence genes [44, 84]. MOB-suite was used to predict and type plasmids and identify putative replicons [85]. Plasmids identified by whole genome sequencing were aligned using BLAST Ring Image generator (BRIG) with an alignment threshold of 85% identity [86]. MASH (v2.3) was used to cluster plasmids based on Jaccard index, and Cytoscape (v3.8.2) was used to create plasmid networks [87]. Plasmid replicons were identified using PlasmidFinder v2.1 and MOB-suite [85, 88].
Mouse studies
Anesthetized 6- to 8-week-old C57BL/6 female mice purchased from Jackson Labs were infected intranasally as described previously [64]. Briefly, sets of mice were anesthetized with Ketamine-xylazine cocktail (100 mg/kg and 20 mg/kg, respectively). Mice were then infected with different doses of K. pneumoniae and monitored for two weeks post-infection for pre-lethal illness. Experimenters were blinded to strains and inoculum sizes. Mice infected with each strain or dose were chosen at random. To minimize pain and distress, mice were euthanized with CO2 when they reached predetermined endpoints of > 20% weight loss, abnormal respiratory rate, or a hunched posture with minimal activity. 50% lethal dose (LD50) values were determined in R using the drc package [89]. Strain dosing, deaths, and total mice inoculated are included in Additional file 20: Table S8. The number of mice used were limited to the minimum number required to calculate an accurate LD50. No experiments were excluded from these data. All procedures were approved by and performed in accordance with the guidelines of the Northwestern University Animal Care and Use Committee (protocol IS00002172).
Statistical analysis
Statistical analysis was performed using GraphPad Prism. Student T-test and analysis of variance (ANOVA) followed by the Bonferroni’s correction for multiple comparisons were performed for parametric variables. For non-parametric variables, the Mann-Whitney U test was used. For comparison of survival curves, the Mantel-Cox log rank test was used. Pearson correlation coefficient was used to determine statistical associations between LD50 values and siderophore production. Simpson’s Diversity index was calcuated in R using the package ‘vegan’.
Results
Microbiological characteristics of Klebsiella bloodstream isolates from a single United States medical center
To determine the prevalence of antibiotic-resistant and hypervirulent K. pneumoniae strains within United States hospitals, we collected 140 consecutive bloodstream isolates identified as KpSC by the Clinical Microbiology Laboratory at NMH. These isolates were cultured from 119 patients between 2015–2017 at NMH [90]. Among these 140 isolates, 119 (85%) were confirmed by whole-genome sequencing to be K. pneumoniae, with the remaining isolates being Klebsiella variicola (n = 16), Klebsiella quasipneumoniae subsp. quasipneumoniae (n = 1), Klebsiella quasipneumoniae subsp. similipneumoniae (n = 3), and a non KpSC species, Klebsiella oxytoca (n = 1) (Additional file 1: Fig S1). The 119 K. pneumoniae isolates were cultured from 101 patients and consisted of 75 (STs) (Simpson’s diversity index = 0.98), 51 unique capsule loci (KL) (Simpson’s diversity index = 0.97), and 10 distinct O-antigen serotypes (Simpson’s diversity index = 0.69). After removing isolates of the same strain cultured from the same patient, we were left with 104 K. pneumoniae isolates that were obtained from 101 patients. Three patients had two separate K. pneumoniae bloodstream infections caused by distinct strains, which were included in this study. Within this collection, 32 (30.7%) isolates had STs associated with carbapenem-resistant high-risk clones such as ST258 (n = 6), ST45 (n = 6), ST29 (n = 5), ST14 (n = 3), ST17 (n = 3), ST15 (n = 3), ST147 (n = 3) (Fig. 2). Two isolates belonged to the ESBL-producing high-risk clone ST307 (Fig. 2; Additional file 2: Fig. S2A). In addition, we identified three STs associated with hvKP: ST23 (n = 2), ST66 (n = 1), and ST380 (n = 1) (Fig. 2; Additional file 2: Fig. S2A). The most common KL-types were KL2 (n = 7), KL107 (n = 7), KL30 (n = 6), KL25 (n = 5), KL24 (n = 4), KL28 (n = 4), KL22 (n = 4), and KL54 (n = 4) (Fig S2B). KL107 is associated with ST258, and KL2 is associated with ST14 and hvKP STs such as ST380 or ST66. The most common O antigen serotypes were O1 (n = 38), O2 (n = 27), and O3b (n = 21) (Additional file 2: Fig S2C). These findings indicate that bloodstream isolates at NMH are highly diverse with respect to their STs, capsule types, and O-antigen serotypes. This set of 104 K. pneumoniae isolates was used in the subsequent analyses.

Core genome phylogenetic tree, genomic content, and antibiotic resistance of 104 K. pneumoniae bloodstream isolates. A Maximum likelihood phylogenetic tree was generated from core genome SNP loci in 104 K. pneumoniae bloodstream isolates. Sequence types, the presence of virulence genes, hypermucoviscosity, and antibiotic resistances are indicated. ybt = yersiniabactin biosynthesis loci, clb = colibactin biosynthesis loci, rmpADC = mucoid regulator operon, rmpA2 = regulator of mucoid phenotype 2, iuc = aerobactin biosynthesis genes, iro = salmochelin biosynthesis genes, Amp = ampicillin, TMP/SMX = trimethoprim-sulfamethoxazole. Strains were considered hypermucoviscous if they were positive by string test. The complete genome of NTUH-K2044 was used as the reference genome
Antimicrobial resistance genes and alleles
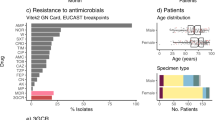
We next examined the resistance profiles of the isolates in our collection of 104 bloodstream isolates to β-lactam antibiotics. Since K. pneumoniae is inherently resistant to ampicillin, it was not surprising that 100% of the isolates had minimal inhibitory concentrations (MICs) above the resistance threshold for this antibiotic (Fig. 2; Additional file 3: S3). Non-susceptibility rates (either resistant or intermediate) were as follows: 39 (37.5%) isolates to ampicillin/sulbactam, 17 (16.3%) to aztreonam, 23 (22.1%) to cefazolin, 18 (17.3%) to ceftriaxone, 17 (16.3%) to cefepime, and 9 (8.7%) to meropenem (Additional file 13: Table S1; Fig. 2; Additional file 3: S3). Of the 104 isolates, 44 (42%) contained at least one acquired antimicrobial resistance gene (Additional file 13: Table S1). Twelve isolates (11.5%) contained extended-spectrum beta-lactamase genes, the most common of which were blaCTX-M-15 (n = 7, 6.7%) and blaSHV-12 (n = 4, 3.8%) (Additional file 13: Table S1; Additional file 4: Fig S4). Strains carrying blaCTX-M-15 were highly resistant to antibiotics, including all beta-lactams tested except meropenem (Additional file 4: Fig S4A-F). Strains carrying blaSHV-12 also carried genes predicted to confer carbapenem resistance (Fig. 2; Additional file 4: S4A–F). Nine (8.7%) isolates contained a carbapenemase gene (8 blaKPC-3, 1 blaNDM-1) (Additional file 4: Fig S4). blaNDM-1 was found in an ST147 isolate, while blaKPC-3 carbapenemase genes were found in ST258 (n = 5), ST15 (n = 2), and ST45 (n = 1) isolates (Additional file 13: Table S1). One ST258 isolate, KPN88, did not contain a carbapenemase gene and was sensitive to several beta-lactams (Additional file 13: Table S1; Fig. 2). Four of the NMH bloodstream isolates contained both ESBL and carbapenamase genes (Fig. 2; Additional file 13: Table S1). Isolates containing one or both of these genes also carried genes predicted to confer resistance to aminoglycosides, trimethoprim, sulfonamides, tetracycline, fluoroquinolones, and phenicols, suggesting these strains contain one or more antimicrobial resistance plasmids (Additional file 13: Table S1).
Resistance to other antibiotics was also quite common in the overall collection. Thirty-four isolates (32.6%) were non-susceptible to the combination of trimethoprim and sulfamethoxazole (TMP/SMX) (Additional file 3: Fig. S3). Of these isolates 31 (29.8%) contained both the dfrA and sul1/2/3 genes, which encode resistance to TMP/SMX (Additional file 13: Table S1). Two resistant isolates contained only dfrA and one contained only sul2 (Additional file 13: Table S1). Eighteen isolates (17.3%) were resistant to ciprofloxacin (Additional file 3: Fig S3). Each of these isolates contained either the qnrB gene or a mutation in the gyrA gene (Additional file 13: Table S1). Non-susceptibility rates to tobramycin, gentamicin, and amikacin were 17.3%, 10.5%, and 1.9%, respectively (Additional file 3: Fig S3). The overall prevalence of aminoglycoside modifying enzyme (AME) genes was 31.7% (n = 33) (Additional file 13: Table S1). The most common AMEs were streptomycin resistance genes strA and strB (n = 22) and the streptomycin/spectinomycin adenyltransferase aadA2 (n = 10); however, isolates containing only these AMEs were sensitive to tobramycin, gentamicin, and amikacin (Additional file 13: Table S1). The acetyltransferases aac(6')-Ib-cr.V2 (n = 7), aac(3)-IId (n = 2), and aac(3)-IIa (n = 2), and the phosphotransferases aph(3)-Ia.v1 (n = 5) and aph(3’)-Ia (n = 2) were associated with nonsusceptibility to gentamicin and tobramycin (Additional file 13: Table S1). Collectively, 17 (16.3%) of the 104 isolates were MDR, including 12 (11.5%) that were XDR (Fig. 2). These findings indicate the MDR bacteria comprise a substantial proportion of K. pneumoniae strains causing bloodstream infections at NMH and suggest that many strains carry mobile elements with multiple resistance genes.
Antimicrobial resistance plasmids
Next, we sought to characterize the ESBL or carbapenamase plasmids harbored by the 18 NMH isolates resistant to ceftriaxone. To define plasmid sequences, we used MOB-suite to predict and reconstruct plasmid sequences in silico from short-read draft genome assemblies. We presumptively identified 16 plasmids that contained ESBL (n = 8) or carbapenamase genes (n = 8) (Additional file 14: Table S2). These 16 plasmids were predicted to belong to 8 plasmid groups that together formed a plasmid network of 5 clusters based on genomic similarity, replicon family, and antimicrobial resistance gene content (Fig. 3A; Additional file 14: Table S2). One plasmid, from strain KPN103, clustered with ESBL containing plasmids, but did not contain an ESBL (Fig. 3; Additional file 14: Table S2). To clarify the identity of these plasmid clusters, we performed Nanopore long-read sequencing on four of the ceftriaxone-resistant isolates: KPN94, KPN103, KPN107, and KPN132. These four isolates cumulatively contained five plasmids with either ESBL (n = 3) or carbapenamase (n = 2) genes (Additional file 15: Table S3). Each of these plasmids belonged to a distinct MOB cluster (Additional file 15: Table S3). While inferred plasmid content was accurate, specific clustering of the plasmids based on hybrid assemblies did not correlate well with those inferred by MOB-suite (Additional file 14: Table S2, Additional file 15: Table S3). This is likely due to the extensive plasmid diversity in K. pneumoniae and the tendency of these plasmids to recombine [45, 91]. These results suggest that ESBL and carbapenemase genes are carried by a variety of plasmids in our collection.

Prediction and analysis of antimicrobial-resistant plasmids in ceftriaxone-resistant K. pneumoniae isolates. MOB-suite was used to infer the presence and sequence of plasmids containing ESBL and carbapenemase genes in ceftriaxone-resistant isolates. Inferred plasmids were then assigned to plasmid groups (represented by different colors) based on replicon family, relaxase type, and matepair formation and transferability (A). Clustering of inferred plasmids was performed using Mash. Each node represents a plasmid, and two plasmids are connected by an edge if their Jaccard index is ≥ 0.95 (B). Networks were graphed using Cytoscape
Virulence traits
We next searched the K. pneumoniae genomes for genes associated with enhanced virulence. The siderophore yersiniabactin promotes pulmonary infections in animal models, is associated with more severe disease in human infections, and is produced by both hvKP and cKP strains [52, 54, 55, 92]. We identified yersiniabactin biosynthesis loci (ybt) in 37.5% (n = 39) of the 104 NMH bloodstream isolates (Fig. 2; Table 1). Of these 39 isolates, 10 (26%) contained ESBL or carbapenamase genes. Eleven distinct variants of ybt were detected, the most common being ybt10 (n = 12), ybt17 (n = 11) and ybt4 (n = 4) (Additional file 13: Table S1). Although yersiniabactin biosynthetic genes are carried by several different integrative conjugative elements (ICEs), ybt and colibactin (clb) biosynthesis loci are found together on ICEKp10 [31]. We identified 12 (11.5%) isolates representing five STs (ST258, ST29, ST66, ST15, and ST234) that contained ICEKp10 (Additional file 13: Table S1). Although ICEKp10 elements containing ybt1 and clb2 lineages are found in the globally distributed CG23-I sub-lineage, our isolates primarily contained ICEKp10 elements with ybt17 and clb3, which are found in the epidemic ST258 lineage [31, 54, 93]. Indeed, in our collection 6 (50%) of the 12 isolates with ICEKp10 were ST258, and 7 (58%) contained ESBL or carbapenemase genes (Additional file 13: Table S1). Only one potential hvKP isolate (ST66) contained ICEKp10 (Additional file 13: Table S1). These findings suggest that yersiniabactin and colibactin genes were predominantly associated with cKP isolates in our collection.
In addition to ybt and clb, we screened our collection for the following hvKP associated factors: iuc, iro, rmpA, and rmpA2. iuc was identified in 5 (4.8%), iro in 4 (3.8%), rmpA in 4 (3.8%), and rmpA2 in 2 (1.9%) of the isolates (Fig. 2; Table 1). These virulence factors tended to be clustered within the same few isolates. For example, four isolates contained iuc, iro, and rmpA (Table 2). Two of these four (KPN8 and KPN115) were ST23 and the other two (KPN49 and KPN165) were ST66 and ST380, respectively. A single isolate, KPN23, was ST881 and contained iuc but not iro or rmpA. (Fig. 2; Table 2). Three distinct lineage variants of the iuc locus were identified: iuc1 (n = 2, ST23), iuc2 (n = 2, ST66 and ST380), and iuc3 (n = 1, ST881) (Table 2). Although the rmpA2 gene was found in two strains (KPN8 and KPN115), both genes contained a frameshift mutation in a previously identified homopolymer region [94]. All iuc+ isolates were sensitive to every antibiotic tested except ampicillin (Additional file 13: Table S1), consistent with the highly susceptible nature of hvKP strains.
We next screened the collection for hmv by both the string test and a centrifugation assay. A total of 10 (9.6%) and 18 (17.3%) isolates, respectively, were positive for hmv by these two tests (Table 1), including all four of the isolates containing iuc, iro, and rmpA (KPN8, KPN49, KPN115, KPN165) but not the isolate containing only iuc (KPN23) (Table 2; Additional file 13: Table S1). These data confirm previous reports that the string test identifies fewer strains as hmv than centrifugation [64, 81]. Several isolates (e.g., KPN80) were hmv but lacked iuc, iro, rmpA, and rmpA2 (Table 2). These findings are consistent with published studies showing that rmpA and rmpA2 are not necessary for hmv [40, 93].
Isolates were also tested for tellurite resistance, a phenotype commonly encoded by pK2044 hypervirulence plasmids (Fig. 1). Five isolates (4.8%) grew on tellurite, including the two hmv ST23 isolates that contained rmpA, rmpA2, iro1 and iuc1 (Table 1). These results suggest that these two isolates contained pK2044-like plasmids. In contrast, the two isolates containing iro2, iuc2 and rmpA were susceptible to tellurite, suggesting that they contain KP52.145pII-like plasmids. Collectively, these findings indicate that clinical isolates with genomic features of hvKP were present at NMH.
Analysis of virulence plasmid content
To identify potential virulence plasmids, we used MOB-suite to predict plasmids from Illumina-generated sequences of all 104 isolates. This analysis identified the presence of 397 potential plasmids (Additional file 14: Table S2). Five isolates were predicted to harbor plasmids containing iuc (four of these also contained iro and rmpA) and five were predicted to harbor plasmids containing ybt (Additional file 14: Table S2). These predicted plasmids were from four distinct groups that formed four different plasmid network clusters based on replicon family, relaxase type, and matepair formation and transferability (Additional file 5: Fig S5; Additional file 14: Table S2). No other plasmids from our collection clustered with these plasmids. These data suggest that three distinct types of iuc+ plasmids and one type of ybt plasmid were present in our collection.
We next focused on the iuc + plasmids. We performed Nanopore sequencing on the isolates that were predicted to harbor them, which allowed complete plasmid sequences to be generated. KPN8 and KPN115 contained plasmids pKPN8_1 and pKPN115_1, which had near complete alignment with pK2044, including plasmid replicons repB and IncHI1B and pathogenicity loci PAL-1 and PAL-2 (Fig. 4A; Table 2). A third isolate, KPN165, harbored pKPN165_1, an IncFIB(K) plasmid with close alignments with KP52.145pII but not pK2044 (Fig. 4B; Additional file 6: Fig. S6). Similar to KP52.145pII, this plasmid contained a version of PAL-1 with iucABCD and iutA but lacking rmpA2 (Figs. 1, 4B). Closer examination, however, revealed that pKPN165_1 contained a ~ 40 kB insertion (absent in KP52.145pII) consisting of a tra conjugation locus similar to the tra locus of KP52.145pI, a second (non-virulence) plasmid harbored by KP52.145 (Fig. 5B) [48, 95]. We also identified a similar chimeric hypervirulence plasmid (99.0% identity) in the previously published ST380 hypervirulent strain hvKP4 (Fig. 5B) [96, 97]. The fourth isolate, KPN49, contained a plasmid pKPN49_1, which also had an IncFIB(K) replicon and similarity to KP52.145pII and a ~ 40-kb insert of tra genes (Figs. 4B, 5B). Interestingly, this isolate contained a second plasmid, pKPN49_2, with similarity to KP52.145pI and that may have served as the source of the ~ 40 kb tra insertion in pKPN49_1. Consistent with these alignments, pKPN49_1, pKPN165_1, KP52.145pII, and the plasmid from hvKP4 group closely with each other based on genomic similarity (Jaccard index > 0.98) (Fig. 5A). The fifth isolate, KPN23, carried pKPN23_1, a two-replicon plasmid (IncFIB(K) and IncFII(pKP91)) with conjugation genes, iucABCD, and iutA but lacking rmpA, rmpA2, and iro (Table 2). This plasmid did not resemble known hypervirulence plasmids but was highly similar to a plasmid harbored by a swine isolate KPCTRSRTH01_p2 collected in Thailand in 2016 (Additional file 7: Fig S7; Table 2). For comparison, we also included in our analysis pTK421_2, a unique hypervirulence plasmid we had previously identified from a bloodstream isolate collected at NMH in 2013 [49]. pTK421_2 contains PAL-1, PAL-2, and conjugation genes (Fig. 1). This plasmid was distinct from pK2044, KP52.145pII, and pKPN23_1 (Fig. 4B; Additional file 6: S6).

The bloodstream isolates KPN115 and KPN8 harbor pK2044-like plasmids. pKPN115_1 and pKPN8_1 sequences were aligned to the pK2044 sequence (NC_006625.1) using blast ring image generator (BRIG) (A). The indicated plasmids sequences were aligned to KP52.145pII using BRIG (B). A sequence identity threshold of 85% was used. Aerobactin biosynthesis genes are indicated with green arrows, salmochelin with orange arrows, rmpA with a blue arrow, and rmpA2 with a red arrow

The bloodstream isolates KPN49 and KPN165 harbor KP52.145pII-like plasmids. De novo clustering of plasmids using Mash (A). Individual nodes represent a sequenced plasmid. The thickness of the edge connecting plasmids is determined by Jaccard index as indicated. Networks were graphed using Cytoscape. The indicated plasmid sequences were aligned to pKPN49_1 using BRIG (B). A sequence identity threshold of 85% was used. The ~ 40 kb tra locus insertion is labeled. Aerobactin biosynthesis genes are indicated with green arrows, salmochelin with orange arrows, and rmpA with a blue arrow. Plasmid accession numbers for previously published plasmids are listed in Table S7
Overall, among the 104 K. pneumoniae bloodstream isolates we identified four isolates that contained two distinct hypervirulence plasmids, a fifth isolate that contained a plasmid containing aerobactin biosynthetic genes, and five additional isolates containing presumptive yersiniabactin plasmids.
Phylogenetic analysis of bloodstream isolates containing hvKP pathogenicity loci
To examine the phylogenetic relationships of the five NMH bloodstream isolates containing aerobactin biosynthesis genes, we generated a core genome phylogenetic tree of these isolates along with previously published hvKP isolates. These results confirmed previous findings that KL1 hvKP isolates tend to be phylogenetically clustered, whereas KL2 hvKP isolates tend to be more dispersed (Additional file 8: Fig S8). While KPN115 and KPN8 are both ST23 isolates, they are not part of a previously described and globally distributed CG23-I clade and, as such, neither contained the combination of yersiniabactin and colibactin biosynthetic genes observed in ICEKp10 (Additional file 8: Fig S8) [31]. KPN49 is part of a rarely described ST66-K2 lineage that has recently re-emerged around the globe (Additional file 8: Fig S8) [95, 98, 99]. KPN165 is phylogenetically close to hvKP4, and both isolates harbor similar KP52.145pII-like plasmids containing additional putative conjugation loci. However, KPN49 harbors a very similar plasmid (99.6% identity) despite being quite divergent genetically (Additional file 8: Fig S8; Table 2). These findings suggest that this plasmid has spread by horizontal transmission, perhaps aided by the presence of the insertion containing tra genes. Together, these results suggest that multiple lineages with features of hvKP are present at NMH.
Virulence in a murine model of pneumonia
Studies to date suggest that hvKP strains share the common feature of being highly virulent in mouse models of infection [40, 51, 97, 100]. To more definitively determine whether the five bloodstream isolates with features of hvKP were indeed hypervirulent, we measured the virulence of representative isolates in a mouse pneumonia model. To determine the level of virulence of the isolates containing characteristics of hvKP, LD50 measurements were made. For reference, we first determined the virulence of a published hvKP isolate, hvKP5, which is an ST23 KL1 strain that harbors pK2044 (iuc1, iro1, rmpA, a disrupted allele of rmpA2) and is hmv (Table 2; Additional file 8: Fig S8) [60]. As expected, hvKP5 was extremely virulent and had a LD50 of 102.0 CFU (Fig. 6A). hvKP5 was slightly more virulent than previously published hvKP isolates hvKP1 and hvKP4, which have an LD50 of 103.2 and 104.6 CFU, respectively [96]. As additional controls, we tested classical isolates CRE-229 (KL107, ST258) and CRE-163 (KL112, ST15) from our previously published collection of carbapenem-resistant isolates [101]. CRE-229 and CRE-163 had LD50s of 107.4 and 108.1 CFU, respectively (Fig. 6B, C). From the NMH collection, we selected KPN49 and KPN8 as representative pK2044 and KP52.145pII isolates, respectively. These isolates had LD50 values of 101.5 and 102.2 CFU, respectively, indicating high levels of virulence (Fig. 6D, E). In comparison, KPN23, which is non-hmv and has an iuc3-containing plasmid lacking other hvKP virulence genes, had an intermediate LD50 value of 106.1 CFU (Fig. 6F). Next, we tested KPN80, an hmv bloodstream isolate from our collection lacking iuc, iro, rmpA, and rmpA2 (Table 2). The hmv phenotype of KPN80 is likely due to missense mutations identified in its wzc gene (G132D, L147I, N248D, L607I, S629F), a part of the capsule biosynthesis loci [102]. This isolate had an LD50 value of 108.1 CFU which is consistent with our cKP controls and previously published LD50 values for cKP (Fig. 6G) [96]. We also included in the analysis TK421, the previously identified hmv+ hvKP-like strain that contained iuc2, iro2, and rmpA (Table 2). TK421 had an LD50 of 104.5 CFU (Fig. 6H). Collectively, these data confirm that KPN8, KPN49, and TK421 are hvKP strains and suggest that KPN115 and KPN165 are also hvKP. KPN23 has an intermediate LD50 value between that of hvKP and cKP.

Virulence of representative NMH K. pneumoniae isolates containing hvKP pathogenicity loci in a murine model of pneumonia. C57BL/6 mice were infected by nasal aspiration with the indicated doses of hvKP5 (A), CRE-229 (B), CRE-163 (C), KPN49 (D), KPN8 (E), KPN23 (F), KPN80 (G), and TK421 (H). The estimated log10 LD50 values are listed in blue. The number of mice used for each dose are listed in Additional file 20: Table S8
Siderophore production
hvKP virulence has been attributed to the secretion of unusually large amounts of siderophores [59]. For this reason, we quantified siderophore concentrations in bacterial supernatants using a chrome-azurol S (CAS) assay. We again used a set of representative isolates for these assays. Cell-free supernatants from cultures of iuc+ isolates (KPN8, KPN23, KPN49, TK421, and hvKP5) had the greatest iron chelation activity, equivalent to > 150 mM dipyridyl (Additional file 9: Fig S9A). CRE-229 and KPN80 produced somewhat less activity (> 100 mM dipyridyl equivalent), while CRE-163 produced ~ 50 mM dipyridyl equivalent (Additional file 9: Fig S9A). In these select isolates, there was a strong inverse correlation between siderophore production and log(LD50) (Pearson correlation coefficient − 0.86, R2 0.76) (Additional file 9: Fig S9). These findings suggest that the amount of siderophore secretion is associated with K. pneumoniae virulence.
Infections caused by hvKP strains
Since KPN8, KPN49, KPN115, and KPN165 had microbiological, genetic, and virulence properties of hvKP, we next examined the clinical context of the infections they caused. None of the four patients infected by these strains had a known travel history to areas where hvKP is endemic. Three of the four patients had infections that were community-acquired (Table 3). Two patients had hepatic abscesses, and one had a lung abscess. Two patients had diabetes mellitus, a known risk factor for hvKP infection [103,104,105]. None of the patients had other common manifestations of hvKP infections, such as endophthalmitis, meningitis, or necrotizing fasciitis. We also looked at the infections cause by TK421, KPN23, and KPN80. The patient infected with TK421, a slightly less virulent strain with features of hvKP, had a history of diabetes mellitus and presented with a community-acquired liver abscess, although this patient had predisposing conditions including a previous Whipple procedure and a history of prior liver abscesses. The patient infected with KPN23, the iuc3+ , non-hmv strain with intermediate levels of virulence, did not have features of or risk factors for hvKP infection. Likewise, the patient infected with KPN80, the hmv isolate that lacked hvKP-associated genes, also did not have features of or risk factors for hvKP infection. Although the very small number of patients prevents conclusions from being drawn, the characteristics of the infections caused by KPN8, KPN49, KPN115, and KPN165 are consistent with their classification as hvKP.
Genomic analysis of bloodstream isolates from the United States
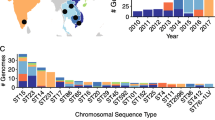
We next sought to examine a larger set of K. pneumoniae bloodstream isolates from more diverse sites across the United States for evidence of hvKP strains. For this purpose, we downloaded Illumina sequencing reads for all available bloodstream K. pneumoniae genomes (n = 963) from the NCBI database. Among these 963 sequences, we identified 139 unique STs, 94 unique KL, and 10 unique O-antigen serotypes (Fig. 7; Additional file 10: Fig. S10). The most common STs were endemic high-risk clones ST258 (n = 499) and ST307 (n = 63) (Additional file 10: Fig S10A, Additional file 16: Table S4). STs associated with hvKP were detected at a low prevalence (n = 6, 0.62%): ST23 (n = 4, 0.42%), ST380 (n = 1, 0.10%), and ST65 (n = 1, 0.10%). The most common KL-types were KL107 (n = 299), KL106 (n = 159), and KL102 (n = 69). KL106 and KL107 are associated with ST258, and KL102 is associated with ST307. The most common O antigen serotype was O2 (n = 640) which is associated with ST258, ST307, and ST147 (Additional file 10: Fig S10C, Additional file 16: Table S4). The diversity of this collection of isolates is relatively low for ST, KL, and O-antigen, with Simpson’s diversity indices of 0.72, 0.85, and 0.53, respectively. In comparison with the NMH collection, NCBI isolates are highly clonal and likely the result of reporting bias of clonal outbreaks and highly antibiotic-resistant isolates. For example, 43 (68%) of ST307 bloodstream isolates were from a single hospital system (Houston-Methodist) [25]. Several temporal trends were apparent among the 963 isolates. Since 2008, ST258 isolates have comprised a substantial number of the sequences deposited in NCBI, but the proportional representation of this ST has dropped in recent years (Additional file 11: Fig S11). ST307 isolates were first submitted in 2011 and continued to be submitted through 2020 (Additional file 11: Fig S11). ST147 isolates were first submitted in 2015 but accounted for 35% of sequences uploaded in 2021 (Additional file 11: Fig S11). It is unclear whether these differences represent changes in the epidemiology of K. pneumoniae or simply reporting biases.

Phylogenetic tree of United States K. pneumoniae bloodstream isolates. Maximum likelihood phylogenetic tree generated from core genome SNP loci in 1067 K. pneumoniae bloodstream isolates (NMH and NCBI). Sequence types, capsule loci, and the presence of virulence and antibiotic resistance genes are indicated. iuc = aerobactin biosynthesis genes, CR = contains a carbapenemase, ESBL = contains an extended-spectrum beta-lactamase gene, NMH = isolate was collected from Northwestern Memorial Hospital. The complete genome of NTUH-K2044 was used as the reference genome. Branch lengths are not to scale
Among these publicly available United States bloodstream sequences, antimicrobial resistance was remarkably common; 91.7% of isolates contained an extended-spectrum beta-lactamase (ESBL) gene or a carbapenemase gene, compared to 17.3% of isolates in the NMH collection (Fig. 7; Additional file 16: Table S4). The large proportion of antimicrobial-resistant high-risk clones suggests that this collection suffers from sampling bias, with preferential submission of drug-resistant isolates potentially from clonal outbreaks.
We searched the genomes of the 963 isolates for acquired virulence genes. We identified genes required for the biosynthesis of yersiniabactin in 345 (35.8%) isolates, with the most common variants being ybt17 (n = 179), ybt10 (n = 42), and ybt9 (n = 32) (Tables 1; Additional file 16: Table S4). Five isolates were predicted to contain a ybt4-containing plasmid (Additional file 12: Fig S12C). Colibactin biosynthetic genes were identified in 194 (20%) isolates, with clb3 variants being the most common. NMH isolates had more diverse variants of ybt than those found in the NCBI collection, suggesting that the ybt variants in the NCBI collection were largely driven by ICEKp10-containing ST258 isolates.
In addition to ybt and clb, the hvKP-associated loci iuc, iro, rmpADC, and rmpA2 were also examined. iuc was identified in 32 (3.3%), iro in 9 (0.9%), rmpADC in 14 (1.5%), and rmpA2 in 21 (2.2%) of the genomes (Table 1; Additional file 16: Table S4). The 32 iuc+ isolates were all predicted to contain a plasmid that fell into one of seven distinct groups, which in turn formed five plasmid network clusters (Additional file 12: Fig S12). Examination of the first of these clusters (Additional file 12: Fig. S12A) indicated that it was closely related to pK2044. A second cluster corresponded to KP52.145pII, a third to pTK421, and a fourth to aerobactin-encoding Escherichia coli plasmids (Additional file 12: Fig S12). A total of 25 (2.6%) isolates contained hypervirulence-associated lineages of iuc (iuc1 or iuc2), and either rmpA or rmpA2 indicating they are likely hvKP (Additional file 12: Fig S12A; Table 1; Additional file 16: Table S4). Among these, 18 isolates contained carbapenamase genes, indicating that these isolates are potential “convergent” isolates or carbapenem resistant-hvKP (Tables 1; Additional file 16: Table S4). Fourteen isolates were predicted to contain a hybrid antibiotic resistance-virulence plasmid (Additional file 12: Fig S12A) similar to those described previously in Europe [106, 107]. These data support the conjectures that hvKP strains are circulating in the United States and that some of these strains are both antibiotic-resistant and hypervirulent [67].
Discussion
We examined consecutive K. pneumoniae bloodstream isolates at a single United States medical center and found that they had significant diversity in sequence type, capsule type, antimicrobial resistance genes, and virulence factors. In total, there were 75 distinct sequence types in our collection of 104 isolates, demonstrating that genetically diverse K. pneumoniae strains have the potential to cause invasive infections. As expected, the majority (96.2%) were cKP isolates, of which 16.3% were MDR. However, hvKP isolates comprised 3.8% of this collection. These findings indicate that most K. pneumoniae strains at NMH are antibiotic-susceptible but that a substantial number of MDR and hvKP strains are also present.
MDR cKP pose a considerable therapeutic challenge to clinicians in that the delay in administration of active antimicrobial therapy may lead to poor outcomes [108, 109]. The prevalence of MDR cKP bloodstream infections varies dramatically globally ranging from 18% to -61% [110,111,112,113,114,115,116]. Our finding that 16.3% of K. pneumoniae bloodstream isolates were MDR and 11.5% XDR underscores the difficulty in choosing empiric antibiotic therapy for patients at risk for K. pneumoniae bacteremia. Much of this resistance was driven by ESBL (11.5%) and carbapenemase (8.7%) genes found on mobile elements. These proportions agree with a report from the National Healthcare Safety Network, which found that 5–25% of Klebsiella species cultured from patients in the United States between 2015 and 2017 were resistant to carbapenems [117]. In our study, a substantial number of MDR isolates (70.6%) were high-risk clones, confirming the role of these widely disseminated lineages in driving antibiotic resistance [13, 118]. Recently, two large studies evaluated the molecular characteristics of either ESBL or carbapenem-resistant isolates from U.S. hospitalized patients and found that high-risk clones ST258 and ST307 accounted for 27–64% and 7–36% of these isolates, respectively [25, 119]. At NMH, ST258 was the most abundant ST and accounted for 6% of isolates, yet only two ST307 isolates were identified among bloodstream isolates. ST258 isolates are split into two distinct sub-lineages based on capsule type and KPC allele; KL107 is associated with KPC-3 while KL106 is associated with KPC-2 [120]. Both sub-lineages are found in high abundance in the NCBI collection (Fig. 7). At NMH, ST258 isolates are primarily part of the KL107, KPC-3 sub-lineage (Additional file 13: Table S1, Fig. 7). These observations illustrate the burden of antibiotic resistance and clonal outbreaks of high-risk clones among U.S. K. pneumoniae isolates.
Intriguingly, four (3.8%) of the isolates in the NMH collection had features of hvKP. These isolates belonged to the established hvKP lineages ST23 and ST380 and to the re-emerging lineage ST66. Each harbored a plasmid similar to one of the two previously described hvKp virulence plasmids (pK2044 or KP52.145pII) [41,42,43]. These plasmids contained multiple hvKP-associated genetic loci (iuc, iro, and rmpA). The isolates were hmv and had LD50 values in mice consistent with hvKp (Fig. 6; Table 2). Three of the isolates (KPN49, KPN115, and KPN165) caused severe community-acquired infections with abscess formation (2 hepatic and 1 lung). Taken together, these data indicate that these four isolates are indeed hvKP and that multiple strains of hvKp are circulating in the Chicago region.
One strength of our approach is the use of a mouse model to quantify the in vivo virulence of potential hvKP isolates. Virulence in mice accurately distinguishes hvKP from cKP strains [40]. This approach allowed us to show that KPN23, a non-hmv isolate containing iuc but not rmpA, rmpA2, or iro, had an intermediate level of virulence between hvKP and cKP (Fig. 6). These findings suggest that some K. pneumoniae strains may not fall neatly into the categories of cKP or hvKP but may have more nuanced genotypes and phenotypes. Our experiments also demonstrated that KPN80, an hmv isolate lacking iuc, rmpA, rmpA2, and iro, had a low level of virulence consistent with cKP strains (Fig. 6). The characteristics of this isolate support previous reports indicating that hmv alone does not accurately predict hvKP [64, 81]. Together, these observations suggest that inferences about hvKP should be made with caution when examining only a single bacterial gene or trait.
Our findings also have implications for the pathogenesis of hvKP. They suggest that hmv caused by rmpA (e.g., KPN49) has dramatically different pathogenic consequences than hmv caused by a capsule locus mutation (KPN80) (Fig. 6; LD50 of 101.5 vs. 108.1 CFU, respectively). Likewise, our finding that increased levels of siderophore secretion were associated with increased in vivo virulence (Additional file 9: Fig S9) suggests that siderophores also contribute to the overall hvKP virulence phenotype. We note that these results are only associations, and that experiments using genetically defined mutants will be necessary to unravel the relative contributions of these genetically linked hvKP genes to virulence.
Knowledge of the epidemiology of hvKP has been hampered by the lack of a consensus definition for these strains. The two previously published U.S. surveillance studies dealt with this difficulty in different ways. In 2010, a study of 64 K. pneumoniae bloodstream isolates from two hospitals in the greater Houston area screened for isolates containing rmpA or wzy_K1 (a biomarker for a K1 capsule type) [39]. They identified 4 (6.3%) potential hvKP isolates using this approach. In 2018, a larger study in New York City examined K. pneumoniae isolates from 462 patients for the presence of both rmpA and iucA [37]. This study identified 17 (3.7%) potential hvKP isolates containing these two genes. In contrast, a 2013 report from China, where hvKP is endemic, described an hvKP prevalence of 37.8% using the presence of the iuc locus to define hvKP [65]. Similarly, a recent genomic surveillance study of seven countries in South and Southeast Asia analyzed the sequences of 365 K. pneumoniae bloodstream isolates collected from 2010–2017 [69]. Sequencing revealed that 95 (26%) were iuc+ . Of these, 63 (17%) also contained either rmpA or rmpA2. Precise definitions that accurately distinguish hvKP from cKP and at the same time capture the diversity of hvKP will be necessary in future studies to evaluate the prevalence and clinical consequences of hvKP infections.
Thus far, antimicrobial-resistant hvKp infections appear to be rare in the United States, as only 1.9% of bloodstream isolates from NCBI had features of carbapenem-resistant hvKP and none of the iuc+ isolates identified at NMH contained accessory antimicrobial resistance genes. However, the KP52.145pII-like plasmids of KPN49 and KPN165 have acquired putative conjugation genes, suggesting that these hypervirulence plasmids are capable of horizontal transmission. Indeed, KPN49 and KPN165 are phylogenetically distinct (ST66 and ST380, Additional file 8: Fig S8), consistent with horizontal transfer of this plasmid. This finding is worrisome and suggests that these plasmids may be prone to transfer to other K. pneumoniae strains, including MDR strains. Additional studies are necessary to demonstrate whether these conjugation genes are functional and indeed confer transmissibility on this hypervirulence plasmid.
Conclusions
In this study, consecutive K. pneumoniae bloodstream isolates were collected from 2015-2017 at a single U.S. medical center. Whole genome sequencing revealed that these isolates are diverse in sequence type, capsule type, and virulence gene content. The majority (79.9%) of isolates were cKP strains susceptible to most antibiotics. However, MDR cKP isolates and hvKP isolates comprised 16.3% and 3.8% of this collection, respectively. These findings confirm previous reports that MDR cKP are common in the U.S. and that several hvKP strains are also circulating in the Chicago region. Examination of the larger NCBI database confirmed that strains likely to be hvKP are circulating in the U.S.
Availability of data and materials
The whole-genome assemblies have been deposited at GenBank under BioProject number PRJNA788509. Assembly accession numbers are included in Additional file 16: Table S4, Additional file 17: Table S5, and Additional file 18: Table S6. Kleborate and MIC data are included in Additional file 13: Table S1.
Abbreviations
- ST:
-
Sequence type
- KL:
-
Capsule loci
- MDR:
-
Multidrug-resistance
- XDR:
-
Extensively drug-resistant
- ESBL:
-
Extended-spectrum beta-lactamase
- cKP:
-
Classical Klebsiella pneumoniae
- hvKP:
-
Hypervirulent Klebsiella pneumoniae
- NMH:
-
Northwestern Memorial Hospital
- PAL-1:
-
Pathogenicity loci 1
- PAL-2:
-
Pathogenicity loci 2
- ICE:
-
Integrative and conjugative element
- hmv:
-
Hypermucoviscous
- KpSC:
-
Klebsiella pneumoniae species complex
- MIC:
-
Minimum inhibitory concentration
- TMP/SMX:
-
Trimethoprim and sulfamethoxazole
- AME:
-
Aminoglycoside modifying enzyme
- LD50:
-
50% Lethal dose
- CAS:
-
Chrome-azurol S
- NCBI:
-
National Center for Biotechnology Information
References
Podschun R, Ullmann U. Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin Microbiol Rev. 1998;11:589–603.
Dhesi Z, Enne VI, Brealey D, Livermore DM, High J, Russell C, Colles A, Kandil H, Mack D, Martin D, Page V, Parker R, Roulston K, Singh S, Wey E, Swart AM, Stirling S, Barber JA, O’Grady J, Gant VA. Organisms causing secondary pneumonias in COVID-19 patients at 5 UK ICUs as detected with the FilmArray test. medRvix. 2020. https://doi.org/10.1101/2020.06.22.20131573.
Li J, Ren J, Wang W, Wang G, Gu G, Wu X, Wang Y, Huang M, Li J. Risk factors and clinical outcomes of hypervirulent Klebsiella pneumoniae induced bloodstream infections. Eur J Clin Microbiol Infect Dis. 2018;37:679–89.
Meatherall BL, Gregson D, Ross T, Pitout JD, Laupland KB. Incidence, risk factors, and outcomes of Klebsiella pneumoniae bacteremia. Am J Med. 2009;122:866–73.
Girometti N, Lewis RE, Giannella M, Ambretti S, Bartoletti M, Tedeschi S, Tumietto F, Cristini F, Trapani F, Gaibani P, Viale P. Klebsiella pneumoniae bloodstream infection: epidemiology and impact of inappropriate empirical therapy. Medicine (Baltimore). 2014;93:298–309.
Russo TA, Marr CM. Hypervirulent Klebsiella pneumoniae. Clin Microbiol Rev. 2019;32.
Marcade G, Brisse S, Bialek S, Marcon E, Leflon-Guibout V, Passet V, Moreau R, Nicolas-Chanoine MH. The emergence of multidrug-resistant Klebsiella pneumoniae of international clones ST13, ST16, ST35, ST48 and ST101 in a teaching hospital in the Paris region. Epidemiol Infect. 2013;141:1705–12.
Meng X, Yang J, Duan J, Liu S, Huang X, Wen X, Huang X, Fu C, Li J, Dou Q, Liu Y, Wang J, Yan Q, Zou M, Liu W, Peng Z, Chen L, Li C, Wu A. Assessing molecular epidemiology of carbapenem-resistant Klebsiella pneumoniae (CR-KP) with MLST and MALDI-TOF in Central China. Sci Rep. 2019;9:2271.
Roulston KJ, Bharucha T, Turton JF, Hopkins KL, Mack DJF. A case of NDM-carbapenemase-producing hypervirulent Klebsiella pneumoniae sequence type 23 from the UK. JMM Case Rep. 2018;5: e005130.
Shen D, Ma G, Li C, Jia X, Qin C, Yang T, Wang L, Jiang X, Ding N, Zhang X, Yue L, Yin Z, Zeng L, Zhao Y, Zhou D, Chen F. Emergence of a multidrug-resistant hypervirulent Klebsiella pneumoniae sequence type 23 strain with a rare bla CTX-M-24-harboring virulence plasmid. Antimicrob Agents Chemother. 2019;63.
Shu L, Dong N, Lu J, Zheng Z, Hu J, Zeng W, Sun Q, Chan EW, Zhou H, Hu F, Chen S, Zhang R. Emergence of OXA-232 carbapenemase-producing Klebsiella pneumoniae that carries a pLVPK-like virulence plasmid among elderly patients in China. Antimicrob Agents Chemother. 2019;63.
Lester CH, Olsen SS, Jakobsen L, Arpi M, Fuursted K, Hansen DS, Heltberg O, Holm A, Hojbjerg T, Jensen KT, Johansen HK, Justesen US, Kemp M, Knudsen JD, Roder B, Frimodt-Moller N, Hammerum AM. Emergence of extended-spectrum beta-lactamase (ESBL)-producing Klebsiella pneumoniae in Danish hospitals; this is in part explained by spread of two CTX-M-15 clones with multilocus sequence types 15 and 16 in Zealand. Int J Antimicrob Agents. 2011;38:180–2.
Navon-Venezia S, Kondratyeva K, Carattoli A. Klebsiella pneumoniae: a major worldwide source and shuttle for antibiotic resistance. FEMS Microbiol Rev. 2017;41:252–75.
Magiorakos AP, Srinivasan A, Carey RB, Carmeli Y, Falagas ME, Giske CG, Harbarth S, Hindler JF, Kahlmeter G, Olsson-Liljequist B, Paterson DL, Rice LB, Stelling J, Struelens MJ, Vatopoulos A, Weber JT, Monnet DL. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect. 2012;18:268–81.
Turton J, Davies F, Turton J, Perry C, Payne Z, Pike R. Hybrid Resistance and Virulence Plasmids in "High-Risk" Clones of Klebsiella pneumoniae, Including Those Carrying blaNDM-5. Microorganisms. 2019; 7.
Goncalves GB, Furlan JPR, Vespero EC, Pelisson M, Stehling EG, Pitondo-Silva A. Spread of multidrug-resistant high-risk Klebsiella pneumoniae clones in a tertiary hospital from southern Brazil. Infect Genet Evol. 2017;56:1–7.
Nascimento T, Cantamessa R, Melo L, Fernandes MR, Fraga E, Dropa M, Sato MIZ, Cerdeira L, Lincopan N. International high-risk clones of Klebsiella pneumoniae KPC-2/CC258 and Escherichia coli CTX-M-15/CC10 in urban lake waters. Sci Total Environ. 2017;598:910–5.
Oteo J, Perez-Vazquez M, Bautista V, Ortega A, Zamarron P, Saez D, Fernandez-Romero S, Lara N, Ramiro R, Aracil B, Campos J, Spanish Antibiotic Resistance Surveillance Program Collaborating G. The spread of KPC-producing Enterobacteriaceae in Spain: WGS analysis of the emerging high-risk clones of Klebsiella pneumoniae ST11/KPC-2, ST101/KPC-2 and ST512/KPC-3. J Antimicrob Chemother. 2016;71:3392–9.
Rodriguez I, Novais A, Lira F, Valverde A, Curiao T, Martinez JL, Baquero F, Canton R, Coque TM. Antibiotic-resistant Klebsiella pneumoniae and Escherichia coli high-risk clones and an IncFII(k) mosaic plasmid hosting Tn1 (blaTEM-4) in isolates from 1990 to 2004. Antimicrob Agents Chemother. 2015;59:2904–8.
Oliveira S, Moura RA, Silva KC, Pavez M, McCulloch JA, Dropa M, Matte MH, Mamizuka EM, Sato MI, Pestana de Castro AF, Lincopan N. Isolation of KPC-2-producing Klebsiella pneumoniae strains belonging to the high-risk multiresistant clonal complex 11 (ST437 and ST340) in urban rivers. J Antimicrob Chemother. 2014;69:849–52.
Wyres KL, Lam MMC, Holt KE. Population genomics of Klebsiella pneumoniae. Nat Rev Microbiol. 2020;18:344–59.
Chmelnitsky I, Doniger T, Shklyar M, Naparstek L, Banin E, Edgar R, Carmeli Y. Draft genome sequence of an extremely drug-resistant KPC-producing Klebsiella pneumoniae ST258 epidemic strain. J Bacteriol. 2012;194:6014.
Chen L, Chavda KD, Mediavilla JR, Zhao Y, Fraimow HS, Jenkins SG, Levi MH, Hong T, Rojtman AD, Ginocchio CC, Bonomo RA, Kreiswirth BN. Multiplex real-time PCR for detection of an epidemic KPC-producing Klebsiella pneumoniae ST258 clone. Antimicrob Agents Chemother. 2012;56:3444–7.
Bogdanovich T, Adams-Haduch JM, Tian GB, Nguyen MH, Kwak EJ, Muto CA, Doi Y. Colistin-resistant, Klebsiella pneumoniae carbapenemase (KPC)-producing Klebsiella pneumoniae belonging to the international epidemic clone ST258. Clin Infect Dis. 2011;53:373–6.
Long SW, Olsen RJ, Eagar TN, Beres SB, Zhao P, Davis JJ, Brettin T, Xia F, Musser JM. Population Genomic Analysis of 1,777 Extended-Spectrum Beta-Lactamase-Producing Klebsiella pneumoniae Isolates, Houston, Texas: Unexpected Abundance of Clonal Group 307. mBio 2017;8.
Tacconelli E, Magrini N. Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. https://www.who.int/medicines/publications/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf. Accessed 2 Oct. 2017.
Talbot GH, Bradley J, Edwards JE Jr, Gilbert D, Scheld M, Bartlett JG. Bad bugs need drugs: an update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America. Clin Infect Dis. 2006;42:657–68.
CDC. Antibiotic resistance threats in the United States, 2013. http://www.cdc.gov/drugresistance/threat-report-2013/. Accessed 18 Sept. 2013.
Liu YC, Cheng DL, Lin CL. Klebsiella pneumoniae liver abscess associated with septic endophthalmitis. Arch Intern Med. 1986;146:1913–6.
Fang FC, Sandler N, Libby SJ. Liver abscess caused by magA+ Klebsiella pneumoniae in North America. J Clin Microbiol. 2005;43:991–2.
Lam MMC, Wyres KL, Duchene S, Wick RR, Judd LM, Gan YH, Hoh CH, Archuleta S, Molton JS, Kalimuddin S, Koh TH, Passet V, Brisse S, Holt KE. Population genomics of hypervirulent Klebsiella pneumoniae clonal-group 23 reveals early emergence and rapid global dissemination. Nat Commun. 2018;9:2703.
Turton JF, Englender H, Gabriel SN, Turton SE, Kaufmann ME, Pitt TL. Genetically similar isolates of Klebsiella pneumoniae serotype K1 causing liver abscesses in three continents. J Med Microbiol. 2007;56:593–7.
Siu LK, Fung CP, Chang FY, Lee N, Yeh KM, Koh TH, Ip M. Molecular typing and virulence analysis of serotype K1 Klebsiella pneumoniae strains isolated from liver abscess patients and stool samples from noninfectious subjects in Hong Kong, Singapore, and Taiwan. J Clin Microbiol. 2011;49:3761–5.
Vila A, Cassata A, Pagella H, Amadio C, Yeh KM, Chang FY, Siu LK. Appearance of Klebsiella pneumoniae liver abscess syndrome in Argentina: case report and review of molecular mechanisms of pathogenesis. Open Microbiol J. 2011;5:107–13.
Pastagia M, Arumugam V. Klebsiella pneumoniae liver abscesses in a public hospital in Queens, New York. Travel Med Infect Dis. 2008;6:228–33.
Rahimian J, Wilson T, Oram V, Holzman RS. Pyogenic liver abscess: recent trends in etiology and mortality. Clin Infect Dis. 2004;39:1654–9.
Parrott AM, Shi J, Aaron J, Green DA, Whittier S, Wu F. Detection of multiple hypervirulent Klebsiella pneumoniae strains in a New York City hospital through screening of virulence genes. Clin Microbiol Infect. 2020. https://doi.org/10.1016/j.cmi.2020.05.012.
Peirano G, Pitout JD, Laupland KB, Meatherall B, Gregson DB. Population-based surveillance for hypermucoviscosity Klebsiella pneumoniae causing community-acquired bacteremia in Calgary, Alberta. Can J Infect Dis Med Microbiol. 2013;24:e61–4.
Chou A, Nuila RE, Franco LM, Stager CE, Atmar RL, Zechiedrich L. Prevalence of hypervirulent Klebsiella pneumoniae-associated genes rmpA and magA in two tertiary hospitals in Houston, TX, USA. J Med Microbiol. 2016;65:1047–8.
Russo TA, Olson R, Fang CT, Stoesser N, Miller M, MacDonald U, Hutson A, Barker JH, La Hoz RM, Johnson JR. Identification of biomarkers for differentiation of hypervirulent Klebsiella pneumoniae from Classical K. pneumoniae. J Clin Microbiol. 2018;56.
Chen YT, Chang HY, Lai YC, Pan CC, Tsai SF, Peng HL. Sequencing and analysis of the large virulence plasmid pLVPK of Klebsiella pneumoniae CG43. Gene. 2004;337:189–98.
Nassif X, Fournier JM, Arondel J, Sansonetti PJ. Mucoid phenotype of Klebsiella pneumoniae is a plasmid-encoded virulence factor. Infect Immun. 1989;57:546–52.
Wu KM, Li LH, Yan JJ, Tsao N, Liao TL, Tsai HC, Fung CP, Chen HJ, Liu YM, Wang JT, Fang CT, Chang SC, Shu HY, Liu TT, Chen YT, Shiau YR, Lauderdale TL, Su IJ, Kirby R, Tsai SF. Genome sequencing and comparative analysis of Klebsiella pneumoniae NTUH-K2044, a strain causing liver abscess and meningitis. J Bacteriol. 2009;191:4492–501.
Lam MMC, Wyres KL, Judd LM, Wick RR, Jenney A, Brisse S, Holt KE. Tracking key virulence loci encoding aerobactin and salmochelin siderophore synthesis in Klebsiella pneumoniae. Genome Med. 2018;10:77.
Wyres KL, Wick RR, Judd LM, Froumine R, Tokolyi A, Gorrie CL, Lam MMC, Duchene S, Jenney A, Holt KE. Distinct evolutionary dynamics of horizontal gene transfer in drug resistant and virulent clones of Klebsiella pneumoniae. PLoS Genet. 2019;15: e1008114.
Shon AS, Bajwa RP, Russo TA. Hypervirulent (hypermucoviscous) Klebsiella pneumoniae: a new and dangerous breed. Virulence. 2013;4:107–18.
Vornhagen J, Bassis CM, Ramakrishnan S, Hein R, Mason S, Bergman Y, Sunshine N, Fan Y, Holmes CL, Timp W, Schatz MC, Young VB, Simner PJ, Bachman MA. A plasmid locus associated with Klebsiella clinical infections encodes a microbiome-dependent gut fitness factor. PLoS Pathog. 2021;17:e1009537.
Lery LM, Frangeul L, Tomas A, Passet V, Almeida AS, Bialek-Davenet S, Barbe V, Bengoechea JA, Sansonetti P, Brisse S, Tournebize R. Comparative analysis of Klebsiella pneumoniae genomes identifies a phospholipase D family protein as a novel virulence factor. BMC Biol. 2014;12:41.
Kochan TJ, Ozer EA, Pincus NB, Fitzpatrick MA, Hauser AR. Complete Genome Sequence of Klebsiella pneumoniae Strain TK421, a Conjugative Hypervirulent Isolate. Microbiol Resour Announc. 2020;9.
Hsieh PF, Lin TL, Lee CZ, Tsai SF, Wang JT. Serum-induced iron-acquisition systems and TonB contribute to virulence in Klebsiella pneumoniae causing primary pyogenic liver abscess. J Infect Dis. 2008;197:1717–27.
Russo TA, Olson R, MacDonald U, Beanan J, Davidson BA. Aerobactin, but not yersiniabactin, salmochelin, or enterobactin, enables the growth/survival of hypervirulent (hypermucoviscous) Klebsiella pneumoniae ex vivo and in vivo. Infect Immun. 2015;83:3325–33.
Lawlor MS, Hsu J, Rick PD, Miller VL. Identification of Klebsiella pneumoniae virulence determinants using an intranasal infection model. Mol Microbiol. 2005;58:1054–73.
Xiong H, Carter RA, Leiner IM, Tang YW, Chen L, Kreiswirth BN, Pamer EG. Distinct contributions of neutrophils and CCR2+ monocytes to pulmonary clearance of different Klebsiella pneumoniae strains. Infect Immun. 2015;83:3418–27.
Lam MMC, Wick RR, Wyres KL, Gorrie CL, Judd LM, Jenney AWJ, Brisse S, Holt KE. Genetic diversity, mobilisation and spread of the yersiniabactin-encoding mobile element ICEKp in Klebsiella pneumoniae populations. Microb Genom. 2018;4.
Bachman MA, Oyler JE, Burns SH, Caza M, Lepine F, Dozois CM, Weiser JN. Klebsiella pneumoniae yersiniabactin promotes respiratory tract infection through evasion of lipocalin 2. Infect Immun. 2011;79:3309–16.
Merlet A, Cazanave C, Dutronc H, de Barbeyrac B, Brisse S, Dupon M. Primary liver abscess due to CC23-K1 virulent clone of Klebsiella pneumoniae in France. Clin Microbiol Infect. 2012;18:E338–9.
Patel PK, Russo TA, Karchmer AW. Hypervirulent Klebsiella pneumoniae. Open Forum Infect Dis. 2014;1:ofu028.
Pomakova DK, Hsiao CB, Beanan JM, Olson R, MacDonald U, Keynan Y, Russo TA. Clinical and phenotypic differences between classic and hypervirulent Klebsiella pneumonia: an emerging and under-recognized pathogenic variant. Eur J Clin Microbiol Infect Dis. 2012;31:981–9.
Russo TA, Shon AS, Beanan JM, Olson R, MacDonald U, Pomakov AO, Visitacion MP. Hypervirulent K. pneumoniae secretes more and more active iron-acquisition molecules than “classical” K. pneumoniae thereby enhancing its virulence. PLoS ONE. 2011;6:e26734.
Sachdev DD, Yin MT, Horowitz JD, Mukkamala SK, Lee SE, Ratner AJ. Klebsiella pneumoniae K1 liver abscess and septic endophthalmitis in a U.S. resident. J Clin Microbiol. 2013;51:1049–51.
Shon AS, Russo TA. Hypervirulent Klebsiella pneumoniae: the next superbug? Future Microbiol. 2012;7:669–71.
Fang CT, Chuang YP, Shun CT, Chang SC, Wang JT. A novel virulence gene in Klebsiella pneumoniae strains causing primary liver abscess and septic metastatic complications. J Exp Med. 2004;199:697–705.
Lee CH, Liu JW, Su LH, Chien CC, Li CC, Yang KD. Hypermucoviscosity associated with Klebsiella pneumoniae-mediated invasive syndrome: a prospective cross-sectional study in Taiwan. Int J Infect Dis. 2010;14:e688–92.
Krapp F, Morris AR, Ozer EA, Hauser AR. Virulence characteristics of carbapenem-resistant Klebsiella pneumoniae strains from patients with necrotizing skin and soft tissue infections. Sci Rep. 2017;7:13533.
Zhang Y, Zhao C, Wang Q, Wang X, Chen H, Li H, Zhang F, Li S, Wang R, Wang H. High prevalence of hypervirulent Klebsiella pneumoniae infection in China: geographic distribution, clinical characteristics, and antimicrobial resistance. Antimicrob Agents Chemother. 2016;60:6115–20.
Harada S, Doi Y. Hypervirulent Klebsiella pneumoniae: a Call for Consensus Definition and International Collaboration. J Clin Microbiol. 2018;56.
Karlsson M, Stanton RA, Ansari U, McAllister G, Chan MY, Sula E, Grass JE, Duffy N, Anacker ML, Witwer ML, Rasheed JK, Elkins CA, Halpin AL. Identification of a carbapenemase-producing hypervirulent Klebsiella pneumoniae isolate in the United States. Antimicrob Agents Chemother. 2019;63.
Liu C, Du P, Xiao N, Ji F, Russo TA, Guo J. Hypervirulent Klebsiella pneumoniae is emerging as an increasingly prevalent K. pneumoniae pathotype responsible for nosocomial and healthcare-associated infections in Beijing, China. Virulence. 2020;11:1215–24.
Wyres KL, Nguyen TNT, Lam MMC, Judd LM, van Vinh CN, Dance DAB, Ip M, Karkey A, Ling CL, Miliya T, Newton PN, Lan NPH, Sengduangphachanh A, Turner P, Veeraraghavan B, Vinh PV, Vongsouvath M, Thomson NR, Baker S, Holt KE. Genomic surveillance for hypervirulence and multi-drug resistance in invasive Klebsiella pneumoniae from South and Southeast Asia. Genome Med. 2020;12:11.
Leverstein-van Hall MA, Fluit AC, Paauw A, Box AT, Brisse S, Verhoef J. Evaluation of the Etest ESBL and the BD Phoenix, VITEK 1, and VITEK 2 automated instruments for detection of extended-spectrum beta-lactamases in multiresistant Escherichia coli and Klebsiella spp. J Clin Microbiol. 2002;40:3703–11.
Suravaram S, Hada V, Ahmed SI. Comparison of antimicrobial susceptibility interpretation among Enterobacteriaceae using CLSI and EUCAST breakpoints. Indian J Med Microbiol. 2021;39:315–9.
Heil EL, Johnson JK. Impact of CLSI breakpoint changes on microbiology laboratories and antimicrobial stewardship programs. J Clin Microbiol. 2016;54:840–4.
Huang CC, Chen YS, Toh HS, Lee YL, Liu YM, Ho CM, Lu PL, Liu CE, Chen YH, Wang JH, Tang HJ, Yu KW, Liu YC, Chuang YC, Xu Y, Ni Y, Ko WC, Hsueh PR. Impact of revised CLSI breakpoints for susceptibility to third-generation cephalosporins and carbapenems among Enterobacteriaceae isolates in the Asia-Pacific region: results from the Study for Monitoring Antimicrobial Resistance Trends (SMART), 2002–2010. Int J Antimicrob Agents. 2012;40(Suppl):S4-10.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Prjibelski A, Antipov D, Meleshko D, Lapidus A, Korobeynikov A. Using SPAdes De Novo Assembler. Curr Protoc Bioinformatics. 2020;70: e102.
Cherny KE, Ozer EA, Kochan TJ, Kociolek LK. Complete genome sequence of Clostridium innocuum Strain ATCC 14501. Microbiol Resour Announc. 2020;9.
Cherny KE, Ozer EA, Kochan TJ, Johnson S, Kociolek LK. Complete genome sequence of Clostridium innocuum strain LC-LUMC-CI-001, isolated from a patient with recurrent antibiotic-associated diarrhea. Microbiol Resour Announc. 2020; 9.
Wick RR, Judd LM, Holt KE. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 2019;20:129.
Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, Lomsadze A, Pruitt KD, Borodovsky M, Ostell J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016;44:6614–24.
Hadano Y. String test. BMJ Case Rep. 2013.
Walker KA, Miller VL. The intersection of capsule gene expression, hypermucoviscosity and hypervirulence in Klebsiella pneumoniae. Curr Opin Microbiol. 2020;54:95–102.
Price MN, Dehal PS, Arkin AP. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5: e9490.
Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49:W293–6.
Wick RR, Heinz E, Holt KE, Wyres KL. Kaptive Web: user-friendly capsule and lipopolysaccharide serotype prediction for Klebsiella Genomes. J Clin Microbiol. 2018; 56.
Robertson J, Nash JHE. 2018. MOB-suite: software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb Genom. 2018; 4.
Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12:402.
Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S, Phillippy AM. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016;17:132.
Carattoli A, Zankari E, Garcia-Fernandez A, Voldby Larsen M, Lund O, Villa L, Moller Aarestrup F, Hasman H. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother. 2014;58:3895–903.
Ritz C, Baty F, Streibig JC, Gerhard D. Dose–response analysis using R. PLoS ONE. 2015;10: e0146021.
Holt KE, Wertheim H, Zadoks RN, Baker S, Whitehouse CA, Dance D, Jenney A, Connor TR, Hsu LY, Severin J, Brisse S, Cao H, Wilksch J, Gorrie C, Schultz MB, Edwards DJ, Nguyen KV, Nguyen TV, Dao TT, Mensink M, Minh VL, Nhu NT, Schultsz C, Kuntaman K, Newton PN, Moore CE, Strugnell RA, Thomson NR. Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc Natl Acad Sci USA. 2015;112:E3574–81.
Conlan S, Park M, Deming C, Thomas PJ, Young AC, Coleman H, Sison C, Program NCS, Weingarten RA, Lau AF, Dekker JP, Palmore TN, Frank KM, Segre JA. Plasmid dynamics in KPC-positive Klebsiella pneumoniae during long-term patient colonization. mBio. 2016; 7.
Lawlor MS, O’Connor C, Miller VL. Yersiniabactin is a virulence factor for Klebsiella pneumoniae during pulmonary infection. Infect Immun. 2007;75:1463–72.
Marsh JW, Mustapha MM, Griffith MP, Evans DR, Ezeonwuka C, Pasculle AW, Shutt KA, Sundermann A, Ayres AM, Shields RK, Babiker A, Cooper VS, Van Tyne D, Harrison LH. Evolution of outbreak-causing carbapenem-resistant Klebsiella pneumoniae ST258 at a Tertiary Care Hospital over 8 Years. mBio 2019;10.
Yu WL, Lee MF, Tang HJ, Chang MC, Chuang YC. Low prevalence of rmpA and high tendency of rmpA mutation correspond to low virulence of extended spectrum beta-lactamase-producing Klebsiella pneumoniae isolates. Virulence. 2015;6:162–72.
Rodrigues C, d'Humieres C, Papin G, Passet V, Ruppe E, Brisse S. Community-acquired infection caused by the uncommon hypervirulent Klebsiella pneumoniae ST66-K2 lineage. Microb Genom. 2020;6.
Agard MJ, Ozer EA, Morris AR, Piseaux R, Hauser AR. A genomic approach to identify Klebsiella pneumoniae and acinetobacter baumannii strains with enhanced competitive fitness in the lungs during multistrain pneumonia. Infect Immun. 2019;87.
Russo TA, Olson R, Macdonald U, Metzger D, Maltese LM, Drake EJ, Gulick AM. Aerobactin mediates virulence and accounts for increased siderophore production under iron-limiting conditions by hypervirulent (hypermucoviscous) Klebsiella pneumoniae. Infect Immun. 2014;82:2356–67.
Kamau E, Allyn PR, Beaird OE, Ward KW, Kwan N, Garner OB, Yang S. Endogenous endophthalmitis caused by ST66-K2 hypervirulent Klebsiella pneumoniae, United States. Emerg Infect Dis. 2021;27:2215–8.
Klaper K, Wendt S, Lubbert C, Lippmann N, Pfeifer Y, Werner G. Hypervirulent Klebsiella pneumoniae of Lineage ST66-K2 Caused Tonsillopharyngitis in a German Patient. Microorganisms 2021; 9.
Russo TA, MacDonald U. The Galleria mellonella infection model does not accurately differentiate between hypervirulent and classical Klebsiella pneumoniae. mSphere 2020;5.
Huang Y, Rana AP, Wenzler E, Ozer EA, Krapp F, Bulitta JB, Hauser AR, Bulman ZP. Aminoglycoside-resistance gene signatures are predictive of aminoglycoside MICs for carbapenem-resistant Klebsiella pneumoniae. J Antimicrob Chemother. 2022;77:356–63.
Ernst CM, Braxton JR, Rodriguez-Osorio CA, Zagieboylo AP, Li L, Pironti A, Manson AL, Nair AV, Benson M, Cummins K, Clatworthy AE, Earl AM, Cosimi LA, Hung DT. Adaptive evolution of virulence and persistence in carbapenem-resistant Klebsiella pneumoniae. Nat Med. 2020;26:705–11.
Han SH. Review of hepatic abscess from Klebsiella pneumoniae. An association with diabetes mellitus and septic endophthalmitis. West J Med. 1995;162:220–4.
Huang CR, Lu CH, Chang HW, Lee PY, Lin MW, Chang WN. Community-acquired spontaneous bacterial meningitis in adult diabetic patients: an analysis of clinical characteristics and prognostic factors. Infection. 2002;30:346–50.
Wang JH, Liu YC, Lee SS, Yen MY, Chen YS, Wang JH, Wann SR, Lin HH. Primary liver abscess due to Klebsiella pneumoniae in Taiwan. Clin Infect Dis. 1998;26:1434–8.
Martin MJ, Corey BW, Sannio F, Hall LR, MacDonald U, Jones BT, Mills EG, Harless C, Stam J, Maybank R, Kwak Y, Schaufler K, Becker K, Hubner NO, Cresti S, Tordini G, Valassina M, Cusi MG, Bennett JW, Russo TA, McGann PT, Lebreton F, Docquier JD. Anatomy of an extensively drug-resistant Klebsiella pneumoniae outbreak in Tuscany, Italy. Proc Natl Acad Sci USA. 2021; 118.
Heiden SE, Hubner NO, Bohnert JA, Heidecke CD, Kramer A, Balau V, Gierer W, Schaefer S, Eckmanns T, Gatermann S, Eger E, Guenther S, Becker K, Schaufler K. A Klebsiella pneumoniae ST307 outbreak clone from Germany demonstrates features of extensive drug resistance, hypermucoviscosity, and enhanced iron acquisition. Genome Med. 2020;12:113.
Lambregts MMC, Wijnakker R, Bernards AT, Visser LG, Cessie SL, Boer MGJ. Mortality after delay of adequate empiric antimicrobial treatment of bloodstream infection. J Clin Med. 2020; 9.
Weiss SL, Fitzgerald JC, Balamuth F, Alpern ER, Lavelle J, Chilutti M, Grundmeier R, Nadkarni VM, Thomas NJ. Delayed antimicrobial therapy increases mortality and organ dysfunction duration in pediatric sepsis. Crit Care Med. 2014;42:2409–17.
Xiao SZ, Wang S, Wu WM, Zhao SY, Gu FF, Ni YX, Guo XK, Qu JM, Han LZ. The resistance phenotype and molecular epidemiology of Klebsiella pneumoniae in bloodstream infections in Shanghai, China, 2012–2015. Front Microbiol. 2017;8:250.
Kim YK, Pai H, Lee HJ, Park SE, Choi EH, Kim J, Kim JH, Kim EC. Bloodstream infections by extended-spectrum beta-lactamase-producing Escherichia coli and Klebsiella pneumoniae in children: epidemiology and clinical outcome. Antimicrob Agents Chemother. 2002;46:1481–91.
Edelstein M, Pimkin M, Palagin I, Edelstein I, Stratchounski L. Prevalence and molecular epidemiology of CTX-M extended-spectrum beta-lactamase-producing Escherichia coli and Klebsiella pneumoniae in Russian hospitals. Antimicrob Agents Chemother. 2003;47:3724–32.
Marra AR, Wey SB, Castelo A, Gales AC, Cal RG, Filho JR, Edmond MB, Pereira CA. Nosocomial bloodstream infections caused by Klebsiella pneumoniae: impact of extended-spectrum beta-lactamase (ESBL) production on clinical outcome in a hospital with high ESBL prevalence. BMC Infect Dis. 2006;6:24.
Tumbarello M, Spanu T, Sanguinetti M, Citton R, Montuori E, Leone F, Fadda G, Cauda R. Bloodstream infections caused by extended-spectrum-beta-lactamase-producing Klebsiella pneumoniae: risk factors, molecular epidemiology, and clinical outcome. Antimicrob Agents Chemother. 2006;50:498–504.
Xu M, Fu Y, Kong H, Chen X, Chen Y, Li L, Yang Q. Bloodstream infections caused by Klebsiella pneumoniae: prevalence of blaKPC, virulence factors and their impacts on clinical outcome. BMC Infect Dis. 2018;18:358.
Anonymous. 2019. English Surveillance Programme for Antimicrobial Utilisation and Resistance (ESPAUR). https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/843129/English_Surveillance_Programme_for_Antimicrobial_Utilisation_and_Resistance_2019.pdf.
Weiner-Lastinger LM, Abner S, Benin AL, Edwards JR, Kallen AJ, Karlsson M, Magill SS, Pollock D, See I, Soe MM, Walters MS, Dudeck MA. Antimicrobial-resistant pathogens associated with pediatric healthcare-associated infections: summary of data reported to the National Healthcare Safety Network, 2015–2017. Infect Control Hosp Epidemiol. 2020;41:19–30.
Andrade LN, Vitali L, Gaspar GG, Bellissimo-Rodrigues F, Martinez R, Darini AL. Expansion and evolution of a virulent, extensively drug-resistant (polymyxin B-resistant), QnrS1-, CTX-M-2-, and KPC-2-producing Klebsiella pneumoniae ST11 international high-risk clone. J Clin Microbiol. 2014;52:2530–5.
van Duin D, Arias CA, Komarow L, Chen L, Hanson BM, Weston G, Cober E, Garner OB, Jacob JT, Satlin MJ, Fries BC, Garcia-Diaz J, Doi Y, Dhar S, Kaye KS, Earley M, Hujer AM, Hujer KM, Domitrovic TN, Shropshire WC, Dinh A, Manca C, Luterbach CL, Wang M, Paterson DL, Banerjee R, Patel R, Evans S, Hill C, Arias R, Chambers HF, Fowler VG Jr, Kreiswirth BN, Bonomo RA, Multi-Drug Resistant Organism Network I. Molecular and clinical epidemiology of carbapenem-resistant Enterobacterales in the USA (CRACKLE-2): a prospective cohort study. Lancet Infect Dis. 2020;20:731–41.
Deleo FR, Chen L, Porcella SF, Martens CA, Kobayashi SD, Porter AR, Chavda KD, Jacobs MR, Mathema B, Olsen RJ, Bonomo RA, Musser JM, Kreiswirth BN. Molecular dissection of the evolution of carbapenem-resistant multilocus sequence type 258 Klebsiella pneumoniae. Proc Natl Acad Sci U S A. 2014;111:4988–93.
Acknowledgements
The authors thank Adam Ratner for graciously providing us with the strain hvKP5. This work was uploaded as a preprint (https://doi.org/10.1101/2020.12.04.410209).
Funding
This work was funded by American Heart Association grant 837089 (T.K.), Chicago Biomedical Consortium Catalyst Award (A.H.), and National Institute of Health grants: T32 AI007476 (T.K.), R01 AI118257 (A.H.), R21 AI153953 (A.H.), K24 AI104831 (A.H.), R21 AI164254 (A.H.), U19 AI35964 (A.H.), T32 AI095207 (R.M.). The funding sources had no influence on the design of the study and collection, analysis, interpretation of data, and writing of the manuscript.
Author information
Authors and Affiliations
Contributions
TK and AH designed the study. TK, SN, BC, SG, MLC SM, and NK performed the phenotypic experiments. TK and EO performed the computational analyses. RM and BC analyzed and interpreted the patient data. FK and CQ collected and identified the isolates as Klebsiella spp. TK, SN, SM, EO, and AR analyzed the data. TK and AH wrote the paper, and all other authors contributed to the writing. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The following information was extracted retrospectively from the electronic health records of patients from which the blood cultures were obtained: antibiotic susceptibility, age, type of infection, medical comorbidities, travel history, and community vs. hospital acquisition of the K. pneumoniae infection (i.e., onset prior to or after 48 h of hospitalization). All data were collected as part of routine clinical care, and all samples and data were de-identified to protect patient confidentiality. This study was approved and granted a waiver of the consent process by the Northwestern Institutional Review Board (protocol STU00211033).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Figure S1. A variety of Klebsiella spp. were identified among NMH bloodstream isolates. Maximum likelihood phylogenetic tree generated from core genome SNP loci in 140 Klebsiella spp. bloodstream isolates. The tree has a truncated outlier branch for Klebsiella oxytoca (yellow). The scale bar represents genetic distance (number of substitutions per site).
Additional file 2:
Figure S2. NMH K. pneumoniae bloodstream isolates are highly diverse in ST, KL, and O-antigen. Numbers of genomes with each corresponding ST (A), KL (B), or O-antigen type (C) were determined using Kleborate and Kaptive.
Additional file 3:
Figure S3. Antimicrobial resistance phenotypes of K. pneumoniae bloodstream isolates from NMH. The percentage of isolates resistant (solid bars) or intermediately susceptible (cross-hatched bars) to the indicated antibiotics are shown. Antibiotic classes are grouped by color.
Additional file 4:
Figure S4. Beta-lactam-resistant isolates contain a variety of extended-spectrum beta-lactamase or carbapenemase genes. Numbers of genomes containing the indicated beta-lactamase genes present in isolates resistant to ceftriaxone (A), meropenem (B), ampicillin/sulbactam (C), aztreonam (D), cefazolin (E), or cefepime (F) are plotted. “- “ indicates isolates without an extended-spectrum beta-lactamase gene.
Additional file 5:
Figure S5. Predicted plasmids containing virulence genes among NMH bloodstream isolates. Putative plasmids predicted to contain iuc or ybt loci were inferred and clustered using MOB-suite. Individual nodes represent a plasmid, and different colors represent a different plasmid group. Two plasmids are connected by an edge if their Jaccard index is ≥ 0.95. Networks were graphed using Cytoscape.
Additional file 6:
Figure S6. pKPN49_2, pKPN165_1, pTK421_2, and pKPN23_1 have little similarity to pK2044. Plasmid sequences were aligned to pK2044 using blast ring image generator (BRIG). A sequence identity threshold of 85% was used. Aerobactin biosynthesis genes are indicated with green arrows, salmochelin with orange arrows, rmpA with a blue arrow, and rmpA2 with a red arrow.
Additional file 7: Figure S7.
The bloodstream isolate KPN23 harbors a plasmid containing aerobactin biosynthesis genes. The indicated plasmids were aligned to a plasmid identified in strain KPCTRSRTH01_p2 (A) or to pKPN23_1 (B) using BRIG. A sequence identity threshold of 85% was used. Aerobactin biosynthesis genes are indicated with green arrows. Plasmid accession numbers for previously published plasmids are listed in Table S7.
Additional file 8:
Figure S8. Core genome phylogenetic tree of hypervirulent K. pneumoniae isolates. A maximum likelihood phylogenetic tree was generated from core genome SNP loci in hvKP isolates from United States hospitals and selected global reference isolates. Isolates labeled in red text are NMH bloodstream isolates from this study. Clonal group 23 sublineage 1 (CG23-1) isolates are indicated in a red box. Sequence types (ST) and capsule types (KL) are indicated. The presence of virulence genes is indicated next to each isolate: ybt = yersiniabactin biosynthesis loci, clb = colibactin biosynthesis loci, rmpADC = mucoid regulator operon, rmpA2 = regulator of mucoid phenotype 2, iuc = aerobactin biosynthesis genes, iro = salmochelin biosynthesis genes. KPPR1, KP52.145, SB5881, K180005, NCTC9494, NTUH-K2044, RJF999, and ED2 are isolates used as references (accession numbers are listed in Table S7).
Additional file 9
: Figure S9. Siderophore activity and virulence of NMH bloodstream isolates. Siderophore activity and virulence is plotted for each isolate (A) and against each other (B): left y-axis (A, purple triangles) or x-axis (B). In (B), aerobactin-positive (iuc+) strains are labeled in purple (B). Virulence was measured by LD50 values in a mouse model of pneumonia. Siderophore activity was measured by dipyridyl equivalents (mM) detected in cell free supernatants of bacterial cultures. Number of mice and doses used to determine LD50 values are listed in Table S8.
Additional file 10: Figure S10.
Sequence, capsule, and O-antigen types of NCBI K. pneumoniae bloodstream isolates. Numbers of genomes with each corresponding ST (A), KL (B), or O-antigen type (C) were determined using Kleborate and Kaptive. 1LV, 2LV, and 3LV indicate 1, 2, or 3 SNPs from a previously published ST.
Additional file 11: Figure S11.
Proportion of bloodstream genomes deposited to NCBI that were high-risk clones. Percent of genomes with the indicated sequence types are graphed for each year from 2007 – 2021. Numbers of total sequences deposited each year are listed directly above the graph. Sequence types were determined using Kleborate.
Additional file 12: Figure S12.
Predicted virulence plasmids carried by NCBI bloodstream isolates. Putative plasmids predicted to contain iuc or ybt genes were inferred and clustered using MOB-suite: iuc1 (A), iuc5 and iuc unknown (B), ybt4 (C), and iuc2 (D). Individual nodes represent a plasmid, and different colors represent a different plasmid group. Two plasmids are connected by an edge if their Jaccard index is ≥ 0.95. Networks were graphed using Cytoscape. Hybrid ESBL-virulence or carbapenemase-virulence plasmids are outlined with a dashed line and labeled with the type of beta-lactamase. Networks were graphed using Cytoscape.
Additional file 13
: Table S1. Detailed characteristics of Klebsiella pneumoniae isolates.
Additional file 14
: Table S2. Features of in silico-predicted plasmids.
Additional file 15
: Table S3. Characteristics of plasmids predicted to contain ESBL or carbapenemase genes.
Additional file 16
: Table S4. Characteristics of NCBI bloodstream isolates.
Additional file 17
: Table S5. Sequenced isolates from this study.
Additional file 18
: Table S6. Complete genomes from this study.
Additional file 19
: Table S7. Reference genomes.
Additional file 20
: Table S8. Number of mice dosing and mortality.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kochan, T.J., Nozick, S.H., Medernach, R.L. et al. Genomic surveillance for multidrug-resistant or hypervirulent Klebsiella pneumoniae among United States bloodstream isolates. BMC Infect Dis 22, 603 (2022). https://doi.org/10.1186/s12879-022-07558-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-022-07558-1