
Abstract
Background
Activation of Adenosine 5′-monophosphate-activated protein kinase/Sirtuin1 (AMPK/SIRT1) exerts an effect in alleviating obesity and gut damage. Sodium nitroprusside (SNP), a nitric oxide (NO) donor, has been reported to activate AMPK. This study was to investigate the effect of SNP on HFD induced gut dysfunction and the mechanism.
Methods
SNP was applied on lipopolysaccharide (LPS) stimulated Caco-2 cell monolayers which mimicked intestinal epithelial barrier dysfunction and HFD-fed mice which were complicated by gut dysfunction. Then AMPKα/SIRT1 pathway and gut barrier indicators were investigated.
Results
SNP rescued the loss of tight junction proteins ZO-1 and occludin, the inhibition of AMPKα/SIRT1 in LPS stimulated Caco-2 cell monolayers, and the effects were not shown when AMPKa1 was knocked-down by siRNA. SNP also alleviated HFD induced obesity and gut dysfunction in mice, as indicated by the decreasing of intestinal permeability, the increasing expression of ZO-1 and occludin, the decreasing levels of pro-inflammatory cytokine IL-6, and the repairing of gut microbiota dysbiosis. These effects were complicated by the increased colonic NO content and the activated AMPKα/SIRT1 signaling.
Conclusions
The results may imply that SNP, as a NO donor, alleviates HFD induced gut dysfunction probably by activating the AMPKα/SIRT1 signaling pathway.
Similar content being viewed by others


Background
The gut dysfunction, which is characterized by gut microbiota dysbiosis and intestine epithelium barrier impairment, has a strong link to the development of metabolic disorders such as obesity and type 2 diabetes mellitus [1, 2]. The gut microbiota, which has more than 1200 different bacterial species and 1.5–2.0 kg biomass in the gastrointestinal tract, encodes 500 times more genes than its human host [3]. The gut microbiota plays important roles in ingesta digestion, immunity regulation, and energy equilibrium. The intestine epithelium barrier, which comprises epithelial cells and intercellular tight junction proteins including ZOs, occludins, and claudins, is a multilayered barrier that defends against harmful compounds and pathogens. The tight junction proteins play a vital role in retaining the intestine epithelium barrier. Loss of epithelial tight junction proteins impairs paracellular barrier, resulting in the increasing of the intestine permeability and subsequent intestinal penetration of luminal bacteria and harmful metabolites [4]. Increasing shreds of evidence have shown that gut dysfunction contributes to obesity, and modulation of gut dysfunction is able to improve lipid metabolism and alleviate systemic inflammation in severe obesity [5, 6]. Therefore, amelioration of gut dysfunction is a potential therapeutic measure for obesity and its complicated metabolic syndrome.
Obesity, which is resulted from the high energy intake and low energy expenditure also causes gut dysfunction [5, 7]. AMPK is a crucial kinase that is involved in the regulation of energy metabolism. It has been reported that the activation/phosphorylation of AMPK alleviates obesity and its related metabolic syndrome via activating the downstream effector SIRT1 [8,9,10,11,12]. SIRT1 is a nicotinamide adenine dinucleotide (NAD)-dependent histone deacetylase and its activation by NAD displays an ability to rejuvenate the gut adult stem cells and attenuate the gut damage in aged mice [13]. In addition, AMPK has SIRT1-dependent anti-apoptosis and anti-inflammatory effects [14, 15], and the anti-inflammatory effect of activated/phosphorylated AMPKα is involved in the protection of intestinal barrier [7, 16].
SNP, as a NO donor, is a rapid-acting intravenous vasodilator that has been widely used clinically in hypertensive crises for decades [17,18,19,20,21]. However, recent studies have shown that SNP may also have roles in regulating energy metabolism. SNP increases the phosphorylation of AMPK, specifically AMPKα, then the phosphorylated AMPKα translocates to the nucleus, and then activates gene expression that leads to increasing glucose uptake in the skeletal muscle [22, 23]. NO that is derived from enterocytes has been reported to restore colitis via alleviating tissue damage and macrophage infiltration [24]. However, the role of SNP and NO on obesity-induced gut dysfunction has not been explored.
Due to the crosstalk between gut dysfunction, obesity, AMPK/SIRT1 signaling, and the role of SNP on AMPKα phosphorylation, we hypothesized that SNP protected against HFD induced gut dysfunction via activating AMPKα/SIRT1 signaling. Our experiments on LPS stimulated Caco-2 cell monolayers that mimicked intestine epithelium barrier dysfunction and HFD-fed mice that were complicated by gut dysfunction found that SNP alleviated obesity and gut dysfunction by recovering HFD inhibited AMPKα/SIRT1 signaling.
Method
Animals
The male C57BL/6 mice (6w, 20 ± 2 g), normal diet, and high-fat diet feed were provided by the Guangdong Medical Laboratory Animal Center (Foshan, China). Mice were kept under controlled temperature and light conditions (25 °C,12 h light–dark cycle), with free access to food and water. Mice were randomly distributed into three groups containing six animals each: normal diet (ND), high-fat diet (HFD), HFD with sodium nitroprusside (HFD + SNP). Mice were fed with ND or HFD for 16 weeks. SNP (71778, Sigma, USA) was supplemented in drinking water (1 mg/mL) from the fifth week of feeding to the end of the experiment. The study was carried out in compliance with the ARRIVE guidelines 2.0. All methods were performed in accordance with the relevant guidelines and regulations (Directive 2010/63/EU in Europe), and the animal study protocols were approved by committee review of animal experiments in Guangzhou Medical University (Document no. 2018-084).
Gut microbiota analysis
Gut microbiota analysis was performed as previously described [25]. After 16 weeks of feeding and SNP treatment, feces samples were collected for the 16 S rRNA gene sequencing. Firstly, fecal DNA was extracted using a PowerSoil DNA Isolation kit (12888, Mobio, USA). Then, V3-V4 regions of the 16 S rRNA gene were amplified by PCR with specific primer sets. Next, PCR products were purified and normalized to equal DNA concentration and sequenced using the Illumina Miseq sequencer PE250 (Illumina, USA). The effective reads from all samples were clustered into OTUs based on 99% sequence similarity according to Qiime Uclust. The OTUs were annotated through RDP Classifier (Version 2.2), according to the GreenGene database, then the composition and abundance information of each sample at different classification levels were statistically summarized.
Intestinal permeability assay
The intestinal permeability assay was performed as previously described [26]. Fluorescein isothiocyanate conjugated dextran (FITC-dextran) (68,059, Sigma, USA) was dissolved in sterile saline (100 mg/mL). Mice were fasted for 12 h and deprived of water for 4 h, then mice were orally gavaged with FITC-dextran (500 mg/kg body weight). 4 h after the gavage, mice were anesthetized with sodium pentobarbital (50 mg/kg body weight), and blood samples were collected from the abdominal aorta. The serum was prepared for fluorescence measurements (excitation, 485 nm; emission, 520 nm) using Thermo Scientific ™ Varioskan ™ LUX (Thermo Scientific, USA). Serum FITC-dextran concentration was quantified against a calibration curve.
Histology analysis
Mice colon tissues were collected and fixed in 4% formaldehyde, paraffin-embed, sectioned and stained by hematoxylin and eosin (H&E). Histology analysis was performed as previously described [27].
Cell culture
The human epithelial cell line Caco2 was purchased from American Type Culture Collection (ATCC). Cells were cultured with Dulbecco’s modified eagle medium (DMEM) containing 10% fetal bovine serum in a humidified incubator with 5% CO2 at 37 °C. Caco2 cells were differentiated into polarized epithelial monolayers by culturing on polypropylene membrane on 100 mm culture plates. The Caco2 cells were cultured and passaged successfully before the tests. After passaging for five generations, Caco-2 cells were passaged into 60 mm culture plates at a density of 1 × 106 cells /mL and were subjected to four treatments as follows: Control, lipopolysaccharide(LPS, 50 ng/mL) treatment for 46 h, LPS plus SNP (LPS,50 ng/mL; SNP, 10, 30 ng/mL) for 48 h. After the treatments, cells were harvested for subsequent experiments.
Western blot analysis
Western blot was performed as previously described [28]. Colon tissues were ground and Caco2 cells were lysed in RIPA buffer, and 50 µg total protein was separated by 12% SDS-PAGE, and then transferred to polyvinylidene fluoride membrane (cas# FFP22, Beyotime Biotechnology, China). The membrane was blocked using 5% skimmed milk powder in PBST (PBS with 0.1% Tween-20) and then was incubated overnight at 4℃ with the following primary antibodies: anti-phospho-AMPKα, anti-AMPK, anti-SIRT1 (cas# 2531s, 8469s·2532s, 1:1000, Cell Signaling, USA), anti-ZO-1, anti-β-actin (cas# ab216880, ab227387, 1:1000, Abcam, USA), and anti-occludin (cas# 66378-1, 1:1000, Proteintech Group, USA). After washed with PBST for 3 times, the membrane was incubated with secondary antibodies (cas# E030120-02, E030110-02, EarthOx, USA) in PBST for 2 h. BeyoECL Moon (cas# P0018FM, Beyotime Biotechnology, China) was used for chemiluminescence and ChemiDicTM XRS + Imaging System was used to take pictures (BioRad Laboratories).
Transfection of small interfering RNA
The specific and control siRNA sequences were as follows: AMPKα1, sense 5′-CGGGAUCAGUUAGCAACUATT-3′ and antisense 5′-UAGUUGCUAACUGAUCCCGTT-3′; negative control, sense 5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense 5′-ACGUGACACGUUCGGAGAATT-3′ (GenePharma, China). Caco-2 cells were cultured and passaged in the same way as the above procedures, then cells were seed into 60 mm culture plates at a density of 1 × 106 cells/mL. The cells were incubated in Opti-MEM and transfected with each siRNA (100 nM) using Lipofectamine 3000 Transfection Reagent (L3000015, Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s protocol. Transfection of small interfering RNA was performed as previously described [7]. Following the transfection, LPS and SNP were added and co-incubatied in the same way as the above procedures. After the treatments, cells were harvested for subsequent experiments.
Statistics analysis
Data were subjected to statistical analysis by one-way ANOVA or Student’s T-test using SPSS 16.0. The least significant difference test was selected when the variance was homogeneous, and Dunnett’s T3 test was used when the variance was not homogeneous. Statistical significance was considered when *p < 0.05, **p < 0.01 and ***p < 0.001. All data were expressed as Mean ± S.E.M.
Result
SNP alleviated LPS-induced intestinal barrier dysfunction via AMPKα/SIRT1 signaling in human Caco2 cells
Phosphorylation of AMPKα has been reported to be involved in intestinal barrier function [7], and studies have shown that NO plays a role in the activation of AMPK pathway [29,30,31]. Therefore, we investigated whether SNP, a NO donor, could ameliorate intestinal barrier dysfunction via AMPKα phosphorylation in the human colonic epithelial cell line Caco2. LPS was used to induce intestinal barrier dysfunction in Caco-2 cell monolayers, and as expected, it reduced the expression of tight junction proteins ZO-1 and occludin. 10 µM and 30 µM SNP treatment markedly restored their expression (Fig. 1A–C). LPS also inhibited the phosphorylation of AMPKα in Caco-2 cells, and the inhibition was restored by SNP treatment (Fig. 1D, E). Coincidently, SIRT1, the key player in AMPK mediated anti-inflammatory effects, was also inhibited by LPS and again restored by SNP (Fig. 1D, F). However, when AMPKα was knocked down by siRNA, LPS decreased SIRT1 and occludin could no longer be restored by SNP (Fig. 1G–K). Taken together, these data indicated that SNP alleviated LPS-induced intestinal barrier dysfunction via AMPKα/SIRT1 signaling.

SNP alleviated LPS-induced intestinal barrier dysfunction via AMPKα-SIRT1 signaling. Intestinal barrier dysfunction of Caco-2 cell monolayers was stimulated by 50 ng/mL LPS treatment for 46 h and LPS plus 10 µM/30 µM SNP treatment for 48 h. A–C Protein levels of intestinal barrier indicators ZO-1 and occludin. (D-F) Protein levels of phosphorylated AMPKα and SIRT1. G–I Protein levels of SIRT1 and phosphorylated AMPKα when AMPKα was knocked down. J–K Protein levels of occludin when AMPKα was knocked down; Data were presented as the mean ± S.E.M. (*p < 0.05, **p < 0.01, ***p < 0.001)
SNP alleviated obesity of HFD-fed mice
The impaired intestinal epithelial barrier has been reported to be associated with the development of metabolic disorders, especially obesity [2, 32,33,34,35]. Since SNP alleviated intestinal barrier dysfunction in human Caco2 cells, we then investigated whether SNP could alleviate obesity of HFD-fed mice. As shown in Fig. 2, SNP significantly reduced body weight, mesenteric fat mass and fasting blood glucose in HFD-fed mice (Fig. 2A–C). These results suggested that SNP alleviated obesity and improved metabolic parameters resulted from HFD.

SNP alleviated obesity of HFD-fed mice. Mice (n = 6) were fed with ND or HFD for 16 weeks. SNP was supplemented in drinking water (1 mg/mL) from the fifth week of feeding to the end of the experiment. A Body weight changes; B Mesenteric fat mass; C Fasting blood glucose levels. Data were presented as the mean ± S.E.M. (*p < 0.05, **p < 0.01, ***p < 0.001)
SNP ameliorated HFD induced gut barrier dysfunction
Following the results that SNP alleviated obesity of HFD-fed mice, we investigated whether SNP could protect gut barrier function of HFD-fed mice. The results showed that colon length was decreased, intestinal permeability was increased, and content of colonic inflammatory factor IL-6 was increased in HFD-fed mice compared with ND-fed mice, while all the damage was ameliorated by SNP (Fig. 3A-D). Moreover, SNP restored the decreased colonic expression of epithelial tight junction protein ZO-1 and occludin of HFD-fed mice (Fig. 3E-G). Consistent with these results, less intestinal mucosal morphological damage and lamina propria leukocyte infiltration were found in SNP-treated HFD-fed mice than those of HFD-fed mice (Fig. 3 H). These results showed that SNP protected against HFD induced intestinal barrier dysfunction and intestinal inflammation.

SNP ameliorated HFD-induced intestinal barrier dysfunction. Mice (n = 6) were fed with ND or HFD for 16 weeks. SNP was supplemented in drinking water (1 mg/mL) from the fifth week of feeding to the end of the experiment. A Representative pictures of colons of each group; B Colon length; C Serum FITC-dextran levels; D IL-6 levels of colon; E-G Protein levels of intestinal barrier indicators ZO-1 and occludin (n = 4); (H) Representative histological pictures of hematoxylin and eosin staining. Data were presented as the mean ± S.E.M. (*p < 0.05, **p < 0.01, ***p < 0.001)
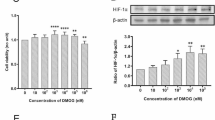
SNP resisted HFD inhibited AMPKα/SIRT1 signaling in mice colon
To verify whether the anti-obesity and intestinal barrier protection effects of SNP on HFD-fed mice were also mediated by AMPKα/SIRT1 signaling, we tested the colonic expression of these signals. Colonic NO level was reduced in HFD-fed mice compared with ND-fed mice, and SNP treatment increased the colonic NO level of HFD-fed mice (Fig. 4A). Protein levels of SIRT1 and phosphorylated AMPKα were lower in the colon of HFD-fed mice, but these alterations were restored by SNP (Fig. 4B-D). Linear regression analyses showed a significant positive correlation of the NO content and the protein levels of SIRT1 and phosphorylated AMPKα (Fig. 4E-F). These results indicated that SNP rescued colonic AMPKα-SIRT1 signaling suppressed by HFD.

SNP resisted HFD inhibited colonic AMPKα/SIRT1 signaling. Mice (n = 6) were fed with ND or HFD for 16 weeks. SNP was supplemented in drinking water (1 mg/mL) from the fifth week of feeding to the end of the experiment. A NO levels of colon; B–D Protein levels of phosphorylated AMPKα and SIRT1 (n = 4); E Correlation between colonic NO content and phosphorylated AMPKα; F Correlation between colonic NO content and SIRT1. Data were presented as the mean ± S.E.M. (* p < 0.05, **p < 0.01, ***p < 0.001)
SNP ameliorated HFD induced gut dysbiosis
Previous studies have shown that obesity and gut barrier dysfunction is associated with gut microbiota dysbiosis [36, 37]. We next investigated whether SNP had effects on gut microbiota dysbiosis of HFD-fed mice. As shown in Fig. 5A, HFD caused significant changes of gut miacrobiota profiles compared with ND while SNP treatment modulated the profiles of gut microbiota. HFD significantly decreased microbiota richness and diversity, SNP reversed the changes of the diversity but not richness (Fig. 5B). At the genus level, HFD significantly reduced the relative abundance of Lactobacillus and increased the relative abundance of Paraprevotella, Prevotella, and Helicobacter, whereas SNP reversed the changes of these microbes (Fig. 5 C-F). So, SNP rescued part of HFD induced gut microbiota dysbiosis.

SNP alleviated HFD-induced gut microbiota dysbiosis. Mice (n = 6) were fed with ND or HFD for 16 weeks. SNP was supplemented in drinking water (1 mg/mL) from the fifth week of feeding to the end of the experiment. A Principal coordinate analysis (PCoA) plots of fecal microbiota; B Richness (Chao1 and ACE indexes) and diversity (Shannon and Simpson indexes) of microbial communities; C–F Relative abundance of Lactobacillus, Paraprevotella, Prevotella, and Helicobacter. Data were presented as the mean ± S.E.M. (* p < 0.05, **p < 0.01, ***p < 0.001)
Discussion
NO is not only a key regulator of vascular endothelial homeostasis, but may also play roles in intestinal epithelial function. Previous studies have demonstrated that enhancing the bioavailability of NO in enterocytes attenuates colitis by meliorating colorectal inflammation and gut microbiota dysbiosis [38]. A recent study reported that endogenous NO derived from enterocytes ameliorated colitis by alleviating inflammation, macrophage infiltration, and tissue damage. Moreover, induction of NO production in enterocytes with NO precursors inhibited colon cancer by alleviating epithelial barrier damage and inflammation [39]. Our results showed that NO content was significantly decreased by HFD compared with ND, whereas SNP supplementation increased NO content in colon tissue of HFD-fed mice, and then confered protective effects on HFD induced gut dysfunction. The role of SNP in alleviating intestinal barrier damage was also reported in ischemia/reperfusion injury mice models [40,41,42].
Tight junction proteins are key components of the epithelial barrier and loss of them leads to elevated intestinal permeability and inflammation [43]. Loss of tight junction proteins was found in endotoxin LPS stimulated enterocyte [44,45,46,47,48,49]. HFD, which produces LPS also leads to loss of tight junction proteins. Our results showed that SNP not only restored LPS-induced loss of the tight junction proteins ZO-1 and occludin in Caco-2 cell monolayers, but also restored HFD induced loss of the tight junction proteins ZO-1 and Occludin in mice, which reversed the increased intestinal barrier permeability.
The impaired intestinal epithelum barrier has been considered to be a pathophysiological factor of metabolic diseases, such as obesity [50,51,52]. Gut microbiota dysbiosis, which promotes intestinal barrier disruption, contributes to the development of metabolic disease [53,54,55]. Meanwhile, the metabolic disease also causes gut dysfunction and then leads to gut problems such as colitis. So, gut dysfunction and obesity may interact as both cause and effect. Anyhow, NO is a pleiotropic signaling molecule that involves in regulation of both the intestinal barrier function and the metabolic deseases.[56, 57]. Consistently, we found that NO donor SNP protects against gut dysfunction, as well as obesity.
AMPK signaling has been reported to be involved in endothelial homeostasis [58], Inhibition of AMPK promotes epithelial barrier dysfunction [7], while the activated/phosphorylated AMPKα protects against colitis via alleviating intestinal barrier dysfunction and anti-inflammation [7, 16]. The effect of prebiotics, fructo-oligosaccharides (FOS) for example, in protecting intestinal epithelial tight junction has been reported to be mediated by AMPK activation[59]. The role of NO in endothelial homeostasis has been reported to be mediated by AMPK signaling. Increased NO derived from low-concentration SNP increases levels of phosphorylated AMPK and its activity in endothelial cells [60]. L-arginine, another precursor for NO synthesis, has been reported to increase the expression of tight junction proteins and improve intestinal barrier function by activating of AMPK signaling in rat small intestines and IEC-6 cells during heat stress [61]. Consistent with the previous reports, we found that NO donor SNP restored LPS and HFD induced loss of tight junction proteins via AMPKα-SIRT1 signaling in Caco-2 cell monolayers and HFD-fed mice. What’s more, NO content and the activated AMPKα/SIRT1 signaling were positively correlated in mice’s colons.
The present study didn’t include a SNP control on ND-fed mice. So if SNP reduced the basal mesenteric fat mass and blood glucose could not been shown here. Effects of SNP or other NO-generating compounds in regulating blood glucose and insulin sensitivity have been tested sufficiently on cell and animal models of diabetes and human with diabetes, and all the results have shown that treatment of NO donor SNP or NO precursor L-Arginine reduce blood glucose levels, improve insulin resistance and lipid profiles in diabetic models [62,63,64,65,66,67,68], however, they do not affect blood glucose, lipid profile in non-diabetic group [69, 70]. So, we inferred that SNP might act specifically on subjects with metabolic problems while have little effects on normal subjects, however, this assumption need further investigation.
In conclusion, our study indicated that SNP, as a NO donor, which has been applied as an anti-hypertensive drug, had protective effects on epithelial barrier dysfunction and gut microbiota dysbiosis resulted from HFD. It was noted that SNP alleviated HFD induced intestinal epithelium barrier dysfunction and inflammation by activating the AMPKα/SIRT1 signal, and these effects were dependent on the increased NO content derived from SNP. These results implied that SNP might be a potential drug for patients with gut dysfunction.
Availability of data and materials
Sequence data of the raw paired-end reads were deposited to NCBI/Sequence Read Archive and are accessible through the accession number PRJNA674745, or http://www.ncbi.nlm.nih.gov/bioproject/674745.
Abbreviations
- AMPK:
-
Adenosine 5′-monophosphate-activated protein kinase
- SIRT1:
-
Sirtuin1
- SNP:
-
Sodium nitroprussiate
- NO:
-
Nitric oxide
- IV:
-
Intravenous
- HFD:
-
High-fat diet
- LPS:
-
Lipopolysaccharide
- ZO-1:
-
Zonula occludens 1
- siRNA:
-
Small interfering Ribonucleic Acid
- IL-6:
-
Interleukin- 6
- NAD+ :
-
nicotinamide adenine dinucleotide
- ND:
-
Normal diet
- PCR:
-
Polymerase chain reaction
- OTUs:
-
Operational taxonomic units
- RDP:
-
Ribosomal Database Project
- FITC-dextran:
-
Fluorescein isothiocyanate conjugated dextran
- H&E:
-
Hematoxylin and eosin
- siNC:
-
Negative control small interfering RNA
- siAMPKα1:
-
AMPKα1 small interfering RNA
References
Monk JM, Wu W, Lepp D, Wellings HR, Hutchinson AL, Liddle DM, Graf D, Pauls KP, Robinson LE, Power KA. Navy bean supplemented high-fat diet improves intestinal health, epithelial barrier integrity and critical aspects of the obese inflammatory phenotype. J Nutr Biochem. 2019;70:91–104.
Thaiss CA, Levy M, Grosheva I, Zheng D, Soffer E, Blacher E, Braverman S, Tengeler AC, Barak O, Elazar M, et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science (New York, NY). 2018;359(6382):1376–83.
Schroeder BO, Bäckhed F. Signals from the gut microbiota to distant organs in physiology and disease. Nat Med. 2016;22(10):1079–89.
Buckley A, Turner JR. Cell biology of tight junction barrier regulation and mucosal disease. Cold Spring Harbor Perspect Biol. 2018;10(1).
Huang YC, Huang LT, Sheen JM, Hou CY, Yeh YT, Chiang CP, Lin IC, Tiao MM, Tsai CC, Lin YJ, et al. Resveratrol treatment improves the altered metabolism and related dysbiosis of gut programed by prenatal high-fat diet and postnatal high-fat diet exposure. J Nutr Biochem. 2020;75:108260.
Zhou AL, Ward RE. Milk polar lipids modulate lipid metabolism, gut permeability, and systemic inflammation in high-fat-fed C57BL/6J ob/ob mice, a model of severe obesity. J Dairy Sci. 2019;102(6):4816–31.
Deng J, Zeng L, Lai X, Li J, Liu L, Lin Q, Chen Y. Metformin protects against intestinal barrier dysfunction via AMPKα1-dependent inhibition of JNK signalling activation. J Cell Mol Med. 2018;22(1):546–57.
Manna P, Achari AE, Jain SK. Vitamin D supplementation inhibits oxidative stress and upregulate SIRT1/AMPK/GLUT4 cascade in high glucose-treated 3T3L1 adipocytes and in adipose tissue of high fat diet-fed diabetic mice. Arch Biochem Biophys. 2017;615:22–34.
Salomone F, Barbagallo I, Godos J, Lembo V, Currenti W, Cinà D, Avola R, D’Orazio N, Morisco F, Galvano F, et al. Silibinin Restores NAD+ levels and induces the SIRT1/AMPK pathway in non-alcoholic fatty liver. Nutrients 2017;9(10).
Edmunds LR, Huckestein BR, Kahn M, Zhang D, Chu Y, Zhang Y, Wendell SG, Shulman GI, Jurczak MJ. Hepatic insulin sensitivity is improved in high-fat diet-fed Park2 knockout mice in association with increased hepatic AMPK activation and reduced steatosis. Physiol Rep. 2019;7(21):e14281.
Sun Y, Zhou S, Guo H, Zhang J, Ma T, Zheng Y, Zhang Z, Cai L. Protective effects of sulforaphane on type 2 diabetes-induced cardiomyopathy via AMPK-mediated activation of lipid metabolic pathways and NRF2 function. Metabolism Clin Exp. 2020;102:154002.
Paraíso AF, Sousa JN, Andrade JMO, Mangabeira ES, Lelis DF, de Paula AMB, Martins A, Lima WJN, Guimarães ALS, Melo GA, et al. Oral gallic acid improves metabolic profile by modulating SIRT1 expression in obese mice brown adipose tissue: a molecular and bioinformatic approach. Life Sci. 2019;237:116914.
Igarashi M, Miura M, Williams E, Jaksch F, Kadowaki T, Yamauchi T, Guarente L. NAD(+) supplementation rejuvenates aged gut adult stem cells. Aging Cell. 2019;18(3):e12935.
Ren H, Shao Y, Wu C, Ma X, Lv C, Wang Q. Metformin alleviates oxidative stress and enhances autophagy in diabetic kidney disease via AMPK/SIRT1-FoxO1 pathway. Mol Cell Endocrinol. 2020;500:110628.
Fu C, Hao S, Xu X, Zhou J, Liu Z, Lu H, Wang L, Jin W, Li S. Activation of SIRT1 ameliorates LPS-induced lung injury in mice via decreasing endothelial tight junction permeability. Acta Pharmacol Sin. 2019;40(5):630–41.
Jang HM, Han SK, Kim JK, Oh SJ, Jang HB, Kim DH. Lactobacillus sakei alleviates high-fat-diet-induced obesity and anxiety in mice by inducing AMPK activation and SIRT1 expression and inhibiting gut microbiota-mediated NF-κB activation. Mol Nutr Food Res. 2019;63(6):e1800978.
Olesen ND, Fischer M, Secher NH. Sodium nitroprusside dilates cerebral vessels and enhances internal carotid artery flow in young men. J Physiol. 2018;596(17):3967–76.
Huang H, Zhang YJ, Fan YZ, Wu X, Bourantas CV. Use of sodium nitroprusside in retrograde percutaneous coronary intervention for chronic total occlusion: a case report. Medicine (Baltimore). 2018;97(29):e11498.
Deliu AG, Sanneerappa PBJ, Franklin O, Letshwiti J. Sodium nitroprusside, a lifesaving treatment for neonatal hypertension: an Irish experience. BMJ Case Rep. 2018;2018.
Cobb A, Thornton L. Sodium nitroprusside as a hyperinflation drug and therapeutic alternatives. J Pharm Pract. 2018;31(4):374–81.
Ranadive SM, Eugene AR, Dillon G, Nicholson WT, Joyner MJ. Comparison of the vasodilatory effects of sodium nitroprusside vs. nitroglycerin. J Appl Physiol (1985). 2017;123(2):402–6.
Lira VA, Soltow QA, Long JH, Betters JL, Sellman JE, Criswell DS. Nitric oxide increases GLUT4 expression and regulates AMPK signaling in skeletal muscle. Am J Physiol Endocrinol Metab. 2007;293(4):E1062-1068.
Aguirre E, López-Bernardo E, Cadenas S. Functional evidence for nitric oxide production by skeletal-muscle mitochondria from lipopolysaccharide-treated mice. Mitochondrion. 2012;12(1):126–31.
Serre H, Simon L, Thevenet A, Barjon MC. Hyperparathyroidiem and articular chondrocalcinosis]. Journal belge de rhumatologie et de medecine physique = Belgisch tijdschrift voor reumatologie en fysische geneeskunde. 1966;21(5):223–42.
Chen R, Xu Y, Wu P, Zhou H, Lasanajak Y, Fang Y, Tang L, Ye L, Li X, Cai Z, et al. Transplantation of fecal microbiota rich in short chain fatty acids and butyric acid treat cerebral ischemic stroke by regulating gut microbiota. Pharmacol Res. 2019;148:104403.
Birchenough GM, Johansson ME, Gustafsson JK, Bergstrom JH, Hansson GC. New developments in goblet cell mucus secretion and function. Mucosal Immunol. 2015;8(4):712–9.
Wang C, Yue F, Kuang S. Muscle histology characterization using H&E staining and muscle fiber type classification using immunofluorescence staining. Bio Protoc. 2017;7(10).
Lee C. Western blotting. Methods Mol Biol. 2007;362:391–9.
Shin MJ, Lee YP, Kim DW, An JJ, Jang SH, Cho SM, Sheen SH, Lee HR, Kweon HY, Kang SW, et al. Transduced PEP-1-AMPK inhibits the LPS-induced expression of COX-2 and iNOS in Raw264.7 cells. BMB Rep. 2010;43(1):40–5.
Li JM, Lu W, Ye J, Han Y, Chen H, Wang LS. Association between expression of AMPK pathway and adiponectin, leptin, and vascular endothelial function in rats with coronary heart disease. Eur Rev Med Pharmacol Sci. 2020;24(2):905–14.
Wang Z, Wu Y, Zhang S, Zhao Y, Yin X, Wang W, Ma X, Liu H. The role of NO-cGMP pathway inhibition in vascular endothelial-dependent smooth muscle relaxation disorder of AT1-AA positive rats: protective effects of adiponectin. Nitric Oxide. 2019;87:10–22.
Araújo JR, Tomas J, Brenner C, Sansonetti PJ. Impact of high-fat diet on the intestinal microbiota and small intestinal physiology before and after the onset of obesity. Biochimie. 2017;141:97–106.
Chelakkot C, Ghim J, Ryu SH. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp Mol Med. 2018;50(8):103.
Ye C, Liu L, Ma X, Tong H, Gao J, Tai Y, Huang L, Tang C, Wang R. Obesity aggravates acute pancreatitis via damaging intestinal mucosal barrier and changing microbiota composition in rats. Sci Rep. 2019;9(1):69.
Park C, Cheung KP, Limon N, Costanzo A, Barba C, Miranda N, Gargas S, Johnson AMF, Olefsky JM, Jameson JM. Obesity modulates intestinal intraepithelial T cell persistence, CD103 and CCR9 expression, and outcome in dextran sulfate sodium-induced colitis. J Immunol. 2019;203(12):3427–35.
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–31.
Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008;3(4):213–23.
Dyson JK, Rutter MD. Colorectal cancer in inflammatory bowel disease: what is the real magnitude of the risk? World J Gastroenterol. 2012;18(29):3839–48.
Stettner N, Rosen C, Bernshtein B, Gur-Cohen S, Frug J, Silberman A, Sarver A, Carmel-Neiderman NN, Eilam R, Biton I, et al. Induction of nitric-oxide metabolism in enterocytes alleviates colitis and inflammation-associated colon cancer. Cell Rep. 2018;23(7):1962–76.
Chen FH, Li K, Yin L, Chen CQ, Yan ZW, Chen GM. Protective effect of sodium nitroprusside on the rat small intestine transplanted mucosa. Biochem Res Int. 2015;2015:786010.
Payne D, Kubes P. Nitric oxide donors reduce the rise in reperfusion-induced intestinal mucosal permeability. Am J Physiol. 1993;265(1 Pt 1):G189-195.
Takizawa Y, Kishimoto H, Kitazato T, Tomita M, Hayashi M. Effects of nitric oxide on mucosal barrier dysfunction during early phase of intestinal ischemia/reperfusion. Eur J Pharmaceut Sci Off J Eur Feder Pharm Sci. 2011;42(3):246–52.
Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, Said HM. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol. 2004;286(3):G367-376.
Bein A, Zilbershtein A, Golosovsky M, Davidov D, Schwartz B. LPS induces hyper-permeability of intestinal epithelial cells. J Cell Physiol. 2017;232(2):381–90.
Ghosh SS, Wang J, Yannie PJ, Ghosh S. Intestinal barrier dysfunction, LPS translocation, and disease development. J Endocr Soc. 2020;4(2):bvz039.
Han F, Lu Z, Liu Y, Xia X, Zhang H, Wang X, Wang Y. Cathelicidin-BF ameliorates lipopolysaccharide-induced intestinal epithelial barrier disruption in rat. Life Sci. 2016;152:199–209.
Cao Y, Chen Q, Wang Z, Yu T, Wu J, Jiang X, Jin X, Lu W. PLK1 protects against sepsis-induced intestinal barrier dysfunction. Sci Rep. 2018;8(1):1055.
Guo S, Al-Sadi R, Said HM, Ma TY. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am J Pathol. 2013;182(2):375–87.
Nighot M, Rawat M, Al-Sadi R, Castillo EF, Nighot P, Ma TY. Lipopolysaccharide-induced increase in intestinal permeability ismMediated by TAK-1 activation of IKK and MLCK/MYLK gene. Am J Pathol. 2019;189(4):797–812.
Allam-Ndoul B, Castonguay-Paradis S, Veilleux A. Gut microbiota and intestinal trans-epithelial permeability. Int J Mol Sci. 2020;21(17).
Nakadate K, Hirakawa T, Tanaka-Nakadate S. Small intestine barrier function failure induces systemic inflammation in monosodium glutamate-induced chronically obese mice. Appl Physiol Nutr Metab. 2019;44(6):587–94.
Ouyang J, Lin J, Isnard S, Fombuena B, Peng X, Marette A, Routy B, Messaoudene M, Chen Y, Routy JP. The bacterium Akkermansia muciniphila: a sentinel for gut permeability and its relevance to HIV-related inflammation. Front Immunol. 2020;11:645.
Takiishi T, Fenero CIM, Câmara NOS. Intestinal barrier and gut microbiota: shaping our immune responses throughout life. Tissue Barriers. 2017;5(4):e1373208.
Rolig AS, Mittge EK, Ganz J, Troll JV, Melancon E, Wiles TJ, Alligood K, Stephens WZ, Eisen JS, Guillemin K. The enteric nervous system promotes intestinal health by constraining microbiota composition. PLoS Biol. 2017;15(2):e2000689.
Boulangé CL, Neves AL, Chilloux J, Nicholson JK, Dumas ME. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 2016;8(1):42.
Lundberg JO, Carlström M, Weitzberg E. Metabolic effects of dietary nitrate in health and disease. Cell Metab. 2018;28(1):9–22.
Oleson BJ, Broniowska KA, Yeo CT, Flancher M, Naatz A, Hogg N, Tarakanova VL, Corbett JA. The role of metabolic flexibility in the regulation of the DNA damage response by nitric oxide. Mol Cell Biol. 2019;39(18).
Cao S, Wang C, Yan J, Li X, Wen J, Hu C. Curcumin ameliorates oxidative stress-induced intestinal barrier injury and mitochondrial damage by promoting Parkin dependent mitophagy through AMPK-TFEB signal pathway. Free Radic Biol Med. 2020;147:8–22.
Wongkrasant P, Pongkorpsakol P, Ariyadamrongkwan J, Meesomboon R, Satitsri S, Pichyangkura R, Barrett KE, Muanprasat C. A prebiotic fructo-oligosaccharide promotes tight junction assembly in intestinal epithelial cells via an AMPK-dependent pathway. Biomed Pharmacother. 2020;129:110415.
Zhang J, Xie Z, Dong Y, Wang S, Liu C, Zou MH. Identification of nitric oxide as an endogenous activator of the AMP-activated protein kinase in vascular endothelial cells. J Biol Chem. 2008;283(41):27452–61.
Li Q, Chen H, Zhang M, Wu T, Liu R. Altered short chain fatty acid profiles induced by dietary fiber intervention regulate AMPK levels and intestinal homeostasis. Food Funct. 2019;10(11):7174–87.
Mohamadin AM, Hammad LN, El-Bab MF, Gawad HS. Can nitric oxide-generating compounds improve the oxidative stress response in experimentally diabetic rats? Clin Exp Pharmacol Physiol. 2007;7:586–93.
Popov D, Costache G, Georgescu A, Enache M. Beneficial effects of L-arginine supplementation in experimental hyperlipemia-hyperglycemia in the hamster. Cell Tissue Res. 2002;308(1):109–20.
Henstridge DC, Kingwell BA, Formosa MF, Drew BG, McConell GK, Duffy SJ. Effects of the nitric oxide donor, sodium nitroprusside, on resting leg glucose uptake in patients with type 2 diabetes. Diabetologia. 2005;48(12):2602–8.
Wellhauser L, Chalmers JA, Belsham DD. Nitric oxide exerts basal and insulin-dependent anorexigenic actions in POMC hypothalamic neurons. Mol Endocrinol 2016;30(4):402–16.
Duplain H, Burcelin R, Sartori C, Cook S, Egli M, Lepori M, Vollenweider P, Pedrazzini T, Nicod P, Thorens B, Scherrer U. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation. 2001;104(3):342-5.
Shankar RR, Wu Y, Shen HQ, Zhu JS, Baron AD. Mice with gene disruption of both endothelial and neuronal nitric oxide synthase exhibit insulin resistance. Diabetes. 2000;49(5):684–7.
Cook S, Scherrer U. Insulin resistance, a new target for nitric oxide-delivery drugs. Fundam Clin Pharmacol. 2002;6:441–53.
Mendez JD, Balderas F. Regulation of hyperglycemia and dyslipidemia by exogenous L-arginine in diabetic rats. Biochimie. 2001;83(5):453–8.
El-Missiry MA, Othman AI, Amer MA. L-Arginine ameliorates oxidative stress in alloxan-induced experimental diabetes mellitus. J Appl Toxicol. 2004;24(2):93–7.
Acknowledgements
The authors would like to thank Mr. Duxiang Xiao (from Guangzhou IGE Biotechnology Ltd.) for the kind assistance with microbiota sequencing and analysis.
Funding
The work is supported by grants 201831828 from the Team Innovation Project of Guangzhou Education Bureau, grants 2018A030310168, and 2018A030310290 from the Natural Science Foundation of Guangdong Province, and grants 201905010004 from Guangzhou Key Laboratory Fund.
Author information
Authors and Affiliations
Contributions
Xiaomei Li and Zhihua Liu conceived and designed the experiments. Xiaomei Li, Chen Li, Yuanqi Li, and Cong Liu performed the experiments, analyzed and interpreted the data. Xiaomei Li drafted the manuscript. Zhihua Liu, Ting Liu, and Xue Liang revised and approved the final manuscript. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was carried out in compliance with the ARRIVE guidelines 2.0. All methods were performed in accordance with the relevant guidelines and regulations (Directive 2010/63/EU in Europe), and the animal study protocols were approved by committee review of animal experiments in Guangzhou Medical University (Document no. 2018-084).
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, X., Li, C., Li, Y. et al. Sodium nitroprusside protects HFD induced gut dysfunction via activating AMPKα/SIRT1 signaling. BMC Gastroenterol 21, 359 (2021). https://doi.org/10.1186/s12876-021-01934-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12876-021-01934-y