
Abstract
Background
Indolent T-cell lymphoproliferative disorder of the gastrointestinal tract is a rare low-grade clonal lymphoid proliferation, included as a provisional entity in the current World Health Organization classification. The disease is generally localized to the gastrointestinal tract, mainly small bowel and colon. Involvement of other organs is infrequently reported. The majority of patients show a protracted clinical course with persistent disease. A prolonged survival, even without treatment, is common.
Case presentation
A 28-year-old woman had a 2-year history of dyspepsia and lactose intolerance. Autoimmune diseases and celiac disease were excluded. No gross lesions were identified by endoscopy. Multiple gastric biopsies showed a small-sized lymphoid infiltrate, expanding the lamina propria, with a non-destructive appearance. The lymphoid cells were positive for CD3, CD4, CD5, CD7 and negative for CD20, CD8, CD56, CD103, PD1, CD30, ALK1, CD10, BCL6, perforin, TIA-1, Granzyme B and Epstein-Barr virus-encoded RNA. KI-67 index was low (5%). Molecular analysis revealed a clonal T-cell receptor γ rearrangement. Bone marrow was microscopically free of disease, but molecular testing identified the same T-cell receptor γ rearrangement present in the gastric biopsies. After the diagnosis of indolent T-cell lymphoproliferative disorder of the gastrointestinal tract, the patient received steroid therapy, only for 2 months. She is alive, with a stable disease restricted to the stomach, at 12 months from diagnosis.
Conclusions
Indolent T-cell lymphoproliferative disorder is usually a disease of adulthood (median age: 51 yrs). The small bowel and colon are the sites most commonly involved. Our case occurred in a young woman and affected the stomach, sparing small intestine and colon. Clonality testing identified involvement of bone marrow, a site infrequently affected in this disease. Our aim is focusing on the main diagnostic issues. If appropriate immunostainings and molecular analysis are not performed, the subtle infiltrate may be easily overlooked. The risk of misdiagnosis as more aggressive lymphomas, causing patient overtreatment, needs also to be considered.
Similar content being viewed by others

Background
The gastrointestinal (GI) tract is the most common extra-nodal site of involvement for non-Hodgkin lymphomas [1]. T-cell lymphomas primarily or secondarily involving the GI tract are aggressive diseases in the vast majority of cases [1]. Indolent T-cell lymphoproliferative disorder (ITLPD) is a non-aggressive, clonal, mature T-cell disorder, which can affect almost any part of the GI tract, although it is more uncommon in stomach, esophagus and oral cavity [1,2,3,4,5,6,7]. Patients often show prolonged survival even without any treatment [1,2,3,4,5,6,7]. In daily routine, GI biopsies evaluation represents a common challenge for pathologists, who have to make timely and accurate diagnoses, in order to establish the adequate treatment.
We report a case of ITLPD in a young female with mild gastric symptoms. The disease was limited to the stomach, without small bowel and colon involvement. Clonal T-cell receptor γ rearrangement was identified in the bone marrow. The disease remained stable without chemotherapy, at 12 months from diagnosis. We focus on the main potential diagnostic pitfalls, which can cause inappropriate patient management.
Case presentation
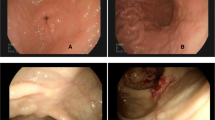
A 28-year-old immunocompetent woman presented with a 2-year history of lactose intolerance and dyspepsia which had been worsening in the last 6 months. Blood exams and lactate dehydrogenase (LDH) level were within normal limits. Laboratory tests ruled out celiac disease as well as other autoimmune diseases. No hepatosplenomegaly and lymphadenopathy were present. By upper gastrointestinal endoscopy, the esophagus showed normal caliber and morphology and the overlying mucosa was unremarkable. A mild incontinence of the cardia was observed. The stomach was normally distensible and mainly the gastric body mucosa appeared mildly hyperemic and edematous (Fig. 1). The pylorus was symmetric; the bulb as well as the second duodenal portion were unremarkable.

Endoscopic view showing hyperemic gastric mucosa
Microscopically, the lamina propria showed a subtle, non-destructive and monotonous lymphoid infiltrate composed of small lymphocytes (Figs. 2, 3), focally invading the glandular epithelium and realizing rare lymphoepithelial lesions (LELs). Few eosinophils and plasma cells were admixed. The lymphoid cells were diffusely positive for T-cell markers such as CD3, CD4 (Fig. 4), CD5 and CD7 and negative for CD20, CD8, CD56, CD103, PD1, CD30, ALK1, CD10, BCL6, perforin, TIA-1 and Granzyme B. In situ hybridization for Epstein-Barr virus (EBV)-encoded RNA (EBER) was negative. The Ki67 proliferative fraction was low (5%). PCR-based T-cell receptor (TCR) clonality testing was performed [8] and, according to EuroClonality/BIOMED-2 guidelines, clonal T-cell receptor γ (TCRγ) rearrangement was reported. The case was interpreted as ITLPD. Bone marrow biopsy showed a subtle and mild, interstitial lymphoid infiltrate with a mixed immunohistochemical phenotype. However, molecular testing identified an identical TCRγ rearrangement. Colonoscopy was negative. Multiple biopsies from small intestine and colon were taken. Immunohistochemistry and molecular analysis ruled out the presence of disease in small and large bowel. Computerized tomography (CT) scan and positron emission tomography (PET) scan were negative. Prednisone (50 mg daily) was administered for 2 months. GI biopsies performed 12 months after the initial diagnosis, confirmed a stable and persistent disease limited to the gastric mucosa. Due to lack of symptoms, the steroid therapy was stopped and a wait and see policy adopted.

Medium power view showing lymphocytes expanding the lamina propria of gastric mucosa. The glands are preserved with few lymphocytes invading the epithelium (HE 200 × magnification)

High power view highlighting cytological details of the infiltrate made up of small-sized, mature-appearing lymphocytes (HE 400 × magnification)

Mucosal lymphoid infiltrate diffusely CD4-positive (immunostain)
Discussion and conclusions
ITLPD of the GI tract is a rare low-grade clonal lymphoid proliferation, included as a provisional entity in the current World Health Organization (WHO) classification [1]. The etiology is unknown, although few cases are reported in patients with inflammatory bowel disease and autoimmune disorders [2,3,4,5,6,7]. Any part of the GI tract can be involved with single or multiple lesions [2,3,4,5,6,7]. The small intestine and colon are the sites most frequently affected. Involvement of the stomach, as occurred in our case, is uncommon as well as esophageal and oral involvement [7]. ITLPD can show heterogeneous phenotype: most cases express CD8, some express CD4 and rare are double negative [1]. The identification of clonal rearrangement of TCR (αβ or γ) is essential for the diagnosis [1]. ITLPD remains localized to the GI tract often for a long duration, with chronic relapsing clinical course. Prolonged survival with persistent disease is common; conventional chemotherapy is usually not effective [1, 7]. Dissemination to other organs can be observed with disease progression [1, 4]. Bone marrow involvement has been infrequently reported [5, 9, 10]. Higher-grade T-cell lymphomas may rarely develop in a subset of cases [1].
Our case underlines how challenging can be the recognition of this entity for both clinicians and pathologists. ITLPD of the GI tract can be either overlooked or misdiagnosed as an aggressive lymphoma. Firstly, the symptoms are non-specific (abdominal pain, diarrhea, vomiting, food intolerance, dyspepsia, bleeding) and commonly lasting for long time before diagnosis. ITLPD is often clinically misinterpreted as refractory celiac disease or inflammatory bowel disease (IBD) with delay in diagnosis [1,2,3,4,5,6,7]. Secondly, the endoscopic features are non-specific as well. The mucosa can appear normal or slightly hyperemic, as in our case. Sometimes prominent folds, erosions or nodules are present [2,3,4,5,6,7]. Lastly, the mucosal lymphoid infiltrate is subtle, generally limited to the mucosa and rarely extending to the submucosa, without forming tumor masses [2,3,4,5,6,7]. The infiltrate can be easily overlooked, if appropriate immunohistochemical and molecular analyses are not performed. A misleading aberrant expression of B-cell markers, such as CD20, has been reported in one case of ITLPD by Wang et al. [9]. It needs to be emphasized that ITLPD diagnosis always requires clonality testing. All cases of ITLPD show clonal rearrangement of TCR genes, either TCRβ or TCRγ [1,2,3,4,5]. Recent data suggest that ITLPD are genetically heterogeneous and share some pathogenetic mechanisms with other intestinal T-cell lymphomas as mutations in the JAK-STAT signaling pathway [11]. Genetic abnormalities involving TET2, DNMT3A, KMT2D genes have also been recently reported [11].
It is mandatory to distinguish ITLPD from aggressive T-cell lymphomas, which can involve the GI tract, in order to avoid unnecessary therapy. In aggressive lymphomas, such as monomorphic epitheliotropic intestinal T-cell lymphoma (MEITL) and enteropathy associated T-cell lymphoma (EATL), about half of the patients present acutely with intestinal obstruction or perforation, although others have an insidious presentation with abdominal pain and diarrhea, more similar to ITLPD [1, 6, 7]. In EATL and MEITL, the lymphoid infiltrate is generally transmural, with a destructive pattern of growth. Pleomorphism is more common in EATL, whereas in MEITL the cells have a more uniform appearance. The proliferation index is high [1, 6, 7]. Both EATL and MEITL are aggressive neoplasms not responding to current therapy. Differently, in ITLPD the infiltrate is often limited to the mucosa, without forming a mass, it is bland-looking with a low proliferative fraction. Extranodal, NK/T-cell lymphoma, nasal-type, needs to be ruled out. It arises mostly in the upper aerodigestive tract. GI involvement can occur generally later in the course of the disease or more rarely the GI tract represents the primary site of the disease [1, 5]. Necrosis, angiocentricity, angioinvasion and EBER positivity are characteristics features of this aggressive lymphoma, typically absent in ITLPD. To be mentioned the indolent NK-cell proliferation of the GI tract, variably labelled as NK-cell enteropathy and lymphomatoid gastropathy [7]. The clinical course is often indolent and protracted, like in ITLPD. EBER negativity is helpful to rule out extranodal-NK/T-cell lymphoma, nasal type, which represents the major diagnostic pitfall. Other entities, typically presenting outside the GI tract, as peripheral T-cell lymphoma not otherwise specified or anaplastic large cell lymphoma may sometimes present as intestinal disease [1]. Clinico-pathological correlation is essential to reach the correct diagnosis.
In conclusion, we report a case of CD4-positive ITLPD with gastric presentation, an uncommon site compared to small and large bowel. In absence of chemotherapy, the disease remained confined to the stomach, at 12 months from the initial biopsy. Despite bone marrow appeared disease-free histologically, T-cell clonal rearrangement was identified in bone marrow. ITLPD usually remains localized to the GI tract for years and involvement of other sites such as bone marrow is rare and usually observed with disease progression. Clinicians and pathologists need to be aware of ITLPD as, in order to make the correct diagnosis, clinico-pathological correlation is essential. After having ruled out autoimmune disorders, a crucial point in the diagnosis of ITLPD is the identification of a bland T-cell infiltrate, usually limited to the mucosa, resulting monoclonal with molecular analysis.
Availability of data and materials
All the original data supporting our research are described in the Case presentation section and in the figures' legends.
Abbreviations
- GI:
-
Gastrointestinal
- ITLPD:
-
Indolent T-cell lymphoproliferative disorder
- LDH:
-
Lactate dehydrogenase
- LEL:
-
Lymphoepithelial lesion
- EBV:
-
Epstein-Barr virus
- EBER:
-
EBV-encoded RNA
- WHO:
-
World Health Organization
- IBD:
-
Inflammatory bowel disease
- MEITL:
-
Monomorphic epitheliotropic intestinal T-cell lymphoma
- EATL:
-
Enteropathy associated T-cell lymphoma
References
Swerdlow SH. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: International Agency for Research on Cancer; 2017.
Carbonnel F, Lavergne A, Messing B, Tsapis A, Berger R, Galian A, Nemeth J, Brouet JC, Rambaud JC. Extensive small intestinal T-cell lymphoma of low-grade malignancy associated with a new chromosomal translocation. Cancer. 1994;73(4):1286–91.
Perry AM, Warnke RA, Hu Q, Gaulard P, Copie-Bergman C, Alkan S, Wang HY, Cheng JX, Bacon CM, Delabie J, Ranheim E, Kucuk C, Hu X, Weisenburger DD, Jaffe ES, Chan WC. Indolent T-cell lymphoproliferative disease of the gastrointestinal tract. Blood. 2013;122(22):3599–606. https://doi.org/10.1182/blood-2013-07-512830.
Matnani R, Ganapathi KA, Lewis SK, Green PH, Alobeid B, Bhagat G. Indolent T- and NK-cell lymphoproliferative disorders of the gastrointestinal tract: a review and update. Hematol Oncol. 2017;35(1):3–16. https://doi.org/10.1002/hon.2317.
Soderquist CR, Bhagat G. Gastrointestinal T- and NK-cell lymphomas and indolent lymphoproliferative disorders. Semin Diagn Pathol. 2020;37(1):11–23. https://doi.org/10.1053/j.semdp.2019.08.001.
Weindorf SC, Smith LB, Owens SR. Update on gastrointestinal lymphomas. Arch Pathol Lab Med. 2018;142(11):1347–51. https://doi.org/10.5858/arpa.2018-0275-RA.
van Vliet C, Spagnolo DV. T- and NK-cell lymphoproliferative disorders of the gastrointestinal tract: review and update. Pathology. 2020;52(1):128–41. https://doi.org/10.1016/j.pathol.2019.10.001.
Langerak AW, Groenen PJ, Brüggemann M, Beldjord K, Bellan C, Bonello L, Boone E, Carter GI, Catherwood M, Davi F, Delfau-Larue MH, Diss T, Evans PA, Gameiro P, Garcia Sanz R, Gonzalez D, Grand D, Håkansson A, Hummel M, Liu H, Lombardia L, Macintyre EA, Milner BJ, Montes-Moreno S, Schuuring E, Spaargaren M, Hodges E, van Dongen JJ. EuroClonality/BIOMED-2 guidelines for interpretation and reporting of Ig/TCR clonality testing in suspected lymphoproliferations. Leukemia. 2012;26(10):2159–71. https://doi.org/10.1038/leu.2012.246.
Montes-Moreno S, King RL, Oschlies I, Ponzoni M, Goodlad JR, Dotlic S, Traverse-Glehen A, Ott G, Ferry JA, Calaminici M. Update on lymphoproliferative disorders of the gastrointestinal tract: disease spectrum from indolent lymphoproliferations to aggressive lymphomas. Virchows Arch. 2020;476(5):667–81. https://doi.org/10.1007/s00428-019-02704-8.
Wang X, Ng CS, Chen C, Yu G, Yin W. An unusual case report of indolent T-cell lymphoproliferative disorder with aberrant CD20 expression involving the gastrointestinal tract and bone marrow. Diagn Pathol. 2018;13(1):82. https://doi.org/10.1186/s13000-018-0762-4.
Soderquist CR, Patel N, Murty VV, Betman S, Aggarwal N, Young KH, Xerri L, Leeman-Neill R, Lewis SK, Green PH, Hsiao S, Mansukhani MM, Hsi ED, de Leval L, Alobeid B, Bhagat G. Genetic and phenotypic characterization of indolent T-cell lymphoproliferative disorders of the gastrointestinal tract. Haematologica. 2020;105(7):1895–906. https://doi.org/10.3324/haematol.2019.230961.
Acknowledgements
None.
Funding
The authors have no financial ties to disclose.
Author information
Authors and Affiliations
Contributions
ZaM and AS designed the study and wrote the manuscript; ZiM, SA, CC and AV collected clinical data; SF, MG, RS and AS performed hystophatological examination; ZiM was involved in review, editing and validation of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Local ethics committee (Comitato Etico dell'Area Vasta Emilia Nord, Italy) ruled that no formal ethics approval was required in this particular case. Patient gave consent to participate.
Consent for publication
Written informed consent was obtained from patient for publication of this Case Report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zanelli, M., Zizzo, M., Sanguedolce, F. et al. Indolent T-cell lymphoproliferative disorder of the gastrointestinal tract: a tricky diagnosis of a gastric case. BMC Gastroenterol 20, 336 (2020). https://doi.org/10.1186/s12876-020-01488-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12876-020-01488-5