
Abstract
Background
Arrhythmogenic cardiomyopathy (AC) is a rare, heritable myocardial disorder that is a leading cause of ventricular arrhythmia and sudden cardiac death (SCD) in young people. Desmoplakin (DSP) mutations account for 3–20% of AC cases. However, the number of patients with DSP mutations is extremely small in all published reports and genotype–phenotype correlations are scant and mostly non-gene-specific.
Case presentation
A 45-year-old man was admitted after an out-of-hospital cardiac arrest, with documented ventricular fibrillation. He had no previous history of heart disease or family history of SCD or cardiomyopathy. The cardiac magnetic resonance showed a mildly dilated left ventricle with an ejection fraction of 30% and a non-dilated right ventricle with mildly depressed systolic function, and extensive subepicardial late gadolinium enhancement. Genetic screening identified a heterozygote nonsense mutation in DSP (NM_004415.2: c.478 C > T; p.Arg160Ter). Cascade genetic screening of the relatives revealed a high prevalence of the genotype and cutaneous phenotype, but a very low penetrance of the cardiac phenotype.
Conclusions
We report a case of SCD and an autosomal dominant mutation in DSP that causes arrhythmogenic dilated cardiomyopathy/AC. Like the recessive mutation in DSP known to cause Carvajal syndrome, Arg160Ter may be associated with cutaneous abnormalities.
Similar content being viewed by others


Background
Arrhythmogenic cardiomyopathy (AC) is a rare, heritable myocardial disorder that is a leading cause of ventricular arrhythmia and sudden cardiac death (SCD) in people age ≤ 35 years [1, 2]. Progressive loss of myocardium and its replacement by fibrofatty tissue is the pathological hallmark of the disease [2]. AC is most well recognized in its classic subtype with right-sided preponderance, arrhythmogenic right ventricular cardiomyopathy [3]. More recently, a broader spectrum of disorders affecting either or both ventricles and an increased propensity to ventricular arrhythmias has been recognized, arousing many questions regarding pathogenesis and clinical management of AC [4, 5].
There is no single gold standard for the diagnosis, which is mainly based on demonstrating characteristic electrical, structural, and/or histological abnormalities. In addition, a positive family history for a pathogenic genetic mutation also contributes to the diagnosis [6]. Mutations in the genes encoding proteins of the desmosomal complex account for 30 to 70% of cases, namely plakoglobin (JUP), plakophilin-2 (PKP2), desmoplakin (DSP), desmocollin-2 (DSC2), and desmoglein-2 (DSG2) [1, 7]. Noteworthy, pathogenic desmosomal mutations are also common in patients with dilated cardiomyopathy (DCM), including 3% of cases in a recent large DCM cohort [8]. Inheritance of AC is classically considered autosomal dominant with age-related, reduced penetrance, and variable expressivity [9]. Autosomal recessive forms are rare but recognized, most prominently in the cardiocutaneous syndromes of Naxos and Carvajal [1, 10, 11]. Nonetheless, emerging evidence suggests that many cases of AC are oligogenic or even multifactorial with both genomic and environmental factors contributing to pathogenesis [12]. DSP mutations account for 3–20% of AC cases [1, 9, 12, 13]. However, the number of patients with DSP mutations is extremely small in all published reports and genotype–phenotype correlations are scant and mostly non-gene-specific [13].
Here, we describe a novel heterozygote nonsense mutation c.478C > T (p.Arg160Ter) in the gene encoding DSP leading to DCM/AC presenting as SCD.
Case presentation
A 45-year-old man was admitted after an out-of-hospital cardiac arrest, with documented ventricular fibrillation. He had no viral prodrome, previous history of heart disease, or family history of SCD or cardiomyopathy. Besides interventricular conduction delay, the baseline electrocardiogram showed T wave inversion in the lateral leads (Fig. 1A). Myocardial ischemia due to epicardial obstructive coronary artery disease was excluded by coronary angiography. The echocardiogram performed on admission revealed a dilated left ventricle (LV) with severely depressed LV ejection fraction (LVEF of 29%); a right ventricle (RV) with preserved function; and cardiac valves and pericardium were normal. The patient evolved into cardiogenic shock and vasopressor and inotropic support was started with norepinephrine and dobutamine. During the first 48 hours of hospitalization, the patient presented one episode of sustained monomorphic ventricular tachycardia with right bundle branch block (RBBB) morphology requiring electrical cardioversion (Fig. 1B). The cardiac magnetic resonance (CMR) showed a mildly dilated LV with an LVEF of 30% due to global hypokinesis; a non-dilated RV with mildly depressed systolic function; and an extensive subepicardial pattern of late gadolinium enhancement (LGE-CMR), almost circumferential in some regions, along with disperse intramyocardial lesions (Fig. 2). No mediastinal lymphadenopathy or pulmonary granulomas were seen. After an implantable cardioverter-defibrillator implantation, the patient was discharged under an angiotensin receptor–neprilysin inhibitor, beta-blocker, mineralocorticoid antagonist, and antiarrhythmic treatment with amiodarone.

A Twelve-lead electrocardiogram after aborted cardiac arrest. B Rhythm strip showing monomorphic ventricular tachycardia

Cardiac magnetic resonance late gadolinium enhancement images showing typical pattern: intramyocardial (*) linear lesion in (A) and (B), long and correspondent short-axis respectively, here affecting the inferior septum; very extensive subepicardial scar (arrow) in (C) and (D), again long and correspondent short-axis respectively, here affecting particularly the anterior and anterolateral wall, but being almost circumferential and also affecting the right apex (A)
Analysis of the index patient’s genomic DNA revealed a nonsense mutation in DSP (NM_004415.2: c.478C > T; p.Arg160Ter). Cascade genetic screening of the relatives revealed a high prevalence of the genotype and cutaneous phenotype (curly hair and palmoplantar keratoderma), but a very low penetrance of the cardiac phenotype (only the index case) (Fig. 3).

A Pedigree of the family. The index case is indicated by an arrow. Cases with the p.Arg160Ter in the DSP gene mutation are shown as (+), and those without the mutation are shown as (−). Squares represent males, and circles represent females. Black-filled symbols represent individuals with cardiac phenotype or symptoms. B Palmoplantar keratoderma affecting index patient’s sister
During the next 24 months of follow-up, neither symptoms nor implantable cardioverter-defibrillator therapy recurred and a LVEF recovery to 45–50% was observed.
Discussion and conclusions
Genetic testing is widely recommended in SCD when there is a cardiac phenotype, including only genes with robust gene-disease association [14]. Nonetheless, growing evidence supports a broad, multi-phenotype genetic testing even without a suspected phenotype, as there is the notion that pathogenic variants in cardiomyopathy genes may result in SCD without overt structural changes [15,16,17].
The combination of aborted cardiac death/ventricular tachycardia with RBBB morphology and a CMR with biventricular dysfunction and a non-ischemic pattern of LGE-CMR raised the suspicion of sarcoidosis, myocarditis, AC, and DCM. Undeniably, genetic testing was crucial for the final diagnosis.
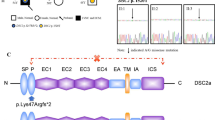
We identified a nonsense mutation in the DSP gene (c.478C > T; p.Arg160Ter), which was absent from large population studies. This nonsense variant leads to a premature termination codon at position 160, which is predicted to lead to a truncated protein. The DSP is a large protein (2871 AA) composed of three domains characterized by an N-terminal plakoglobin/plakophilin binding domain, the central coiled-coil rod dimerization domain, and a C-terminal intermediate filament binding domain [18]. The DSP gene encodes two different splice isoforms produced by alternate splicing (DSP1 and DSP2). The longer peptide DSP-1 is the dominant isoform in heart [19], whereas DSP-2 uses an internal splice donor site and therefore has two-thirds fewer amino acids within the central domain compared to DSP-1 [20]. The Arg160Ter mutation is located in the first domain and hence appears in both isoforms (Fig. 4). This pathogenic variant in the DSP gene has been reported several times in association with AC and DCM [21].

Structure of the DSP gene and the Arg160Ter mutation
DSP is an essential component of the desmosome in epithelial cells and cardiomyocytes. Several human DSP variants have been linked to inherited diseases that variably result in skin fragility, palmoplantar keratoderma, woolly hair, and cardiomyopathies [22]. Nevertheless, a clear phenotype-genotype correlation regarding DSP mutations has not yet been drawn.
There are many diagnostic challenges associated with AC, particularly with the predominant LV involvement (ALVC) form of the disease, that may not display the classic revised 2010 Task Force Criteria and exhibits considerable genetic and phenotypic overlap with DCM [23, 24]. More recently, the “Padua criteria” for the diagnosis of AC included specific criteria for ALVC [25]. Nevertheless, in the absence of RV involvement that meets arrhythmogenic right ventricular cardiomyopathy criteria, the diagnosis relies heavily on a pathogenic mutation combined with CMR results [25]. The distinction between the AC and DCM in our patient was difficult. First, the mutation identified in DSP has been described in both cardiomyopathies [21]. Second, the ECG failed to show low voltage in limb leads, a low specificity but characteristic pattern of ALVC [23]. Additionally, while it revealed T wave inversion in lateral leads which may be present in ALVC, the interventricular conduction delay is more frequently present in DCM [24]. Third, the clinical presentation of SCD in a previously asymptomatic patient favors ALVC diagnosis, as well as the monomorphic sustained ventricular tachycardia with RBBB morphology, the latter is a minor criterion for ALVC [24, 25]. Lastly, although our patient presented severe systolic dysfunction, he had only mild LV dilatation and a large amount of non-ischemic LGE-CMR, a finding more typical in ALVC, as impairment of the LV is often present without scar in DCM [24, 26]. Remarkably, the morpho-functional and structural LV abnormalities are minor and major criteria for ALVC diagnosis, respectively [25].
AC is characterized by incomplete and age-dependent penetrance coupled with variable phenotypic expression [1]. DSP mutations are associated with more penetrant phenotypes with an increased arrhythmic propensity, which is often the first manifestation of the disease [13], as was the case with our patient. Secondly, DSP mutations seem to be more often associated with a predominant LV phenotype or biventricular involvement [13]. Lastly, although autosomal dominant AC forms have been reported as cardiac-specific, a cutaneous phenotype has been described in DSP mutation carriers [22, 27]. A highly penetrant cutaneous phenotype of curly hair and to a lesser extent, palmoplantar keratoderma, is particularly associated with dominant, nonsense, or frameshift mutations falling to the N-terminal domain of DSP [27], just like the mutation described in this family, that besides curly hair also presented the palmoplantar features (Fig. 3B). Increased awareness of this phenotype may facilitate a timely diagnosis of AC in the absence of overt cardiac features [27].
Importantly, cascade screening revealed a high prevalence of the genotype and cutaneous phenotype, but a very low penetrance of the cardiac phenotype. Indeed, the index patient was the only family member with overt cardiac disease. Remarkably, he was the only member of the family who had practiced high-intensity exercise, which is in agreement with the concept that the volume of exercise has a critical influence on the evolution of AC [12].
Nevertheless, the crucial question is how to predict the risk of a major arrhythmic event among asymptomatic mutation carriers [28], as SCD may be the form of presentation. The most important factors to consider when determining arrhythmic risk in AC include: (1) electrical instability, (2) proband status, (3) manifest structural disease, (4) cardiac syncope, (5) male gender, (6) multiple mutations or a mutation in TMEM 43, and (7) vigorous exercise [29].
The arrhythmic risk in AC-causing desmosomal mutation carriers significantly increases when the AC phenotype becomes overt [30, 31]. The major challenge is the detection of structural disease in ALVC, particularly in DSP-mutation carriers [30]. The so-called ‘concealed phase’ of the disease may be a result of the low sensitivity of routine clinical tests, namely ECG and echocardiography [28]. The value of LGE-CMR to detect fibrosis as an early finding of LV involvement in still asymptomatic and not fulfilling criteria for AC diagnosis is emerging [5]. The most distinctive feature of ALVC phenotype is the large amount of LV myocardial fibrosis, which is directly related to the LV systolic dysfunction [24, 26], and is characteristically in the posterolateral and septal LV wall [32,33,34]. More recently, a subepicardial ring-like scar pattern was found to be characteristic in the DSP genotype and is likely to have diagnostic value in ALVC [26]. Interestingly, our index patient showed such a typical pattern, which allowed suspicion of AC. The non-invasive early LV tissue characterization highlights the LGE-CMR potential in broadening the diagnostic criteria for AC and improving risk stratification [6, 32, 35]. However, further studies are warranted to assess whether and when systematic evaluation of DSP-mutation carriers with LGE-CMR in addition to traditional tests may improve our ability to stratify the arrhythmic risk in AC mutation carriers [26, 28, 30].
This case highlights the importance of genetic testing in the determination of arrhythmic cardiac arrest etiology. Here, we present an autosomal dominant mutation in DSP that causes arrhythmogenic DCM/AC. Like the recessive mutation in DSP known to cause Carvajal syndrome, Arg160Ter may be associated with cutaneous abnormalities. Further studies are needed to improve our understanding of AC, and ultimately, provide a more accurate diagnostic algorithm and improved genetics counseling and management strategies.
Availability of data and materials
Not applicable.
Abbreviations
- AC:
-
Arrhythmogenic cardiomyopathy
- ALVC:
-
Arrhythmogenic cardiomyopathy with predominant LV involvement
- CMR:
-
Cardiac magnetic resonance
- DCM:
-
Dilated cardiomyopathy
- DSP:
-
Desmoplakin
- LGE-CMR:
-
Late gadolinium enhancement—cardiac magnetic resonance
- LV:
-
Left ventricle
- LVEF:
-
Left ventricular ejection fraction
- RBBB:
-
Right bundle branch block
- RV:
-
Right ventricle
- SCD:
-
Sudden cardiac death
References
Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace. 2011;13(8):1077–109.
Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318(3):129–33.
Corrado D, Link MS, Calkins H. Arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2017;376(1):61–72.
Elliott PM, Anastasakis A, Asimaki A, Basso C, Bauce B, Brooke MA, et al. Definition and treatment of arrhythmogenic cardiomyopathy: an updated expert panel report. Eur J Heart Fail. 2019;21(8):955–64.
Sen-Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, et al. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol. 2008;52(25):2175–87.
Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J. 2010;31(7):806–14.
Sen-Chowdhry S, Syrris P, Ward D, Asimaki A, Sevdalis E, McKenna WJ. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation. 2007;115(13):1710–20.
Gigli M, Merlo M, Graw SL, Barbati G, Rowland TJ, Slavov DB, et al. Genetic risk of arrhythmic phenotypes in patients with dilated cardiomyopathy. J Am Coll Cardiol. 2019;74(11):1480–90.
Pilichou K, Thiene G, Bauce B, Rigato I, Lazzarini E, Migliore F, et al. Arrhythmogenic cardiomyopathy. Orphanet J Rare Dis. 2016;11:33.
Protonotarios N, Tsatsopoulou A. Naxos disease: cardiocutaneous syndrome due to cell adhesion defect. Orphanet J Rare Dis. 2006;1:4.
Baykan A, Olgar Ş, Argun M, Özyurt A, Pamukçu Ö, Üzüm K, et al. Different clinical presentations of Naxos disease and Carvajal syndrome: case series from a single tertiary center and review of the literature. Anatol J Cardiol. 2015;15(5):404–8.
James CA, Syrris P, van Tintelen JP, Calkins H. The role of genetics in cardiovascular disease: arrhythmogenic cardiomyopathy. Eur Heart J. 2020;41(14):1393–400.
Castelletti S, Vischer AS, Syrris P, Crotti L, Spazzolini C, Ghidoni A, et al. Desmoplakin missense and non-missense mutations in arrhythmogenic right ventricular cardiomyopathy: genotype-phenotype correlation. Int J Cardiol. 2017;249:268–73.
Stiles MK, Wilde AAM, Abrams DJ, Ackerman MJ, Albert CM, Behr ER, et al. 2020 APHRS/HRS expert consensus statement on the investigation of decedents with sudden unexplained death and patients with sudden cardiac arrest, and of their families. Heart Rhythm. 2021;18(1):e1–50.
Isbister JC, Nowak N, Butters A, Yeates L, Gray B, Sy RW, et al. “Concealed cardiomyopathy” as a cause of previously unexplained sudden cardiac arrest. Int J Cardiol. 2021;324:96–101.
Ripoll-Vera T, Pérez Luengo C, Borondo Alcázar JC, García Ruiz AB, Sánchez Del Valle N, Barceló Martín B, et al. Sudden cardiac death in persons aged 50 years or younger: diagnostic yield of a regional molecular autopsy program using massive sequencing. Rev Esp Cardiol. 2021;74(5):402–13.
Mellor G, Laksman ZWM, Tadros R, Roberts JD, Gerull B, Simpson CS, et al. Genetic testing in the evaluation of unexplained cardiac arrest: from the CASPER (Cardiac Arrest Survivors With Preserved Ejection Fraction Registry). Circ Cardiovasc Genet. 2017;10(3):e001686.
Getsios S, Huen AC, Green KJ. Working out the strength and flexibility of desmosomes. Nat Rev Mol Cell Biol. 2004;5(4):271–81.
Uzumcu A, Norgett EE, Dindar A, Uyguner O, Nisli K, Kayserili H, et al. Loss of desmoplakin isoform I causes early onset cardiomyopathy and heart failure in a Naxos-like syndrome. J Med Genet. 2006;43(2):e5.
Green KJ, Parry DA, Steinert PM, Virata ML, Wagner RM, Angst BD, et al. Structure of the human desmoplakins Implications for function in the desmosomal plaque. J Biol Chem. 1990;265(5):2603–12.
te Riele AS, Bhonsale A, James CA, Rastegar N, Murray B, Burt JR, et al. Incremental value of cardiac magnetic resonance imaging in arrhythmic risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol. 2013;62(19):1761–9.
Favre B, Begré N, Marsili L, van Tintelen JP, Borradori L. Desmoplakin gene variants and risk for arrhythmogenic cardiomyopathy. Circ Genom Precis Med. 2018;11(9):e002241.
Corrado D, van Tintelen PJ, McKenna WJ, Hauer RNW, Anastastakis A, Asimaki A, et al. Arrhythmogenic right ventricular cardiomyopathy: evaluation of the current diagnostic criteria and differential diagnosis. Eur Heart J. 2020;41(14):1414–29.
Cipriani A, Bauce B, De Lazzari M, Rigato I, Bariani R, Meneghin S, et al. Arrhythmogenic right ventricular cardiomyopathy: characterization of left ventricular phenotype and differential diagnosis with dilated cardiomyopathy. J Am Heart Assoc. 2020;9(5):e014628.
Corrado D, Perazzolo Marra M, Zorzi A, Beffagna G, Cipriani A, Lazzari M, et al. Diagnosis of arrhythmogenic cardiomyopathy: the Padua criteria. Int J Cardiol. 2020;319:106–14.
Augusto JB, Eiros R, Nakou E, Moura-Ferreira S, Treibel TA, Captur G, et al. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: a comprehensive genotype-imaging phenotype study. Eur Heart J Cardiovasc Imaging. 2020;21(3):326–36.
Maruthappu T, Posafalvi A, Castelletti S, Delaney PJ, Syrris P, O’Toole EA, et al. Loss-of-function desmoplakin I and II mutations underlie dominant arrhythmogenic cardiomyopathy with a hair and skin phenotype. Br J Dermatol. 2019;180(5):1114–22.
Tsatsopoulou A. Desmoplakin: highlights on a left ventricular arrhythmogenic disorder. Int J Cardiol. 2017;249:324–5.
Calkins H, Corrado D, Marcus F. Risk stratification in arrhythmogenic right ventricular cardiomyopathy. Circulation. 2017;136(21):2068–82.
Zorzi A, Rigato I, Pilichou K, Perazzolo Marra M, Migliore F, Mazzotti E, et al. Phenotypic expression is a prerequisite for malignant arrhythmic events and sudden cardiac death in arrhythmogenic right ventricular cardiomyopathy. Europace. 2016;18(7):1086–94.
Chivulescu M, Lie ØH, Popescu BA, Skulstad H, Edvardsen T, Jurcut RO, et al. High penetrance and similar disease progression in probands and in family members with arrhythmogenic cardiomyopathy. Eur Heart J. 2020;41(14):1401–10.
Segura-Rodríguez D, Bermúdez-Jiménez FJ, Carriel V, López-Fernández S, González-Molina M, Oyonarte Ramírez JM, et al. Myocardial fibrosis in arrhythmogenic cardiomyopathy: a genotype-phenotype correlation study. Eur Heart J Cardiovasc Imaging. 2020;21(4):378–86.
te Riele AS, Tandri H, Bluemke DA. Arrhythmogenic right ventricular cardiomyopathy (ARVC): cardiovascular magnetic resonance update. J Cardiovasc Magn Reson. 2014;16(1):50.
He J, Xu J, Li G, Zhou D, Li S, Zhuang B, et al. Arrhythmogenic left ventricular cardiomyopathy: a clinical and CMR study. Sci Rep. 2020;10(1):533.
Miles C, Finocchiaro G, Papadakis M, Gray B, Westaby J, Ensam B, et al. Sudden death and left ventricular involvement in arrhythmogenic cardiomyopathy. Circulation. 2019;139(15):1786–97.
Acknowledgements
Not applicable.
Funding
This study was supported by grant POCI-01-0145-FEDER-032414 to RB. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
CSF analyzed, interpreted the patient data, performed the literature review, and drafted the manuscript. RB analyzed, interpreted the patient data, and revised the manuscript. TT analyzed, interpreted the patient data, and revised the manuscript. LG participated in data interpretation and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the patient for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Santos-Ferreira, C., Baptista, R., Teixeira, T. et al. A 45-year-old man with sudden cardiac death, cutaneous abnormalities and a rare desmoplakin mutation: a case report and literature review. BMC Cardiovasc Disord 22, 41 (2022). https://doi.org/10.1186/s12872-022-02472-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12872-022-02472-5