
Abstract
Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) is one of the most arrhythmogenic forms of inherited cardiomyopathy and a frequent cause of sudden death in the young. Affected individuals typically present between the second and fourth decade of life with arrhythmias coming from the right ventricle. Pathogenic mutations in genes encoding the cardiac desmosome can be found in approximately 60% of index patients, leading to our current perception of ARVC as a desmosomal disease. Although ARVC is known to preferentially affect the right ventricle, early and/or predominant left ventricular involvement is increasingly recognized. Diagnosis is made by combining multiple sources of diagnostic information as prescribed by the “Task Force” criteria. Recent research suggests that electrical abnormalities precede structural changes in ARVC. Cardiovascular Magnetic Resonance (CMR) is an ideal technique in ARVC workup, as it provides comprehensive information on cardiac morphology, function, and tissue characterization in a single investigation. Prevention of sudden cardiac death using implantable cardioverter-defibrillators is the most important management consideration. This purpose of this paper is to provide an updated review of our understanding of the genetics, diagnosis, current state-of-the-art CMR acquisition and analysis, and management of patients with ARVC.
Similar content being viewed by others
Explore related subjects
Find the latest articles, discoveries, and news in related topics.Introduction
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited cardiomyopathy characterized by fibro-fatty replacement of predominantly the right ventricular (RV) myocardium, which predisposes patients to life-threatening ventricular arrhythmias and RV dysfunction [1]–[3]. ARVC is present in up to 20% of individuals who experience sudden cardiac death (SCD) before the age of 35 years and is even more common among athletes who die suddenly [2],[4]. The disease has been reported to have prevalence of 1 in 2000 to 5000 individuals, although some reports estimate the real prevalence could be as high as 1 in 1000 in certain regions of the world due to under-recognition [5],[6]. Over the past decade, genetic testing for ARVC-associated mutations in five desmosomal genes and several non-desmosomal genes has become clinically available [7]. Inheritance is typically autosomal dominant with incomplete penetrance and variable expressivity [7]–[9]. Affected patients classically present between the second and fourth decade of life with ventricular arrhythmias coming from the RV [3]. However, SCD can occur as early as in adolescence, whereas mutation carriers may also remain asymptomatic throughout life [3],[10],[11].
Imaging modalities commonly used for ARVC evaluation include echocardiography, cardiovascular magnetic resonance (CMR), and RV angiography. Both echocardiography and angiography have significant limitations in assessing the RV due to its complex geometry [12]. Over the last decade, CMR has emerged as the imaging modality of choice in ARVC, allowing for non-invasive morphological and functional evaluation, as well as tissue characterization in a single investigation [13],[14]. In spite of its low prevalence, ARVC accounts for a disproportionately high percentage of referrals for CMR. Unfortunately, many imaging centers have little experience with evaluating ARVC, and gaining experience is difficult because of the low prevalence of disease. The aim of this review is to review current knowledge of ARVC that is useful for CMR interpretation in ARVC. Our emphasis will be on an update of issues relating to CMR diagnosis of ARVC [15], including ARVC diagnostic criteria and common regional morphological and functional abnormalities in this disease.
Update on ARVC diagnosis
Diagnosis of ARVC may be challenging, as no single modality is sufficiently specific to establish ARVC diagnosis. Therefore, multiple sources of diagnostic information are combined in a complex set of diagnostic criteria. The original “Task Force” criteria (TFC), described in 1994 [16], largely relied on qualitative parameters and were shown to be insensitive to the disease especially in early stages [17]–[20]. In addition, imaging criteria were not specific, and led to many false positive diagnoses.
In 2010, modifications to the criteria were proposed (Table 1) [21]. These modifications had two purposes: (1) To improve the specificity of the diagnostic criteria by including quantitative metrics for ARVC diagnosis, and (2) To improve sensitivity of diagnosis in individuals who have a high likelihood of inherited/genetic disease. Specifically, quantitative parameters were included for imaging criteria, endomyocardial biopsy, and (signal-averaged) ECG. In addition, the revised TFC now include an ARVC-associated pathogenic mutation as a major criterion towards ARVC diagnosis. These changes to the TFC have resulted in increased sensitivity for inherited/genetic disease, while maintaining satisfactory specificity [22]–[24].
Role of CMR in ARVC diagnosis
CMR protocol for ARVC
The CMR protocol that we recommend for ARVC evaluation shown in Table 2. The protocol has been designed to evaluate the RV for abnormalities in structure and tissue characterization while enabling quantitative evaluation. For black blood imaging, fast spin echo or turbo spin echo imaging sequences are ideal. The RV free wall and RV outflow tract are best evaluated in the axial black blood images. The stack of axial images should include the entire RV. This can be accomplished in 6–8 slices at intervals (slice thickness + gap) of about 1 cm. Most of the diagnostic information is obtained in the slices centered on the middle of the RV. Obtaining an excessive number of image slices will adversely prolong the examination.
For cine imaging, steady state free precession (SSFP) imaging is preferred at 1.5 Tesla. Insufficient information at 3 Tesla is available to determine if SSFP or fast gradient echo (FGRE) is superior. Quantitative analysis of the RV and left ventricle (LV) is performed on short axis images. Thus, 10–12 slices encompassing the entire ventricular volume must be obtained. We prescribe these images beginning approximately 1 cm above the valve plane and increment towards the apex of the ventricles. Cine images should also be obtained in standard long axis views of the LV. Some sites prefer to also acquire a vertical long axis view of the RV. Finally, we routinely obtain a stack of transaxial images of the RV at the same slice positions as the black blood images described above. Given modern CMR scanners, the temporal resolution of cine images is typically about 40 msec.
Delayed gadolinium images are best obtained using phase selective inversion recovery (PSIR), a sequence which does not depend upon identifying the precise inversion time (TI) [25]. In patients with significant ventricular ectopy, a low dose of a beta-blocker (metoprolol 25–50 mg) is recommended for arrhythmia suppression during the CMR scan.
CMR TFC and their derivation
A major addition to the revised TFC was the inclusion of quantitative measurements for imaging criteria. The revised CMR TFC now require presence of both qualitative findings (RV regional akinesia, dyskinesia, dyssynchronous contraction) and quantitative metrics (decreased ejection fraction or increased indexed RV end-diastolic volume) (Table 1).
Quantitative values for RV volume and function for TFC were derived from a comparison of ARVC probands with normal healthy volunteers that were included in the Multi-Ethnic Study of Atherosclerosis (MESA) [26]. To ascertain cutoff values, RV dimension and function from 462 normal MESA participants were compared to 44 probands in the North American ARVC registry [21]. Major criteria (RV ejection fraction ≤40% or indexed RV end-diastolic volume ≥110 mL/m2 for men and ≥100 mL/m2 for women) were chosen to achieve approximately 95% specificity. Cutoffs with high specificity invariably result in lower sensitivity; major CMR criteria have a sensitivity of 68 to 76% [27]. Minor criteria (RV ejection fraction 40-45% or indexed RV end-diastolic volume 100–110 mL/m2 for men and 90–100 mL/m2 for women) had a higher sensitivity (79 to 89%), but a consequently lower specificity (85 to 97%) [27].
Impact of new TFC on diagnostic yield
Several studies report on the impact of the revised TFC on diagnostic yield specifically for CMR [22],[28]–[30]. Unanimously, these studies showed a decrease in the prevalence of major and minor CMR criteria in the modified TFC compared to the original TFC. This corresponded to a decrease in sensitivity in most of these studies [22],[29]. Interestingly, although sensitivity decreased, the positive predictive value (PPV) increased with the revised TFC, as shown by Femia et al. (PPV increase from 23% in original criteria to 55% in revised criteria) [30]. This may largely be due to the inclusion of quantitative CMR criteria. Vermes et al. report that 97% of subjects with minor changes according to the original CMR criteria did not meet revised CMR criteria [29]. In addition, a low sensitivity of CMR for ARVC diagnosis is understandable in the context of the recently published data indicating that electrical abnormalities precede structural changes detected by CMR in ARVC [31]–[33]. This emphasizes the concept that ARVC evaluation should not be solely based on any one test, in particular CMR.
Limitations of quantitative evaluation of the right ventricle: the revised task force criteria
Including quantitative metrics as a component to the CMR TFC has been an important contribution to ARVC evaluation, however some limitations exist. First, although quantitative measures reduce subjectivity of CMR TFC, there is substantial inter-reader variability for the RV. In our experience, two physician readers had excellent agreement (within ~5% of reference values) only after training on approximately 100 CMR cases [34]. In clinical practice, we expect it would be difficult to achieve reproducibility of less than 10% for RV parameters.
Cutoffs for ARVC criteria were derived from the MESA study that used the FGRE technique whereas a majority of study subjects in the US ARVC study had SSFP cine images. RV volumes by SSFP are least 10% larger than those measured with FGRE technique [35]–[39]. SSFP images provide superior contrast between blood and endocardium at the endocardial border, with less blood flow dependence [35],[36],[40]. Also, the revised TFC used MESA subjects whose mean age was 60 years. ARVC subjects in the Task Force study were 20–30 years younger on average. Since that time, Chahal et al. determined that, among MESA participants aged 45 to 84 years, RV end-diastolic volume decreased 4.6% per decade [34]. A very similar percentage of approximately 4% decrease per decade was obtained by Maceira et al. using the SSFP technique in subjects 20 to 80 years old [41]. Adjusting for body surface area did not remove the dependence of RV volume on age.
Fortuitously, the issues pulse sequence and older reference population in the MESA approximately balance each other. As an example, the CMR cutoff values for RV size in ARVC (Table 1) are ≥110 mL/m2 (male) or ≥100 mL/m2 (female) for major criteria. These values closely correspond to the 95th percentile confidence limits of RV volumes for normal subjects less than 60 years old [41]. Thus, until further studies are available, we feel that the current RV metrics in the revised TFC remain highly relevant. As important, further developments are necessary to improve the reproducibility of RV quantification by CMR. When evaluating younger patients, CMR physicians should keep in mind that the size of the RV is expected to be ~10% larger in a 20 year old compared to a 40 year old patient.
Common findings in ARVC by CMR
Most of our information about structural abnormalities in ARVC comes from studies in subjects with a predominant RV phenotype (Figure 1) [42]–[44]. Abnormalities in the RV in ARVC have been extensively described (reviewed in [15]). Besides global reduction in RV function and enlargement of the RV, more subtle regional disease of the RV has been variously described in the literature using a variety of terms (including focal bulges, microaneurysms, segmental dilatation, regional hypokinesis, etc.). In the current TFC, the terms “akinesia” (lack of motion) and “dyskinesia” (abnormal movement – instead of contracting in systole, that segment of myocardium bulges outward in systole) and “dyssynchronous” (regional peak contraction occurring at different times in adjacent myocardium) are used for all imaging modalities (CMR, echocardiography and angiography) to describe regional wall motion abnormalities in ARVC. Microaneurysms are not explicitly described in the revised TFC for CMR; overuse of this finding was considered by the Task Force members to be misinterpreted by CMR physicians resulting in false positive diagnoses. However, microaneurysms are characterized by regional akinesia or dyskinesia in the revised criteria.

Four-chamber (top panels) and short-axis (bottom panels) bright blood images in an ARVC subject with predominant right ventricular abnormalities. End-diastolic images are shown in the left panels, end-systolic images in the right panels. Note subtricuspid dyskinesia in the end-systolic four-chamber image (arrow), and right ventricular free wall aneurysms (i.e. both systolic and diastolic bulging) in the short-axis image (arrows).
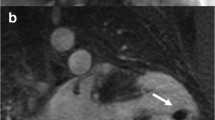
The location of regional wall motion abnormalities of the RV was not addressed in the revised TFC. We now recognize that the distal RV (from the moderator band to the apex in long axis views) shows highly variable contraction patterns in the normal individual. Therefore in ARVC, we emphasize the significance of regional wall motion abnormalities in the subtricuspid region. An excellent example of this is the so-called “accordion sign” that represents a focal “crinkling” of the myocardium (Figure 2) [45],[46]. In terms of TFC, the accordion sign is due to a small region of highly localized myocardium with dyssynchronous contraction.

Regional contraction abnormality in the subtricuspid region. End diastolic (left) and end systolic image (right) show the so-called “accordion sign” in an ARVC mutation carrier. Regional dyssynchronous contraction in the subtricuspid region is a readily recognized qualitative pattern of abnormal RV contraction.
The changing spectrum of ARVC: The triangle of dysplasia displaced
Since the first report in 1982, regional abnormalities in ARVC were thought to locate to the RV inflow tract, outflow tract, and apex, collectively referred to as the “Triangle of Dysplasia” [1]. This concept was based on RV angiographic findings and autopsy data in a series of 24 patients with ARVC. In subsequent years, studies describing structural changes in ARVC focused on abnormalities in these three regions [47]. It is important to note that these observations were made in tertiary centers, without the advantage of genetic testing, and without sensitive TFC.
The last decade has witnessed a paradigm shift in our perception of regional structural involvement in ARVC. Autopsy series have shown predominant fibro-fatty infiltration on the epicardial surface, suggesting that the disease starts in the epicardium and progresses to the endocardium [47]. In 2004, Marchlinski et al. were one of the first to note preferential subtricuspid involvement in ARVC [48]. These results were confirmed in multiple studies using CMR [49],[50], echocardiography [51], and electroanatomic voltage mapping [50],[52],[53].
Recently, Te Riele et al. provided a series of 80 ARVC mutation carriers who underwent CMR and/or endo- and epicardial electroanatomic voltage mapping [54]. Structural abnormalities in this cohort preferentially located to the epicardial subtricuspid region and basal RV free wall, whereas the RV apex and endocardium were relatively spared. In addition, the authors reported that the LV lateral wall was significantly more often involved than the RV apex, especially among subjects with early disease. This led the authors to coin the “displacement” of the RV apex from the Triangle of Dysplasia [54]. Although preferential involvement of the subtricuspid region and LV lateral wall has been described before in separate ARVC reports, the focus on sparing of the RV apex is novel. This is particularly important, as the RV apex is often thin and difficult to assess, resulting in a frequent cause of misdiagnosis in ARVC.
The changing spectrum of ARVC: Left ventricular involvement
The advent of genetic testing and use of sensitive TFC have significantly enhanced our appreciation of the wide phenotypic spectrum of ARVC, and increased our awareness of non-classical (including left-dominant and biventricular) phenotypes. As a result, we now know that some ARVC subjects have early and predominant LV involvement (Figure 3) [19],[55]–[57]. LV involvement has even been reported in 76% of ARVC subjects, of whom the majority had advanced disease [58]. The disease is, therefore, increasingly being referred to as “Arrhythmogenic Cardiomyopathy”.

Horizontal long-axis (top panels) bright blood and late gadolinium enhancement images (bottom panels) in an ARVC subject with predominant left ventricular abnormalities. Note a dilated left ventricle in the bright blood images. Late enhancement is observed in a mid-myocardial pattern in the basal septum and basal lateral wall (arrows, bottom panels).
In 2010, Jain led a study investigating LV regional dysfunction using CMR tagging, and found that LV peak systolic strain was lower in ARVC subjects compared to controls [59]. Sen-Chowdhry et al. recently published data supporting a genetic association between left-dominant ARVC and classical right-sided ARVC [57]. In their study, the authors showed that one-third of genotyped ARVC patients with a left-dominant phenotype have a pathogenic mutation in the ARVC-related desmosomal genes. Phenotypic variations of predominant RV and LV involvement even coexisted in the same family.
LV involvement in ARVC may manifest as late gadolinium enhancement (LGE), often involving the inferior and lateral walls without concomitant wall motion abnormalities [55],[57],[60]. Septal LGE is present in more than 50% of cases with left dominant ARVC, in contrast to the right dominant pattern in which septal involvement is unusual [55]. In addition, LV fatty infiltration was shown to be a prevalent finding in ARVC, often involving the subepicardial lateral LV and resulting in myocardial wall thinning (Figure 4) [54],[61]. Early data by Dalal et al. already showed that LV fat in the lateral wall is very specific for ARVC mutation carriers [45]. Future studies are necessary to confirm these data, and further our understanding of LV abnormalities in ARVC.

Horizontal long-axis bright blood image in an ARVC patient revealing left ventricular lateral wall fatty infiltration with myocardial wall thinning (arrowhead).
Late gadolinium enhancement in ARVC evaluation
Myocardial LGE is a well-validated technique for assessment of myocardial fibrosis. Given that one of the pathologic hallmarks of ARVC is fibro-adipose replacement of the myocardium [47], it is important to note that LGE is not incorporated in the current diagnostic TFC. Although the Task Force did recognize the presence of LGE in many patients with ARVC, several limitations withheld its inclusion in the diagnostic criteria. First, detection of LGE in the RV is greatly hampered by the thin RV wall. A high variability between centers resulted in limitations of LGE in the multi-center US ARVC study. In ARVC, RV wall thinning is pronounced [42], which makes the LGE technique less reliable than for the LV. Second, distinguishing fat from fibrosis by LGE sequences is challenging, which makes its interpretation highly subject to the CMR physician’s experience. Last, LV LGE is non-specific, and has a wide differential diagnosis. LGE may be observed in many mimics of ARVC, such as sarcoidosis [62], myocarditis [63], amyloidosis [64], and dilated cardiomyopathy (DCM) [65].
While these limitations exist, LGE may be very useful in ARVC evaluation (Figure 5). RV LGE has been observed in up to 88% of patients [66]–[68], while LV LGE was reported in up to 61% of cases [50],[69]. Importantly, before LGE can be included in a future iteration of the TFC, more data regarding the specific patterns of LGE that distinguish ARVC from other cardiomyopathies is necessary. Also, improved methods to determine fibrosis in the thin RV wall are needed. Until such a method emerges, use of LGE in clinical practice should be considered as diagnostic confirmation, not sole evidence of ARVC disease expression. LGE CMR is also extremely useful when ARVC is excluded due to other cardiomyopathy such as sarcoidosis.

Right ventricular late gadolinium enhancement in ARVC. The short axis image (left) shows LGE in the RV as well as the LV (black arrows). The lateral wall of the LV shows thinning due to fatty replacement that was confirmed on T1-weighted images. The long axis view (right) shows diffuse LGE involving the free wall of the RV.
LGE may also be useful in management of ARVC patients. Tandri et al. showed excellent correlation of RV LGE with histopathology and inducible ventricular arrhythmias on electrophysiologic study [68]. As such, identification of LGE by CMR may provide guidance for electrophysiologic studies and endomyocardial biopsy. However, it is important to note that recent studies correlating LGE with electroanatomic scar revealed that LGE is less sensitive for the detection of low voltage areas than endocardial mapping during electrophysiologic study [50],[70].
Misdiagnosis of ARVC using CMR
Misdiagnosis of ARVC is a well-recognized problem. A prior study has shown that more than 70% of patients who were referred to Johns Hopkins Hospital from outside institutions with a diagnosis of ARVC did not actually meet diagnostic TFC [71]. In many cases, CMR misinterpretation is the cause of over-diagnosis in ARVC [71],[72]. It is important to realize that, although CMR may be regarded the standard of reference for evaluation of RV morphology and function, the use of CMR alone is not the “gold standard” for ARVC diagnosis. Rather, the TFC prescribe the use of multiple diagnostic tests. Great caution must be employed when the only abnormality in a presumed ARVC patient is found on CMR, as it is uncommon for ARVC patients to have a normal ECG and Holter monitor but an abnormal CMR [32].
A proper understanding of common CMR abnormalities and patterns of disease in ARVC is invaluable for accurate CMR evaluation. Previous reports have extensively focused on fibro-fatty myocardial replacement, wall thinning, RVOT enlargement, and RV dilatation and dysfunction in ARVC [42]–[44]. As one of the pathologic hallmarks of ARVC, intramyocardial fat accumulation was thought to be highly sensitive for the disease. Unfortunately, multiple reports have shown that intramyocardial fat was not reproducible even among experienced readers, constituting an important cause of misdiagnosis in ARVC [44],[71],[73],[74].
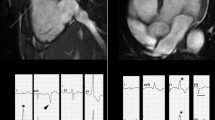
Furthermore, normal variants as well as other pathologic conditions may mimic ARVC. Important normal variants that were previously mistaken for ARVC are pectus excavatum [75], apical-lateral bulging of the RV free wall at the insertion of the moderator band [76], and the “butterfly apex”, a normal anatomical variant of separate RV and LV apices causing the RV apex to look dyskinetic [77]. We have found the butterfly appearance of the apex to be more common on horizontal long axis views at inferior levels (Figure 6). In addition, a prominent band of pericardial connective tissue that joins the RV free wall to the posterior sternum may lead to misinterpretation of RV wall motion: this “tethered” portion of the RV free wall remains static in location and may be misinterpreted as RV dyskinesia (Figure 7). Additionally, pathologic disorders such as myocarditis and sarcoidosis may mimic ARVC [62],[63],[78]. Further testing to specifically exclude these conditions should be strongly considered, especially in the presence of LV dysfunction [79].

Butterfly apex as a normal variation. Stack of horizontal long axis views from inferior (image 1) to superior (image 4) in a control subject. Note the appearance of a butterfly apex on inferior views (arrows in images 1–3). This appearance is not seen on the more superior view (panel 4).

Misdiagnosis of ARVC - Axial and short-axis bright blood images in a control subject. Note the “tethering” of the mid right ventricular free wall to the anterior chest wall (arrows), giving the right ventricle a dyskinetic appearance.
Update on ARVC genetics
ARVC: a desmosomal disease
Beginning with the seminal discovery of plakoglobin in 2000 [80], the past decade has witnessed the identification of mutations in five genes encoding the cardiac desmosome [81]–[84]. In recent reports, desmosomal mutations are found in up to 60% of ARVC cases [8],[23],[24],[85]. Among US ARVC patients, the most common gene involved is plakophilin-2 [85], followed by desmoglein-2, desmocollin-2, and desmoplakin [86]. Prevalence of mutations is similar in Europe [8],[87], although desmoplakin mutations are more prevalent in the United Kingdom and Italy [10],[57]. Desmosomes are complex multiprotein structures providing mechanical [88] and electrical [89] continuity to adjacent cells. Mechanical uncoupling in ARVC is accompanied by cell death and regional fibrosis, which causes the monomorphic arrhythmias typically associated with ARVC. In addition, electrical uncoupling through gap junction remodeling and sodium channel dysfunction may lead to significant activation delay [90],[91], which increases the propensity to functional block and arrhythmia. The exact mechanism by which these mutations cause the highly arrhythmogenic phenotype in ARVC has been subject of many hypotheses, which are extensively reviewed elsewhere [20],[47]. The List of Genes Section shows an overview of genes associated with ARVC.
Genes associated with arrhythmogenic right ventricular dysplasia/cardiomyopathy
Gene
Desmosomal
Desmocollin-2 (DSC2)
Desmoglein-2 (DSG2)
Desmoplakin (DSP)
Junctional Plakoglobin (JUP)
Plakophilin-2 (PKP2)
Non-Desmosomal
Desmin (DES)
Lamin A/C (LMNA)
Phospholamban (PLN)
Ryanodine Receptor (RYR2)
Transmembrane Protein 43 (TMEM43)
Titin (TTN)
ARVC: overlap with other cardiomyopathies
In addition to desmosomal mutations, mutations in non-desmosomal genes have been identified in ARVC [92]–[98]. These non-desmosomal genes include, among others, desmin, titin, lamin A/C, and phospholamban, which are commonly mutated in subjects with DCM [99]–[101]. Although the distinction between ARVC and DCM has important implications for clinical practice, guiding both diagnostics and treatment, a considerable overlap of these conditions is increasingly recognized [102]. Compared to DCM, patients with left-dominant ARVC often have significant ventricular arrhythmias, disproportionate to the morphological abnormalities and impaired LV systolic function [57]. In addition, inflammatory processes such as (viral) myocarditis may mimic left-dominant ARVC [103]. In myocarditis, T2-weighted imaging may detect tissue edema, which is usually absent in ARVC [104]. In addition, fast spin-echo T1-weighted images during the first minutes after contrast injection may be useful to detect myocardial hyperemia and muscular inflammation suggestive of myocarditis [104],[105]. In equivocal cases, invasive studies such as electroanatomic mapping and endomyocardial biopsy may provide a more definite diagnosis [63].
Impact of genetics on clinical ARVC management
With the identification of ARVC-causing mutations, integration of genetic testing into clinical practice is now proliferating. Currently, its main applications are confirmatory testing in index patients and cascade screening of families [106]. ARVC is generally transmitted as an autosomal dominant trait with incomplete penetrance and variable expressivity. A recent study by Cox et al. confirmed that asymptomatic mutation-carrying relatives have a 6-fold increased risk of developing ARVC compared to relatives of a proband without a pathogenic mutation [8]. However, it is important to realize that 50-70% of mutation carriers will never develop disease expression [7],[23],[107], and that severity of disease may vary greatly, even among members of the same family [11] or those carrying the same mutation [9]. In contrast, a negative genetic test result in a proband does not exclude the possibility of disease, nor does it exclude the possibility of a genetic process in the individual or family [106],[108]. Because of the complexities associated with interpreting genetic test results in ARVC, including genetic counseling prior and subsequent to genetic testing has been strongly recommended [106],[109].
CMR in ARVC genotype-phenotype correlations
Over the last decade, several genotype-phenotype correlations in ARVC have been proposed, but large-scale studies confirming these observations are yet to come. Recently, patients with multiple desmosomal mutations were shown to have a more severe clinical course with more ventricular arrhythmias and more heart failure than subjects with a single mutation [10],[86]. In addition, individuals with a mutation in the phospholamban or desmoplakin gene (especially when involving the C-terminus of desmoplakin) have significant left-dominant/biventricular disease expression and a high prevalence of heart failure [19],[98],[110]. LV involvement in these patients often manifests as LGE in a LV circumferential, mid-myocardial pattern extending to the right side of the septum [111]. An example is shown in Figure 3. This left-dominant ARVC pattern should not be confused with LV involvement that occurs in advanced stages of right-dominant ARVC. These right-dominant ARVC subjects (often plakophilin-2 mutation carriers) commonly have focal LV disease involving the lateral LV wall with only mild or moderate LV dysfunction [111]. Large-scale studies from collaborative international registries are necessary to further unravel genotype-phenotype associations in ARVC.
Update on ARVC management
ARVC management is directed towards symptom reduction, delay of disease progression, and prevention of SCD. Because of a lack of randomized trials comparing ARVC treatment options, management recommendations in ARVC are largely based on clinical judgments and results from retrospective registry-based studies. Mainstay therapies consist of conservative measures (exercise restriction), beta-blocking and antiarrhythmic agents, implantable cardioverter-defibrillator (ICD) implantation, and radiofrequency ablation of ventricular arrhythmias.
Evidence for a potential role of exercise in ARVC expression and disease progression is accumulating. Many ARVC patients are highly athletic and those who participate in competitive sports have a 5-fold increased risk of arrhythmic death compared to non-athletes [112]. Recently, James led a study on the role of exercise in ARVC development, showing that endurance exercise and frequent athletics increases the risk of arrhythmias and heart failure in ARVC mutation carriers [113]. This important piece of evidence highlights the importance of exercise restriction in ARVC patients and those at risk of developing disease.
Once the diagnosis of ARVC is established in a patient, the most important decision is whether to implant an ICD for prevention of SCD. It is now standard of care for ARVC subjects with prior sustained ventricular arrhythmia to undergo placement of an ICD [114],[115]. Studies have shown that these patients have a high incidence rate of appropriate ICD discharges of up to 70% during a mean follow-up of 3–5 years [115],[116]. Unfortunately, guidelines for ICD implantation among subjects without prior ventricular arrhythmia are less unambiguous. Recent reports suggest an important role for CMR in risk stratification of these patients [32],[117],[118]. In their study, Deac et al. showed that an abnormal CMR was an independent predictor of arrhythmic events [118]. Also, it was recently shown that the revised CMR TFC have high negative predictive value for arrhythmic occurrence in ARVC [32].
Arrhythmia control in ARVC is often achieved by pharmacologic treatment. Beta-blockers and class III antiarrhythmic drugs (sotalol, amiodarone) have been shown to be successful in reducing arrhythmia burden and likelihood of ICD discharge [119],[120]. In addition, radiofrequency ablation for ventricular arrhythmia in ARVC has gained enormous popularity over the last years. Although the results of endocardial ablation have been moderate [121], good arrhythmia control (but not complete cure) has been obtained using epicardial ablation [52],[122]–[124]. This is understandable, given the primary (sub)epicardial location of the abnormal substrate in ARVC. CMR with LGE may be useful in planning of these procedures, by providing information on the presence and distribution of ventricular scar [125].
Future directions
CMR evaluation in ARVC is a moving target. New CMR sequences such as high-resolution T1 mapping are promising tools to detect early, subtle changes in the RV. In addition, quantification of RV regional wall motion abnormalities and evaluation of inter- and intraventricular dyssynchrony may provide novel tools for early detection of ARVC. The genetic era allowed for ARVC genetic testing using comprehensive cardiomyopathy panels and whole exome sequencing, which are likely to significantly impact our knowledge of the genetic basis of ARVC and the overlap with other cardiomyopathies. Furthermore, genotype-phenotype correlation studies may guide our quest for genetic and environmental modifiers in this disease. Lastly, basic research in in vitro and animal models may have an important impact on our knowledge of ARVC pathophysiology. Results from these studies may open the path to modification of the abnormal substrate in ARVC, allowing for definite prevention of clinical disease manifestation and/or progression.
Conclusion
ARVC is a rare but important cause of SCD in the young and in athletes. The disease is inherited as an autosomal dominant trait with incomplete penetrance and variable expressivity. Because of the inherent risk of potentially lethal arrhythmias, correct diagnosis and early detection of ARVC are essential. This is critically important, because with the advent of genetic testing, the population of at-risk individuals is rapidly increasing. Clinical ARVC diagnosis is facilitated by a complex set of diagnostic criteria which were first described in 1994 and updated in 2010 to increase sensitivity for early disease. As the non-invasive “gold standard” for RV evaluation, CMR plays an important role in clinical ARVC workup. Recent studies have shown that RV involvement in ARVC often manifests as regional wall motion abnormality or global ventricular dysfunction, whereas LV involvement is often observed as LGE and/or fatty infiltration without concomitant wall motion abnormalities. ARVC preferentially affects the basal RV and lateral LV, while sparing the RV apex. Once diagnosis of ARVC is established, the most important management decision is whether to implant an ICD for prevention of SCD. Future studies are necessary to further unravel the pathophysiologic attributes of disease and provide insights into genotype-phenotype correlations in ARVC.
Abbreviations
- ARVC:
-
Arrhythmogenic right ventricular cardiomyopathy
- CMR:
-
Cardiovascular magnetic resonance
- DCM:
-
Dilated cardiomyopathy
- ECG:
-
Electrocardiogram
- FGRE:
-
Fast gradient echo
- ICD:
-
Implantable cardioverter-defibrillator
- LGE:
-
Late Gadolinium enhancement
- LV:
-
Left ventricle
- MESA:
-
Multi-ethnic study of atherosclerosis
- PPV:
-
Positive predictive value
- RV:
-
Right ventricle
- SCD:
-
Sudden cardiac death
- SSFP:
-
Steady state free precession
- TFC:
-
Task Force Criteria
References
Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, Grosgogeat Y: Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982, 65: 384-398.
Corrado D, Thiene G, Nava A, Rossi L, Pennelli N: Sudden death in young competitive athletes: clinicopathologic correlations in 22 cases. Am J Med. 1990, 89: 588-96.
Dalal D, Nasir K, Bomma C, Prakasa K, Tandri H, Piccini J, Roguin A, Tichnell C, James C, Russell SD, Judge DP, Abraham T, Spevak PJ, Bluemke DA, Calkins H: Arrhythmogenic right ventricular dysplasia a United States experience. Circulation. 2005, 112: 3823-32.
Basso C, Corrado D, Thiene G: Cardiovascular causes of sudden death in young individuals including athletes. Cardiol Rev. 1999, 7: 127-35.
Sen-Chowdhry S, Morgan RD, Chambers JC, McKenna WJ: Arrhythmogenic cardiomyopathy: etiology, diagnosis, and treatment. Annu Rev Med. 2010, 61: 233-53.
Basso C, Corrado D, Marcus FI, Nava A, Thiene G: Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009, 373: 1289-300.
Murray B: Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C): a review of molecular and clinical literature. J Genet Couns. 2012, 21: 494-504.
Cox MG, van der Zwaag PA, van der Werf C, van der Smagt JJ, Noorman M, Bhuiyan ZA, Wiesfeld AC, Volders PG, van Langen IM, Atsma DE, Dooijes D, van den Wijngaard A, Houweling AC, Jongbloed JD, Jordaens L, Cramer MJ, Doevendans PA, de Bakker JM, Wilde AA, van Tintelen JP, Hauer RN: Arrhythmogenic right ventricular dysplasia/cardiomyopathy: pathogenic desmosome mutations in index-patients predict outcome of family screening. Circulation. 2011, 123: 2690-700.
Syrris P, Ward D, Asimaki A, Sen-Chowdhry S, Ebrahim HY, Evans A, Hitomi N, Norman M, Pantazis A, Shaw AL, Elliott PM, McKenna WJ: Clinical expression of plakophilin-2 mutations in familial arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006, 113: 356-64.
Rigato I, Bauce B, Rampazzo A, Zorzi A, Pilichou K, Mazzotti E, Migliore F, Marra MP, Lorenzon A, De Bortoli M, Calore M, Nava A, Daliento L, Gregori D, Iliceto S, Thiene G, Basso C, Corrado D: Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet. 2013, 6: 533-42.
Kannankeril PJ, Bhuiyan ZA, Darbar D, Mannens MM, Wilde AA, Roden DM: Arrhythmogenic right ventricular cardiomyopathy due to a novel plakophilin 2 mutation: wide spectrum of disease in mutation carriers within a family. Heart Rhythm. 2006, 3: 939-44.
Boxt LM: Radiology of the right ventricle. Radiol Clin North Am. 1999, 37: 379-400.
Galea N, Carbone I, Cannata D, Cannavale G, Conti B, Galea R, Frustaci A, Catalano C, Francone M: Right ventricular cardiovascular magnetic resonance imaging: normal anatomy and spectrum of pathological findings. Insights Imaging. 2013, 4: 213-23.
Pennell D, Casolo G: Right ventricular arrhythmia: emergence of magnetic resonance imaging as an investigative tool. Eur Heart J. 1997, 18: 1843-5.
Jain A, Tandri H, Calkins H, Bluemke DA: Role of cardiovascular magnetic resonance imaging in arrhythmogenic right ventricular dysplasia. J Cardiovasc Magn Reson. 2008, 10: 32-
McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine G, Camerini F: Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J. 1994, 71: 215-8.
Hamid MS, Norman M, Quraishi A, Firoozi S, Thaman R, Gimeno JR, Sachdev B, Rowland E, Elliott PM, McKenna WJ: Prospective evaluation of relatives for familial arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria. J Am Coll Cardiol. 2002, 40: 1445-50.
Nasir K, Bomma C, Tandri H, Roguin A, Dalal D, Prakasa K, Tichnell C, James C, Spevak PJ, Marcus F, Calkins H: Electrocardiographic features of arrhythmogenic right ventricular dysplasia/cardiomyopathy according to disease severity: a need to broaden diagnostic criteria. Circulation. 2004, 110: 1527-34.
Bauce B, Basso C, Rampazzo A, Beffagna G, Daliento L, Frigo G, Malacrida S, Settimo L, Danieli G, Thiene G, Nava A: Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur Heart J. 2005, 26: 1666-75.
Basso C, Bauce B, Corrado D, Thiene G: Pathophysiology of arrhythmogenic cardiomyopathy. Nat Rev Cardiol. 2012, 9: 223-33.
Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, Fontaine G, Gear K, Hauer R, Nava A, Picard MH, Protonotarios N, Saffitz JE, Sanborn DM, Steinberg JS, Tandri H, Thiene G, Towbin JA, Tsatsopoulou A, Wichter T, Zareba W: Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia proposed modification of the task force criteria. Circulation. 2010, 121: 1533-41.
Cox MG, van der Smagt JJ, Noorman M, Wiesfeld AC, Volders PG, van Langen IM, Atsma DE, Dooijes D, Houweling AC, Loh P, Jordaens L, Arens Y, Cramer MJ, Doevendans PA, van Tintelen JP, Wilde AA, Hauer RN: Arrhythmogenic right ventricular dysplasia/cardiomyopathy diagnostic task force criteria: impact of new task force criteria. Circ Arrhythm Electrophysiol. 2010, 3: 126-33.
Quarta G, Muir A, Pantazis A, Syrris P, Gehmlich K, Garcia-Pavia P, Ward D, Sen-Chowdhry S, Elliott PM, McKenna WJ: Familial evaluation in arrhythmogenic right ventricular cardiomyopathy: impact of genetics and revised task force criteria. Circulation. 2011, 123: 2701-9.
Protonotarios N, Anastasakis A, Antoniades L, Chlouverakis G, Syrris P, Basso C, Asimaki A, Theopistou A, Stefanadis C, Thiene G, McKenna WJ, Tsatsopoulou A: Arrhythmogenic right ventricular cardiomyopathy/dysplasia on the basis of the revised diagnostic criteria in affected families with desmosomal mutations. Eur Heart J. 2011, 32: 1097-104.
Plaisier AS, Burgmans MC, Vonken EP, Prakken NH, Cox MG, Hauer RN, Velthuis BK, Cramer MJ: Image quality assessment of the right ventricle with three different delayed enhancement sequences in patients suspected of ARVC/D. Int J Cardiovasc Imaging. 2012, 28: 595-601.
Tandri H, Daya SK, Nasir K, Bomma C, Lima JA, Calkins H, Bluemke DA: Normal reference values for the adult right ventricle by magnetic resonance imaging. Am J Cardiol. 2006, 98: 1660-4.
Bluemke DA: ARVC: Imaging diagnosis is still in the eye of the beholder. J Am Coll Cardiol Img. 2011, 4: 288-91.
Luijkx T, Velthuis BK, Prakken NH, Cox MG, Bots ML, Mali WP, Hauer RN, Cramer MJ: Impact of revised Task Force Criteria: distinguishing the athlete’s heart from ARVC/D using cardiac magnetic resonance imaging. Eur J Prev Cardiol. 2012, 19: 885-91.
Vermes E, Strohm O, Otmani A, Childs H, Duff H, Friedrich MG: Impact of the revision of arrhythmogenic right ventricular cardiomyopathy/dysplasia task force criteria on its prevalence by CMR criteria. J Am Coll Cardiol Img . 2011, 4: 282-7.
Femia G, Hsu C, Singarayar S, Sy RW, Kilborn M, Parker G, McGuire M, Semsarian C, Puranik R: Impact of new task force criteria in the diagnosis of arrhythmogenic right ventricular cardiomyopathy. Int J Cardiol. 2013, 171: 179-83.
Gomes J, Finlay M, Ahmed AK, Ciaccio EJ, Asimaki A, Saffitz JE, Quarta G, Nobles M, Syrris P, Chaubey S, McKenna WJ, Tinker A, Lambiase PD: Electrophysiological abnormalities precede overt structural changes in arrhythmogenic right ventricular cardiomyopathy due to mutations in desmoplakin-A combined murine and human study. Eur Heart J. 2012, 33: 1942-53.
te Riele AS, Bhonsale A, James CA, Rastegar N, Murray B, Burt JR, Tichnell C, Madhavan S, Judge DP, Bluemke DA, Zimmerman SL, Kamel IR, Calkins H, Tandri H: Incremental value of cardiac magnetic resonance imaging in arrhythmic risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol. 2013, 62: 1761-9.
Baumwart RD, Meurs KM, Raman SV: Magnetic resonance imaging of right ventricular morphology and function in boxer dogs with arrhythmogenic right ventricular cardiomyopathy. J Vet Intern Med. 2009, 23: 271-4.
Chahal H, Johnson C, Tandri H, Jain A, Hundley WG, Barr RG, Kawut SM, Lima JA, Bluemke DA: Relation of cardiovascular risk factors to right ventricular structure and function as determined by magnetic resonance imaging (results from the multi-ethnic study of atherosclerosis). Am J Cardiol. 2010, 106: 110-6.
Malayeri AA, Johnson WC, Macedo R, Bathon J, Lima JA, Bluemke DA: Cardiac cine MRI: Quantification of the relationship between fast gradient echo and steady-state free precession for determination of myocardial mass and volumes. J Magn Reson Imaging. 2008, 28: 60-6.
Moon JC, Lorenz CH, Francis JM, Smith GC, Pennell DJ: Breath-hold FLASH and FISP cardiovascular MR imaging: left ventricular volume differences and reproducibility. Radiology. 2002, 223: 789-97.
Alfakih K, Plein S, Thiele H, Jones T, Ridgway JP, Sivananthan MU: Normal human left and right ventricular dimensions for MRI as assessed by turbo gradient echo and steady-state free precession imaging sequences. J Magn Reson Imaging. 2003, 17: 323-9.
Plein S, Bloomer TN, Ridgway JP, Jones TR, Bainbridge GJ, Sivananthan MU: Steady-state free precession magnetic resonance imaging of the heart: comparison with segmented k-space gradient-echo imaging. J Magn Reson Imaging. 2001, 14: 230-6.
Li W, Stern JS, Mai VM, Pierchala LN, Edelman RR, Prasad PV: MR assessment of left ventricular function: quantitative comparison of fast imaging employing steady-state acquisition (FIESTA) with fast gradient echo cine technique. J Magn Reson Imaging. 2002, 16: 559-64.
Barkhausen J, Ruehm SG, Goyen M, Buck T, Laub G, Debatin JF: MR evaluation of ventricular function: true fast imaging with steady-state precession versus fast low-angle shot cine MR imaging: feasibility study. Radiology. 2001, 219: 264-9.
Maceira AM, Prasad SK, Khan M, Pennell DJ: Reference right ventricular systolic and diastolic function normalized to age, gender and body surface area from steady-state free precession cardiovascular magnetic resonance. Eur Heart J. 2006, 27: 2879-88.
Tandri H, Calkins H, Nasir K, Bomma C, Castillo E, Rutberg J, Tichnell C, Lima JA, Bluemke DA: Magnetic resonance imaging findings in patients meeting task force criteria for arrhythmogenic right ventricular dysplasia. J Cardiovasc Electrophysiol. 2003, 14: 476-82.
Tandri H, Macedo R, Calkins H, Marcus F, Cannom D, Scheinman M, Daubert J, Estes M, Wilber D, Talajic M, Duff H, Krahn A, Sweeney M, Garan H, Bluemke DA: Role of magnetic resonance imaging in arrhythmogenic right ventricular dysplasia: insights from the North American arrhythmogenic right ventricular dysplasia (ARVD/C) study. Am Heart J. 2008, 155: 147-53.
Bluemke DA, Krupinski EA, Ovitt T, Gear K, Unger E, Axel L, Boxt LM, Casolo G, Ferrari VA, Funaki B, Globits S, Higgins CB, Julsrud P, Lipton M, Mawson J, Nygren A, Pennell DJ, Stillman A, White RD, Wichter T, Marcus F: MR Imaging of arrhythmogenic right ventricular cardiomyopathy: morphologic findings and interobserver reliability. Cardiology. 2003, 99: 153-62.
Dalal D, Tandri H, Judge DP, Amat N, Macedo R, Jain R, Tichnell C, Daly A, James C, Russell SD, Abraham T, Bluemke DA, Calkins H: Morphologic variants of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy a genetics-magnetic resonance imaging correlation study. J Am Coll Cardiol. 2009, 53: 1289-99.
Groenink M, Wilde AA: The “accordion sign,” a new tune in arrhythmogenic right ventricular dysplasia/cardiomyopathy magnetic resonance imaging?. J Am Coll Cardiol. 2009, 53: 1300-1.
Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M: Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis?. Circulation. 1996, 94: 983-91.
Marchlinski FE, Zado E, Dixit S, Gerstenfeld E, Callans DJ, Hsia H, Lin D, Nayak H, Russo A, Pulliam W: Electroanatomic substrate and outcome of catheter ablative therapy for ventricular tachycardia in setting of right ventricular cardiomyopathy. Circulation. 2004, 110: 2293-8.
Bomma C, Dalal D, Tandri H, Prakasa K, Nasir K, Roguin A, Tichnell C, James C, Lima JA, Calkins H, Bluemke DA: Regional differences in systolic and diastolic function in arrhythmogenic right ventricular dysplasia/cardiomyopathy using magnetic resonance imaging. Am J Cardiol. 2005, 95: 1507-11.
Marra MP, Leoni L, Bauce B, Corbetti F, Zorzi A, Migliore F, Silvano M, Rigato I, Tona F, Tarantini G, Cacciavillani L, Basso C, Buja G, Thiene G, Iliceto S, Corrado D: Imaging study of ventricular scar in arrhythmogenic right ventricular cardiomyopathy: comparison of 3D standard electroanatomical voltage mapping and contrast-enhanced cardiac magnetic resonance. Circ Arrhythm Electrophysiol. 2012, 5: 91-100.
Teske AJ, Cox MG, Te Riele AS, De Boeck BW, Doevendans PA, Hauer RN, Cramer MJ: Early detection of regional functional abnormalities in asymptomatic ARVD/C gene carriers. J Am Soc Echocardiogr. 2012, 25: 997-1006.
Garcia FC, Bazan V, Zado ES, Ren JF, Marchlinski FE: Epicardial substrate and outcome with epicardial ablation of ventricular tachycardia in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2009, 120: 366-75.
Corrado D, Basso C, Leoni L, Tokajuk B, Turrini P, Bauce B, Migliore F, Pavei A, Tarantini G, Napodano M, Ramondo A, Buja G, Iliceto S, Thiene G: Three-dimensional electroanatomical voltage mapping and histologic evaluation of myocardial substrate in right ventricular outflow tract tachycardia. J Am Coll Cardiol. 2008, 51: 731-9.
Te Riele AS, James CA, Philips B, Rastegar N, Bhonsale A, Groeneweg JA, Murray B, Tichnell C, Judge DP, Cramer MJ, Velthuis BK, Bluemke DA, Zimmerman SL, Kamel IR, Hauer RN, Calkins H, Tandri H: Mutation-positive arrhythmogenic right ventricular dysplasia/cardiomyopathy: the triangle of dysplasia displaced. J Cardiovasc Electrophysiol. 2013, 24: 1311-20.
Sen-Chowdhry S, Syrris P, Ward D, Asimaki A, Sevdalis E, McKenna WJ: Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation. 2007, 115: 1710-20.
Corrado D, Basso C, Thiene G, McKenna WJ, Davies MJ, Fontaliran F, Nava A, Silvestri F, Blomstrom-Lundqvist C, Wlodarska EK, Fontaine G, Camerini F: Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathyysplasia: a multicenter study. cardiomyopathy/dysplasia. 1997, 30: 1512-20.
Sen-Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, Pennell DJ, McKenna WJ: Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol. 2008, 52: 2175-87.
Lindstrom L, Nylander E, Larsson H, Wranne B: Left ventricular involvement in arrhythmogenic right ventricular cardiomyopathy - a scintigraphic and echocardiographic study. Clin Physiol Funct Imaging. 2005, 25: 171-7.
Jain A, Shehata ML, Stuber M, Berkowitz SJ, Calkins H, Lima JA, Bluemke DA, Tandri H: Prevalence of left ventricular regional dysfunction in arrhythmogenic right ventricular dysplasia: a tagged MRI study. Circ Cardiovasc Imaging. 2010, 3: 290-7.
Igual B, Zorio E, Maceira A, Estornell J, Lopez-Lereu MP, Monmeneu JV, Quesada A, Navarro J, Mas F, Salvador A: Arrhythmogenic cardiomyopathy. Patterns of ventricular involvement using cardiac magnetic resonance. Rev Esp Cardiol. 2011, 64: 1114-22.
Rastegar N, Zimmerman SL, James CA, Murray B, te Riele AS, Bhonsale A, Tichnell C, Calkins H, Tandri H, Bluemke DA, Kamel IR: Incidence of Non-Task Force Criteria Findings by CMR in Subjects with Various Arrhythmogenic Right Ventricular Cardiomyopathy Scores. Radiol Soc North Am (RSNA). 2013, ᅟ: SSE04-05. Abstract-
Vasaiwala SC, Finn C, Delpriore J, Leya F, Gagermeier J, Akar JG, Santucci P, Dajani K, Bova D, Picken MM, Basso C, Marcus F, Wilber DJ: Prospective study of cardiac sarcoid mimicking arrhythmogenic right ventricular dysplasia. J Cardiovasc Electrophysiol. 2009, 20: 473-6.
Pieroni M, Dello Russo A, Marzo F, Pelargonio G, Casella M, Bellocci F, Crea F: High prevalence of myocarditis mimicking arrhythmogenic right ventricular cardiomyopathy differential diagnosis by electroanatomic mapping-guided endomyocardial biopsy. J Am Coll Cardiol. 2009, 53: 681-9.
Maceira AM, Joshi J, Prasad SK, Moon JC, Perugini E, Harding I, Sheppard MN, Poole-Wilson PA, Hawkins PN, Pennell DJ: Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2005, 111: 186-93.
Assomull RG, Prasad SK, Lyne J, Smith G, Burman ED, Khan M, Sheppard MN, Poole-Wilson PA, Pennell DJ: Cardiovascular magnetic resonance, fibrosis, and prognosis in dilated cardiomyopathy. J Am Coll Cardiol. 2006, 48: 1977-85.
Pfluger HB, Phrommintikul A, Mariani JA, Cherayath JG, Taylor AJ: Utility of myocardial fibrosis and fatty infiltration detected by cardiac magnetic resonance imaging in the diagnosis of arrhythmogenic right ventricular dysplasia–a single centre experience. Heart Lung Circ. 2008, 17: 478-83.
Hunold P, Wieneke H, Bruder O, Krueger U, Schlosser T, Erbel R, Barkhausen J: Late enhancement: a new feature in MRI of arrhythmogenic right ventricular cardiomyopathy?. J Cardiovasc Magn Reson. 2005, 7: 649-55.
Tandri H, Saranathan M, Rodriguez ER, Martinez C, Bomma C, Nasir K, Rosen B, Lima JA, Calkins H, Bluemke DA: Noninvasive detection of myocardial fibrosis in arrhythmogenic right ventricular cardiomyopathy using delayed-enhancement magnetic resonance imaging. J Am Coll Cardiol. 2005, 45: 98-103.
Santangeli P, Pieroni M, Dello Russo A, Casella M, Pelargonio G, Macchione A, Camporeale A, Smaldone C, Bartoletti S, Di Biase L, Bellocci F, Natale A: Noninvasive diagnosis of electroanatomic abnormalities in arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm Electrophysiol. 2010, 3: 632-8.
Santangeli P, Hamilton-Craig C, Dello Russo A, Pieroni M, Casella M, Pelargonio G, Di Biase L, Smaldone C, Bartoletti S, Narducci ML, Tondo C, Bellocci F, Natale A: Imaging of scar in patients with ventricular arrhythmias of right ventricular origin: cardiac magnetic resonance versus electroanatomic mapping. J Cardiovasc Electrophysiol. 2011, 22: 1359-66.
Bomma C, Rutberg J, Tandri H, Nasir K, Roguin A, Tichnell C, Rodriguez R, James C, Kasper E, Spevak P, Bluemke DA, Calkins H: Misdiagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Cardiovasc Electrophysiol. 2004, 15: 300-6.
Looi KL, Edwards C, Hart H, Christiansen JP: Utility of cardiac magnetic resonance in the evaluation of unselected patients with possible arrhythmogenic right ventricular cardiomyopathy. Clin Med Insights Cardiol. 2012, 6: 153-62.
Tandri H, Castillo E, Ferrari VA, Nasir K, Dalal D, Bomma C, Calkins H, Bluemke DA: Magnetic resonance imaging of arrhythmogenic right ventricular dysplasia: sensitivity, specificity, and observer variability of fat detection versus functional analysis of the right ventricle. J Am Coll Cardiol. 2006, 48: 2277-84.
Macedo R, Prakasa K, Tichnell C, Marcus F, Calkins H, Lima JA, Bluemke DA: Marked lipomatous infiltration of the right ventricle: MRI findings in relation to arrhythmogenic right ventricular dysplasia. AJR Am J Roentgenol. 2007, 188: W423-7.
Oezcan S, Attenhofer Jost CH, Pfyffer M, Kellenberger C, Jenni R, Binggeli C, Faeh-Gunz A, Seifert B, Scharf C, Kretschmar O, Valsangiacomo Buechel ER: Pectus excavatum: echocardiography and cardiac MRI reveal frequent pericardial effusion and right-sided heart anomalies. Eur Heart J Cardiovasc Imaging. 2012, 13: 673-9.
Sievers B, Addo M, Franken U, Trappe HJ: Right ventricular wall motion abnormalities found in healthy subjects by cardiovascular magnetic resonance imaging and characterized with a new segmental model. J Cardiovasc Magn Reson. 2004, 6: 601-8.
Marcus F, Basso C, Gear K, Sorrell VL: Pitfalls in the diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Am J Cardiol. 2010, 105: 1036-9.
Steckman DA, Schneider PM, Schuller JL, Aleong RG, Nguyen DT, Sinagra G, Vitrella G, Brun F, Cova MA, Pagnan L, Mestroni L, Varosy PD, Sauer WH: Utility of cardiac magnetic resonance imaging to differentiate cardiac sarcoidosis from arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2012, 110: 575-9.
Cooper LT, Baughman KL, Feldman AM, Frustaci A, Jessup M, Kuhl U, Levine GN, Narula J, Starling RC, Towbin J, Virmani R: The role of endomyocardial biopsy in the management of cardiovascular disease: a scientific statement from the American Heart Association, the American College of Cardiology, and the European Society of Cardiology. Endorsed by the Heart Failure Society of America and the Heart Failure Association of the European Society of Cardiology. J Am Coll Cardiol. 2007, 50: 1914-31.
McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ: Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet. 2000, 355: 2119-24.
Pilichou K, Nava A, Basso C, Beffagna G, Bauce B, Lorenzon A, Frigo G, Vettori A, Valente M, Towbin J, Thiene G, Danieli GA, Rampazzo A: Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006, 113: 1171-9.
Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, Rossi V, Zimbello R, Simionati B, Basso C, Thiene G, Towbin JA, Danieli GA: Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2002, 71: 1200-6.
Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, Lerman BB, Markowitz SM, Ellinor PT, MacRae CA, Peters S, Grossmann KS, Drenckhahn J, Michely B, Sasse-Klaassen S, Birchmeier W, Dietz R, Breithardt G, Schulze-Bahr E, Thierfelder L: Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004, 36: 1162-4.
Syrris P, Ward D, Evans A, Asimaki A, Gandjbakhch E, Sen-Chowdhry S, McKenna WJ: Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am J Hum Genet. 2006, 79: 978-84.
den Haan AD, Tan BY, Zikusoka MN, Llado LI, Jain R, Daly A, Tichnell C, James C, Amat-Alarcon N, Abraham T, Russell SD, Bluemke DA, Calkins H, Dalal D, Judge DP: Comprehensive desmosome mutation analysis in north americans with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Cardiovasc Genet. 2009, 2: 428-35.
Bhonsale A, Groeneweg JA, James CA, Dooijes D, Tichnell C, Jongbloed JD, Murray B, van der Heijden JF, Tandri H, Wilde AA, Judge D, van Tintelen JP, Calkins H, Hauer RN: Impact of Genotype on Clinical Course in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Associated Mutation Carriers. Circulation. 2012, 126: A13208-
van Tintelen JP, Entius MM, Bhuiyan ZA, Jongbloed R, Wiesfeld AC, Wilde AA, van der Smagt J, Boven LG, Mannens MM, van Langen IM, Hofstra RM, Otterspoor LC, Doevendans PA, Rodriguez LM, van Gelder IC, Hauer RN: Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2006, 113: 1650-8.
Delmar M, McKenna WJ: The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res. 2010, 107: 700-14.
Delmar M: Desmosome-ion channel interactions and their possible role in arrhythmogenic cardiomyopathy. Pediatr Cardiol. 2012, 33: 975-9.
Kaplan SR, Gard JJ, Protonotarios N, Tsatsopoulou A, Spiliopoulou C, Anastasakis A, Squarcioni CP, McKenna WJ, Thiene G, Basso C, Brousse N, Fontaine G, Saffitz JE: Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease). Heart Rhythm. 2004, 1: 3-11.
Sato PY, Musa H, Coombs W, Guerrero-Serna G, Patino GA, Taffet SM, Isom LL, Delmar M: Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ Res. 2009, 105: 523-6.
Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, Larderet G, Brahmbhatt B, Brown K, Bauce B, Muriago M, Basso C, Thiene G, Danieli GA, Rampazzo A: Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet. 2001, 10: 189-94.
Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C, Bauce B, Carraro G, Thiene G, Towbin JA, Danieli GA, Rampazzo A: Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res. 2005, 65: 366-73.
van der Zwaag PA, Cox MG, van der Werf C, Wiesfeld AC, Jongbloed JD, Dooijes D, Bikker H, Jongbloed R, Suurmeijer AJ, van den Berg MP, Hofstra RM, Hauer RN, Wilde AA, van Tintelen JP: Recurrent and founder mutations in the Netherlands : Plakophilin-2 p.Arg79X mutation causing arrhythmogenic right ventricular cardiomyopathy/dysplasia. Neth Heart J. 2010, 18: 583-91.
Merner ND, Hodgkinson KA, Haywood AF, Connors S, French VM, Drenckhahn JD, Kupprion C, Ramadanova K, Thierfelder L, McKenna W, Gallagher B, Morris-Larkin L, Bassett AS, Parfrey PS, Young TL: Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2008, 82: 809-21.
van Tintelen JP, Van Gelder IC, Asimaki A, Suurmeijer AJ, Wiesfeld AC, Jongbloed JD, van den Wijngaard A, Kuks JB, van Spaendonck-Zwarts KY, Notermans N, Boven L, van den Heuvel F, Veenstra-Knol HE, Saffitz JE, Hofstra RM, van den Berg MP: Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm. 2009, 6: 1574-83.
Taylor M, Graw S, Sinagra G, Barnes C, Slavov D, Brun F, Pinamonti B, Salcedo EE, Sauer W, Pyxaras S, Anderson B, Simon B, Bogomolovas J, Labeit S, Granzier H, Mestroni L: Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation. 2011, 124: 876-85.
Groeneweg JA, van der Zwaag PA, Jongbloed JD, Cox MG, Vreeker A, de Boer RA, van der Heijden JF, van Veen TA, McKenna WJ, van Tintelen JP, Dooijes D, Hauer RN: Left-dominant arrhythmogenic cardiomyopathy in a large family: associated desmosomal or nondesmosomal genotype?. Heart Rhythm. 2013, 10: 548-59.
DeWitt MM, MacLeod HM, Soliven B, McNally EM: Phospholamban R14 deletion results in late-onset, mild, hereditary dilated cardiomyopathy. J Am Coll Cardiol. 2006, 48: 1396-8.
Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L, Seidman JG, Seidman CE: Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012, 366: 619-28.
Quarta G, Syrris P, Ashworth M, Jenkins S, Zuborne Alapi K, Morgan J, Muir A, Pantazis A, McKenna WJ, Elliott PM: Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2012, 33: 1128-36.
van der Zwaag PA, van Rijsingen IA, Asimaki A, Jongbloed JD, van Veldhuisen DJ, Wiesfeld AC, Cox MG, van Lochem LT, de Boer RA, Hofstra RM, Christiaans I, van Spaendonck-Zwarts KY, Deprez RH L d, Judge DP, Calkins H, Suurmeijer AJ, Hauer RN, Saffitz JE, Wilde AA, van den Berg MP, van Tintelen JP: Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail. 2012, 14: 1199-207.
Michaels PJ, Kobashigawa JA, Child JS, Fishbein MC: Chronic right-sided myocarditis mimicking arrhythmogenic right ventricular dysplasia. Hum Pathol. 2000, 31: 618-21.
Friedrich MG, Sechtem U, Schulz-Menger J, Holmvang G, Alakija P, Cooper LT, White JA, Abdel-Aty H, Gutberlet M, Prasad S, Aletras A, Laissy JP, Paterson I, Filipchuk NG, Kumar A, Pauschinger M, Liu P: Cardiovascular magnetic resonance in myocarditis: A JACC White Paper. J Am Coll Cardiol. 2009, 53: 1475-87.
Miller DD, Holmvang G, Gill JB, Dragotakes D, Kantor HL, Okada RD, Brady TJ: MRI detection of myocardial perfusion changes by gadolinium-DTPA infusion during dipyridamole hyperemia. Magn Reson Med. 1989, 10: 246-55.
Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, Camm AJ, Ellinor PT, Gollob M, Hamilton R, Hershberger RE, Judge DP, Le Marec H, McKenna WJ, Schulze-Bahr E, Semsarian C, Towbin JA, Watkins H, Wilde A, Wolpert C, Zipes DP: HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies. Heart Rhythm. 2011, 8: 1308-39.
Towbin JA: Arrhythmogenic right ventricular cardiomyopathy: a paradigm of overlapping disorders. Ann Noninvasive Electrocardiol. 2008, 13: 325-6.
Ashley EA, Hershberger RE, Caleshu C, Ellinor PT, Garcia JG, Herrington DM, Ho CY, Johnson JA, Kittner SJ, Macrae CA, Mudd-Martin G, Rader DJ, Roden DM, Scholes D, Sellke FW, Towbin JA, Van Eyk J, Worrall BB: Genetics and cardiovascular disease: a policy statement from the American Heart Association. Circulation. 2012, 126: 142-57.
Charron P, Arad M, Arbustini E, Basso C, Bilinska Z, Elliott P, Helio T, Keren A, McKenna WJ, Monserrat L, Pankuweit S, Perrot A, Rapezzi C, Ristic A, Seggewiss H, van Langen I, Tavazzi L, Pericardial D: Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2010, 31: 2715-26.
Sen-Chowdhry S, Syrris P, McKenna WJ: Desmoplakin disease in arrhythmogenic right ventricular cardiomyopathy: early genotype-phenotype studies. Eur Heart J. 2005, 26: 1582-4.
Te Riele AS, Bhonsale A, Burt JR, Zimmerman SL, Tandri H: Genotype-specific pattern of LV involvement in ARVD/C. J Am Coll Cardiol Img. 2012, 5: 849-51.
Corrado D, Basso C, Rizzoli G, Schiavon M, Thiene G: Does sports activity enhance the risk of sudden death in adolescents and young adults?. J Am Coll Cardiol. 2003, 42: 1959-63.
James CA, Bhonsale A, Tichnell C, Murray B, Russell SD, Tandri H, Tedford RJ, Judge DP, Calkins H: Exercise Increases Age-Related Penetrance and Arrhythmic Risk in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Associated Desmosomal Mutation Carriers. J Am Coll Cardiol. 2013, 62: 1290-7.
Wichter T, Paul M, Wollmann C, Acil T, Gerdes P, Ashraf O, Tjan TD, Soeparwata R, Block M, Borggrefe M, Scheld HH, Breithardt G, Bocker D: Implantable cardioverter/defibrillator therapy in arrhythmogenic right ventricular cardiomyopathy: single-center experience of long-term follow-up and complications in 60 patients. Circulation. 2004, 109: 1503-8.
Corrado D, Calkins H, Link MS, Leoni L, Favale S, Bevilacqua M, Basso C, Ward D, Boriani G, Ricci R, Piccini JP, Dalal D, Santini M, Buja G, Iliceto S, Estes NA 3rd, Wichter T, McKenna WJ, Thiene G, Marcus FI: Prophylactic implantable defibrillator in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia and no prior ventricular fibrillation or sustained ventricular tachycardia. Circulation. 2010, 122: 1144-52.
Piccini JP, Dalal D, Roguin A, Bomma C, Cheng A, Prakasa K, Dong J, Tichnell C, James C, Russell S, Crosson J, Berger RD, Marine JE, Tomaselli G, Calkins H: Predictors of appropriate implantable defibrillator therapies in patients with arrhythmogenic right ventricular dysplasia. Heart Rhythm. 2005, 2: 1188-94.
Keller DI, Osswald S, Bremerich J, Bongartz G, Cron TA, Hilti P, Pfisterer ME, Buser PT: Arrhythmogenic right ventricular cardiomyopathy: diagnostic and prognostic value of the cardiac MRI in relation to arrhythmia-free survival. Int J Cardiovasc Imaging. 2003, 19: 537-43. discussion 545–537
Deac M, Alpendurada F, Fanaie F, Vimal R, Carpenter JP, Dawson A, Miller C, Roussin I, di Pietro E, Ismail TF, Roughton M, Wong J, Dawson D, Till JA, Sheppard MN, Mohiaddin RH, Kilner PJ, Pennell DJ, Prasad SK: Prognostic value of cardiovascular magnetic resonance in patients with suspected arrhythmogenic right ventricular cardiomyopathy. Int J Cardiol. 2013, 168: 3514-21.
Wichter T, Borggrefe M, Haverkamp W, Chen X, Breithardt G: Efficacy of antiarrhythmic drugs in patients with arrhythmogenic right ventricular disease. Results in patients with inducible and noninducible ventricular tachycardia. Circulation. 1992, 86: 29-37.
Marcus GM, Glidden DV, Polonsky B, Zareba W, Smith LM, Cannom DS, Estes NA, Marcus F, Scheinman MM: Multidisciplinary Study of Right Ventricular Dysplasia I. Efficacy of antiarrhythmic drugs in arrhythmogenic right ventricular cardiomyopathy: a report from the North American ARVC Registry. J Am Coll Cardiol. 2009, 54: 609-15.
Dalal D, Jain R, Tandri H, Dong J, Eid SM, Prakasa K, Tichnell C, James C, Abraham T, Russell SD, Sinha S, Judge DP, Bluemke DA, Marine JE, Calkins H: Long-term efficacy of catheter ablation of ventricular tachycardia in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2007, 50: 432-40.
Philips B, Madhavan S, James C, Tichnell C, Murray B, Dalal D, Bhonsale A, Nazarian S, Judge DP, Russell SD, Abraham T, Calkins H, Tandri H: Outcomes of catheter ablation of ventricular tachycardia in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Arrhythm Electrophysiol. 2012, 5: 499-505.
Schmidt B, Chun KR, Baensch D, Antz M, Koektuerk B, Tilz RR, Metzner A, Ouyang F, Kuck KH: Catheter ablation for ventricular tachycardia after failed endocardial ablation: epicardial substrate or inappropriate endocardial ablation?. Heart Rhythm. 2010, 7: 1746-52.
Bai R, Di Biase L, Shivkumar K, Mohanty P, Tung R, Santangeli P, Saenz LC, Vacca M, Verma A, Khaykin Y, Mohanty S, Burkhardt JD, Hongo R, Beheiry S, Dello Russo A, Casella M, Pelargonio G, Santarelli P, Sanchez J, Tondo C, Natale A: Ablation of ventricular arrhythmias in arrhythmogenic right ventricular dysplasia/cardiomyopathy: arrhythmia-free survival after endo-epicardial substrate based mapping and ablation. Circ Arrhythm Electrophysiol. 2011, 4: 478-85.
Andreu D, Ortiz-Perez JT, Boussy T, Fernandez-Armenta J, de Caralt TM, Perea RJ, Prat-Gonzalez S, Mont L, Brugada J, Berruezo A: Usefulness of contrast-enhanced cardiac magnetic resonance in identifying the ventricular arrhythmia substrate and the approach needed for ablation. Eur Heart J. 2014, 35: 1316-26.
Acknowledgments
The authors wish to acknowledge funding from the Alexandre Suerman Stipend (to AT), the National Heart, Lung, and Blood Institute (K23HL093350 to HT), the Dr. Francis P. Chiaramonte Private Foundation, the St. Jude Medical Foundation, Medtronic Inc., and the NIH intramural research program (to DAB). The Johns Hopkins ARVC Program is supported by the Dr. Satish, Rupal, and Robin Shah ARVD Fund at Johns Hopkins, the Bogle Foundation, the Healing Hearts Foundation, the Campanella family, the Patrick J. Harrison Family, the Peter French Memorial Foundation, and the Wilmerding Endowments. We are grateful to the ARVC patients and families who have made this work possible.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AT, HT, and DB conceived the study, performed the background research and review, and drafted the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
te Riele, A.S., Tandri, H. & Bluemke, D.A. Arrhythmogenic right ventricular cardiomyopathy (ARVC): cardiovascular magnetic resonance update. J Cardiovasc Magn Reson 16, 50 (2014). https://doi.org/10.1186/s12968-014-0050-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12968-014-0050-8