
Abstract
Background
Recent studies reported that soluble epoxide hydrolase (sEH) plays an important role in lung diseases. However, the role of sEH in hyperoxia-induced ALI is unclear.
Methods
ALI was induced by exposure to 100% oxygen in an airtight cage for 72 h in wild-type (WT) and sEH gene deletion (EPHX2−/−) mice. ALI was assessed by the lung dry/wet ratio, alveolar capillary protein leak, and the infiltration of inflammatory cells in the lung.
Results
Hyperoxia elevated sEH activity in WT mice. Simultaneously, epoxyeicosatrienoic acids (EETs) levels were decreased in WT mice exposed to hyperoxia. However, the level of EETs was increased in EPHX2−/− mice exposed to hyperoxia. Hyperoxia induced pulmonary edema and inflammation were dampened in EPHX2−/− mice compared with WT mice. Decreased expression of Kelch-like ECH-associated protein 1 (Keap1) was found in EPHX2−/− mice exposed to hyperoxia. Hyperoxia-induced the expression of nuclear-factor erythroid 2-related factor 2 (Nrf2) was enhanced in EPHX2−/− mice compared with WT mice. Simultaneously, the activities of heme oxygenase-1 and superoxide dismutase were elevated in EPHX2−/− mice. The levels of reactive oxygen species were inhibited in EPHX2−/− mice compared with WT mice exposed to hyperoxia.
Conclusions
sEH is a harmful factor for hyperoxic ALI. The beneficial effect of sEH gene deletion is associated with the elevation of EETs and regulation of Nrf2/Keap1 signal pathway.
Similar content being viewed by others

Background
Acute respiratory distress syndrome (ARDS) is a common severe complication of sepsis with a complicated pathophysiological mechanism [1]. A mild form of ARDS was previously known as acute lung injury (ALI) [2]. Consistent with sepsis [3, 4], to date, there are no effective pharmacological interventions for ARDS [1, 5]. A recent animal study reported that inhibition of soluble epoxide hydrolase (sEH) reduces lipopolysaccharide-induced ALI [6]. sEH is a novel pharmacological target for inflammatory disorder [7]. For pulmonary diseases, inhibition or deletion of sEH reduced cigarette smoke-induced pulmonary inflammation [8], bleomycin-induced pulmonary fibrosis [9], angiotensin II-induced ALI [10]. These results suggest that sEH contributes to the pathogenesis of lung inflammatory disorders including ARDS.
Hyperoxia is a very high topic in medicine [11]. Recently, the WHO recommended the use of high fraction of inspired oxygen for the prevention of surgical site infections [12]. However, the hyperoxia may be a double-edged sword. Evidence has shown that hyperoxia causes ARDS [13]. The potential mechanism that is involved in the hyperoxia caused ARDS is complicated. Over generated reactive oxygen species (ROS) products cause an imbalance between oxidant and antioxidant. Antioxidant enzymes such as heme oxygenase (HO)-1 and superoxide dismutase (SOD) scavenge ROS playing an important role in antioxidant defense. The expression of antioxidant enzymes is regulated by nuclear-factor erythroid 2-related factor 2 (Nrf2). Nrf2 defends the lung from hyperoxic ALI [14].
As the sEH is suggested as a pharmacological target for ALI [6], we, therefore, investigate that whether the sEH plays any role in hyperoxic ALI in the present study, and the effect of sEH gene deletion on Nrf2 pathway.
Methods
The current study was performed in accordance with the Guide for the Care and Use of Laboratory Animals of Lanzhou University, and approved by the Animal Care and Use Committee of Lanzhou University (no. 20150520016). Eight- to ten- weeks old male C57BL/6 wild-type (WT) mice and sEH gene deletion (EPHX2−/−) mice were used in the present study. The C57BL/6 WT mice were obtained from Lanzhou University. EPHX2−/− mice were back-crossed onto a C57BL/6 genetic background for more than ten generations as previously described [15]. The mice were housed in a temperature-controlled facility (22 ± 2 °C) and kept under a 12-h light/dark cycle. The animals were given free access to food and water. Hyperoxic ALI was induced by exposure of the mice to 100% oxygen in an airtight cage for 72 h, as described previously [16]. We performed the Evan’s blue (EB) dye technique 30 min prior to sacrifice to measure the pulmonary microvascular albumin-permeability as previously described [17]. The leak of EB dye-labeled albumin into the lung tissue was measured spectrophotometrically and calculated after sacrifice. Animals were euthanized at 72 h after hyperoxia exposure. The trachea was isolated by blunt dissection and a small-caliber tube was inserted into the airway and secured. Then, three volumes of 1 ml of phosphate buffered saline (pH 7.2) were instilled and gently aspirated. The fluid recovery rate was > 90%. The bronchoalveolar lavage fluid (BALF) was centrifuged at 1000×g for 10 min at 4 °C. The supernatant was analyzed for protein and cytokines. Cell counts were measured with a hemocytometer. Then, the lung was stored in liquid nitrogen at − 80 °C. Before use, the frozen lung tissue samples were crushed to powder with a mortar and pestle. A diagram of the protocol was shown in Fig. 1.

A diagram of the protocol. WT, wild-type mice; EPHX2−/−, soluble epoxide hydrolase (sEH) gene knockout mice; D/W ratio, lung dry/wet weight ratio; EETs, epoxyeicosatrienoic acids; TNF-α, tumor necrosis factor-α; MIP-2, macrophage inflammatory protein-2; BALF, bronchoalveolar lavage fluid; Nrf2, nuclear-factor erythroid 2-related factor 2; Keap1, Kelch-like ECH-associated protein 1; HO-1, heme oxygenase-1; SOD, superoxide dismutase; ROS, reactive oxygen species
Pulmonary water content measurement
Pulmonary tissues were weighed and, then, dried in a 70 °C oven for 48 h. Pulmonary water content was calculated as percent H2O = (1 – dry /wet) × 100%.
Enzyme-linked immunosorbent assay (ELISA)
Tumor necrosis factor (TNF)-α and macrophage inflammatory protein (MIP)-2 were quantified using ELISA kits (R&D Systems) according to the manufacturer’s instructions.
The 11,12-epoxyeicosatrienoic acids (EETs) and 14,15-EETs are suggested as effective agents suppressing lung inflammation [18]. ELISA kits (Detroit R&D, Detroit, MI, USA) were used to determine concentrations of 11,12-EETs and 14,15-EETs and their stable metabolites 11,12-dihydroxyeicosatrienoic acids (DHETs) and 14,15-DHETs in lung tissues according to the manufacturer’s instructions.
The level of 8-Isoprostane was assessed by an 8-Isoprostane Oxidative Stress ELISA Kit (Detroit R&D, Michigan, USA) according to the manufacturer’s instructions.
Measurement of SOD activity
The activity of SOD was measured by a SOD activity assay kit according to the manufacturer’s instructions (R&D Systems, Inc., MN, USA). Briefly, total 50 μg of protein samples were incubated with tetrazolium salt and xanthine oxidase enzyme for 20 min at 37 °C. Then, xanthine oxidase solution was added. The absorbance of the formazan salt was detected at 550 nm.
Measurement of HO-1 activity
Lung tissues were harvested. The samples (50 μl of lung cytosol, 200 μl of lung supernatant, 20 μl of 1 mM heme solution, 200 μl of 2.75 mM NADPH solution, and 530 μl of 2 mM MgCl2 in 100 mM phosphate buffered saline (pH 7.4)) were incubated in a 37 °C water bath in the dark for 1 h. Then, the reaction was stopped by placement on ice. The absorbance of the sample was measured by spectrophotometer at 530 nm. The amount of bilirubin formed was calculated from the difference in absorbance at 530 nm. An NADPH-free reaction sample was served as an internal control [19].
Western blot analysis
Homogenized lung tissues were lysed with lyses buffer containing protease inhibitors cocktail (Cell Signaling, Boston, MA, USA). BCA method was used to determine protein concentration (Pierce Chemical Co., TX, and USA). Total 50 μg protein samples were separated by 10% sodium codicil sulfate-PAGE, and were transferred to a hyoid-enhanced chemiluminescence’s (ECL) nitrocellulose membrane. The membrane was blocked in 5% skim milk powder at room temperature for one hour. Then, the membrane was blotted with the primary antibody (1:1000) against Nrf2 (Cambridge, MA, USA) and Kelch-like ECH-associated protein 1 (Keap1) (Santa Cruz Biotechnology Inc., Santa Cruz, CA,USA) overnight at 4 °C and the corresponding secondary antibody (1:5000) at room temperature for one hour. Finally the blots were visualized with an enhanced ECL western blot detection system (Habersham Pharmacia Biotech, Piscataway, NJ, USA). The band intensities were quantified using Image J 1.47v software (NIH.USA). Laming B and β-acting (Cell Signaling, Boston, MA, USA) were used as internal references for determination of nuclear and cryptozoic protein, respectively.
Statistical analysis
Data are expressed as means ± SEM. Data are analyzed by using Graph Pad Prism 3.0 (Graph Pad Software). The two-tailed Student’s t-test for comparison between two groups. The one-way analysis of variance (ANOVA) followed by Bonferroni’s post hoc test for multiple comparisons. A P value of < 0.05 was considered to be statistically significant.
Results
Hyperoxia-triggered lung edema and alveolar leakage were attenuated in sEH knockout mice
The lung edema was measured by the dry to wet ratio of the lung. Our results showed that the dry to wet ratio was the same for both the groups exposed to room air (Fig. 2a). However, lung edema was triggered by hyperoxia exposure (Fig. 2a). As shown in Fig. 2a, the lung dry to wet ratio was inhibited in the sEH knockout mice compared with WT mice under hyperoxia.

Wild-type (WT) and soluble epoxide hydrolase gene knock out (EPHX2−/−) mice exposed to room air or 100% oxygen for 72 h were euthanized. Lung edema (a), alveolar leakage (b), tumor necrosis factor (TNF)-α (c), and macrophage inflammatory protein (MIP)-2 levels (d) were measured. Data are represented as the mean ± SEM of three independent experiments. *P < 0.05 vs. WT mice exposed to room air; # P < 0.05 vs. WT mice exposed to hyperoxia
The alveolar leakage was measured by the Evan’s blue dye technique. The alveolar albumin-permeability increased by 3.9-fold in the WT mice under hyperoxia compared with WT mice under air (Fig. 2b). The hyperoxia-induced alveolar leakage was markedly improved in sEH knockout mice (Fig. 2b).
Hyperoxia-induced upregulation of TNF-α and MIP-2 was suppressed in sEH knockout mice
Seventy-two hours after hyperoxia exposure, the levels of TNF-α and MIP-2 were increased in the WT mice as evaluated by ELISA (Fig. 2c and d). However, the hyperoxia-induced upregulation of TNF-α and MIP-2 was inhibited by 43% and 66% in the sEH knockout mice compared with WT mice, respectively (Fig. 2c and d).
Hyperoxia-induced infiltration of neutrophil was suppressed in sEH knockout mice
Neutrophil plays a vital role throughout the progression of ALI. We found that the neutrophil counts in BALF were increased in the WT mice under hyperoxia compared with the group exposed to room air (Fig. 3a). However, the hyperoxia-induced accumulation of neutrophil was reduced by 44% in the sEH knockout mice compared with WT mice (Fig. 3a).

Wild-type (WT) and soluble epoxide hydrolase gene knock out (EPHX2−/−) mice exposed to room air or 100% oxygen for 72 h were euthanized. Neutrophil counts (a), 8-Isoprostane levels (b), nuclear-factor erythroid 2-related factor 2 (Nrf2) (c), and Kelch-like ECH-associated protein 1 (Keap1) (d) were measured. Lamin B and β-actin were used as internal references for determination of nuclear and cytosolic protein, respectively. Data are represented as the mean ± SEM of three independent experiments. *P < 0.05 vs. WT mice exposed to room air; # P < 0.05 vs. WT mice exposed to hyperoxia
Hyperoxia-induced lipid peroxidation products were decreased in sEH knockout mice
The lipid peroxidation is an indicator for oxidative stress. Hyperoxia increased the incidence of 8-Isoprostan formation, an indicator for lipid peroxidation, in the lung in the WT mice (Fig. 3b). However, we found that the hyperoxia-induced elevation of 8-Isoprostane was inhibited in the sEH knockout mice compared with WT mice (Fig. 3b).
Hyperoxia-induced activation of Nrf2/Keap1 pathway was enhanced in sEH knockout mice
As shown in Fig. 3c, elevated Nrf2 levels in the nucleus were detected by western blot analysis in the WT mice exposed to hyperoxia. The hyperoxia-induced upregulation of Nrf2 in the nucleus was enhanced in the sEH knockout mice compared with WT mice (Fig. 3c).
Keap1 is the inhibitor of Nrf2. Our western blot analysis showed a decrease in Keap1 in the cytoplasm in EPHX2−/− mice compared with WT mice exposed to hyperoxia (Fig. 3d).
Activities of antioxidant enzymes were increased in sEH knockout mice exposed to hyperoxia
In the WT mice, hyperoxia exposure increased the HO-1 activity by 2.8-fold, but reduced the SOD activity by 23%, respectively (Fig. 4a and b). However, both the HO-1 and SOD activities were increased in the sEH knockout mice compared with WT mice exposed to hyperoxia (Fig. 4a and b). There was no significant difference in the activities of HO-1 and SOD between the sEH knockout mice compared with WT mice exposed to room air (Fig. 4a and b).

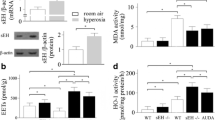
Wild-type (WT) and soluble epoxide hydrolase gene knock out (EPHX2−/−) mice exposed to room air or 100% oxygen for 72 h were euthanized. Heme oxygenase (HO)-1 activity (a), superoxide dismutase (SOD) activity (b), 11,12-dihydroxyeicosatrienoic acids (DHETs) and 14,15-DHETs levels (c), 11,12-epoxyeicosatrienoic acids (EETs) and 14,15-EETs levels (d) were measured. Data are represented as the mean ± SEM of three independent experiments. *P < 0.05 vs. WT mice exposed to room air; # P < 0.05 vs. WT mice exposed to hyperoxia
Hyperoxia-induced elevation of DHETs and reduction of EETs were inhibited in sEH knockout mice
sEH converts EETs to DHETs. The levels of 11,12-DHETs and 14,15-DHETs were used to determine the activity of sEH by an ELISA kit. As shown in Fig. 4c, hyperoxia exposure led to an increase in 11,12-DHETs and 14,15-DHETs products in WT mice. Seventy-two hours after hyperoxia exposure, the levels of 11,12-EETs were reduced; but, the 14,15-EETs levels were increased (Fig. 4d). However, there was no statistically significant in 14,15-EETs levels between WT mice exposed to hyperoxia and room air (Fig. 4d). Our ELISA analysis showed that both 11,12-EETs and 14,15-EETs levels were increased in the sEH knockout mice exposed to hyperoxia (Fig. 4d).
Discussion
Hyperoxia is used to support critically ill patient [20]. However, hyperoxia causes elevation of ROS products, and activates inflammatory mediators [21]. Our data corroborate these findings. The ROS products were markedly increased in WT mice exposed to hyperoxia. It was simultaneously associated with an increase in pulmonary inflammation and edema. However, the hyperoxia-induced ROS products and lung injury were dampened in sEH gene knockout mice. Moreover, the activities of antioxidant enzymes and Nrf2 were enhanced in sEH knockout mice compared with WT mice. These results suggest that sEH is a harmful factor for hyperoxic ALI.
ROS cause toxicity to cellular components including DNA, lipids, and proteins, resulting cell swelling and cell membrane breakdown [22]. ROS generated in normal physiologic conditions did not cause harm, because the ROS can readily detoxified by antioxidants. The balance between oxidants and antioxidants is known as redox homeostasis. However, harmful stimuli such as prolonged hyperoxia exposure can disrupt this equilibrium. Excessive generated oxidants overwhelm the antioxidants leading to oxidative stress [22]. Our results are consistent with previous studies, hyperoxia exposure for 72 h induced ROS products as well as lung inflammation and edema. Antioxidant enzymes such as HO-1 and SOD play an essential role in maintenaning the balance between antioxidants and oxidants. Evidence has shown that SOD gene-modified mesenchymal stem cells attenuate acute radiation-induced ALI [23]. HO-1 is known as a vital antioxidant enzyme [24]. Upregulation of HO-1 protects the lung during ALI [25, 26]. In the present study, the activities of SOD and HO-1 were enhanced in sEH gene deleted mice. Simultaneously, the ROS products were reduced in EPHX2−/− mice.
Nrf2 defends the lung from oxidative stress [14]. Nrf2 deficiency increases susceptibility to hyperoxic lung injury [27], and impairs the resolution of pulmonary inflammation [28, 29]. Likewise, deletion of Nrf2 in airway epithelium also exacerbates hyperoxic ALI and impairs the resolution of pulmonary inflammation [30]. These results suggest that Nrf2 is essential for protection against hyperoxic ALI. Nrf2 is a transcription factor which activates many anti-oxidant genes via phosphorylation and dissociation from the cytoplasmic inhibitor, Keap1. In the present study, the Keap1 was inhibited in sEH knockout mice compared with WT mice exposed to hyperoxia. It was simultaneously associated with an increase in the nuclear level of Nrf2.
Inhibition or deletion of sEH is associated with dampened oxidants products [31, 32]. However, the underlying mechanism has not been fully characterized. sEH saves EETs which has antioxidant role [33]. EETs inhibit Bach-1, a negative regulator of HO-1 expression [34], which in turn activates HO-1 expression. Our data are consistent with previous studies, the EETs levels and HO-1 activity were increased in sEH knockout mice compared with WT mice exposed to hyperoxia. Moreover, our results suggest that the benefit of sEH gene deletion in hyperoxic ALI is associated with activating Nrf2/ Keap1 pathway.
Conclusions
sEH is a harmful factor for hyperoxic ALI. The beneficial effect of sEH gene deletion is associated with the elevation of EETs and regulation of Nrf2/Keap1 signal pathway.
Abbreviations
- ALI:
-
acute lung injury
- ANOVA:
-
analysis of variance
- ARDS:
-
acute respiratory distress syndrome
- BALF:
-
bronchoalveolar lavage fluid
- D/W ratio:
-
lung dry/wet weight ratio
- EETs:
-
epoxyeicosatrienoic acids
- ELISA:
-
enzyme-linked immunosorbent assay
- HO-1:
-
heme oxygenase-1
- Keap1:
-
Kelch-like ECH-associated protein 1
- MIP-2:
-
macrophage inflammatory protein-2
- Nrf2:
-
nuclear-factor erythroid 2-related factor 2
- ROS:
-
reactive oxygen species
- sEH:
-
soluble epoxide hydrolase
- SOD:
-
superoxide dismutase
- TNF-α:
-
tumor necrosis factor-α
References
Leaver SK, Evans TW. Acute respiratory distress syndrome. BMJ. 2007;335(7616):389–94.
Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, Camporota L, Slutsky AS. Acute respiratory distress syndrome: the berlin definition. JAMA. 2012;307(23):2526–33.
Tao W, Li PS, Shen Z, Shu YS, Liu S. Effects of omega-3 fatty acid nutrition on mortality in septic patients: a meta-analysis of randomized controlled trials. BMC Anesthesiol. 2016;16(1):39.
Zhang L, Zhu G, Han L, Fu P. Early goal-directed therapy in the management of severe sepsis or septic shock in adults: a meta-analysis of randomized controlled trials. BMC Med. 2015;13:71.
Sweeney RM, McAuley DF. Acute respiratory distress syndrome. Lancet. 2016;388(10058):2416–30.
Tao W, Li PS, Yang LQ, Ma YB. Effects of a soluble epoxide hydrolase inhibitor on lipopolysaccharide-induced acute lung injury in mice. PLoS One. 2016;11(8):e0160359.
Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc Natl Acad Sci U S A. 2005;102(28):9772–7.
Smith KR, Pinkerton KE, Watanabe T, Pedersen TL, Ma SJ, Hammock BD. Attenuation of tobacco smoke-induced lung inflammation by treatment with a soluble epoxide hydrolase inhibitor. Proc Natl Acad Sci U S A. 2005;102(6):2186–91.
Zhou Y, Yang J, Sun GY, Liu T, Duan JX, Zhou HF, Lee KS, Hammock BD, Fang X, Jiang JX, et al. Soluble epoxide hydrolase inhibitor 1-trifluoromethoxyphenyl-3- (1-propionylpiperidin-4-yl) urea attenuates bleomycin-induced pulmonary fibrosis in mice. Cell Tissue Res. 2016;363(2):399–409.
Tao W, Li PS, Xu G, Luo Y, Shu YS, Tao YZ, Yang LQ. Soluble epoxide hydrolase plays a vital role in angiotensin II-induced lung injury in mice. Ga: Shock (Augusta; 2017.
Akca O, Ball L, Belda FJ, Biro P, Cortegiani A, Eden A, Ferrando C, Gattinoni L, Goldik Z, Gregoretti C, et al. WHO needs high FIO2. Turkish journal of anaesthesiology and reanimation. 2017;45(4):181–92.
Wenk M, Van Aken H, Zarbock A. The new World Health Organization recommendations on perioperative Administration of Oxygen to prevent surgical site infections: a dangerous reductionist approach? Anesth Analg. 2017;125(2):682–7.
Cochrane CG, Spragg R, Revak SD. Pathogenesis of the adult respiratory distress syndrome. Evidence of oxidant activity in bronchoalveolar lavage fluid. J Clin Invest. 1983;71(3):754–61.
Cho HY, Reddy SP, Kleeberger SR. Nrf2 defends the lung from oxidative stress. Antioxid Redox Signal. 2006;8(1–2):76–87.
Luria A, Weldon SM, Kabcenell AK, Ingraham RH, Matera D, Jiang H, Gill R, Morisseau C, Newman JW, Hammock BD. Compensatory mechanism for homeostatic blood pressure regulation in Ephx2 gene-disrupted mice. J Biol Chem. 2007;282(5):2891–8.
Tao W, Shu YS, Miao QB, Zhu YB. Attenuation of hyperoxia-induced lung injury in rats by adrenomedullin. Inflammation. 2012;35(1):150–7.
Wang L, Taneja R, Razavi HM, Law C, Gillis C, Mehta S: Specific role of neutrophil inducible nitric oxide synthase in murine sepsis-induced lung injury in vivo. Shock (Augusta, Ga) 2012, 37(5):539–547.
Li Y, Yu G, Yuan S, Tan C, Xie J, Ding Y, Lian P, Fu L, Hou Q, Xu B, et al. 14,15-Epoxyeicosatrienoic acid suppresses cigarette smoke condensate-induced inflammation in lung epithelial cells by inhibiting autophagy. Am J Physiol Lung Cell Mol Physiol. 2016;311(5):L970–l980.
Jin SW, Zhang L, Lian QQ, Liu D, Wu P, Yao SL, Ye DY. Posttreatment with aspirin-triggered lipoxin A4 analog attenuates lipopolysaccharide-induced acute lung injury in mice: the role of heme oxygenase-1. Anesth Analg. 2007;104(2):369–77.
Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353(16):1685–93.
Bhandari V, Elias JA. Cytokines in tolerance to hyperoxia-induced injury in the developing and adult lung. Free Radic Biol Med. 2006;41(1):4–18.
Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24(5):981–90.
Chen HX, Xiang H, Xu WH, Li M, Yuan J, Liu J, Sun WJ, Zhang R, Li J, Ren ZQ, et al. Manganese superoxide dismutase gene-modified mesenchymal stem cells attenuate acute radiation-induced lung injury. Hum Gene Ther. 2017;28(6):523–32.
Raval CM, Lee PJ. Heme oxygenase-1 in lung disease. Curr Drug Targets. 2010;11(12):1532–40.
Chi X, Guo N, Yao W, Jin Y, Gao W, Cai J, Hei Z. Induction of heme oxygenase-1 by hemin protects lung against orthotopic autologous liver transplantation-induced acute lung injury in rats. J Transl Med. 2016;14:35.
Joo Choi R, Cheng MS, Shik Kim Y. Desoxyrhapontigenin up-regulates Nrf2-mediated heme oxygenase-1 expression in macrophages and inflammatory lung injury. Redox Biol. 2014;2:504–12.
Cho HY, Jedlicka AE, Reddy SP, Kensler TW, Yamamoto M, Zhang LY, Kleeberger SR. Role of NRF2 in protection against hyperoxic lung injury in mice. Am J Respir Cell Mol Biol. 2002;26(2):175–82.
Reddy NM, Kleeberger SR, Kensler TW, Yamamoto M, Hassoun PM, Reddy SP: Disruption of Nrf2 impairs the resolution of hyperoxia-induced acute lung injury and inflammation in mice. Journal of immunology (Baltimore, Md : 1950) 2009, 182(11):7264–7271.
Chan K, Kan YW. Nrf2 is essential for protection against acute pulmonary injury in mice. Proc Natl Acad Sci U S A. 1999;96(22):12731–6.
Reddy NM, Potteti HR, Mariani TJ, Biswal S, Reddy SP. Conditional deletion of Nrf2 in airway epithelium exacerbates acute lung injury and impairs the resolution of inflammation. Am J Respir Cell Mol Biol. 2011;45(6):1161–8.
Shrestha A, Krishnamurthy PT, Thomas P, Hammock BD, Hwang SH. Soluble epoxide hydrolase inhibitor, t-TUCB, protects against myocardial ischaemic injury in rats. J Pharm Pharmacol. 2014;66(9):1251–8.
Elmarakby AA, Faulkner J, Al-Shabrawey M, Wang MH, Maddipati KR, Imig JD. Deletion of soluble epoxide hydrolase gene improves renal endothelial function and reduces renal inflammation and injury in streptozotocin-induced type 1 diabetes. American journal of physiology Regulatory, integrative and comparative physiology. 2011;301(5):R1307–17.
Chen W, Zheng G, Yang S, Ping W, Fu X, Zhang N, Wang DW, Wang J. CYP2J2 and EETs protect against oxidative stress and apoptosis in vivo and in vitro following lung ischemia/reperfusion. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2014;33(6):1663–80.
Vanella L, Kim DH, Sodhi K, Barbagallo I, Burgess AP, Falck JR, Schwartzman ML, Abraham NG. Crosstalk between EET and HO-1 downregulates Bach1 and adipogenic marker expression in mesenchymal stem cell derived adipocytes. Prostaglandins Other Lipid Mediat. 2011;96(1–4):54–62.
Acknowledgements
We thank Dr. Xiaodong Tang for a critical reading of this manuscript. The authors appreciate Qin Chen and Jin Chen for technical assistance.
Author’ contributions
LPL conceived and designed the study, performed experiments, analyzed and interpreted data, and drafted the manuscript. BL acquired and interpreted data, prepared figures, and revised the manuscript. TKS edited and revised manuscript. LZ performed experiments and drafted manuscript. YML conceived and designed the study. The final manuscript was reviewed and approved by all authors.
Funding
Natural Science Foundation of Gansu Province (1506RJZA255, 1308RJZA240–01); the Natural Science Foundation of China (81572437) and the International Science & Technology Cooperation Program of China (2015DFA31650).
Availability of data and materials
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
The current study was approved by the Animal Care and Use Committee of Lanzhou University (no. 20150520016).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Liu, LP., Li, B., Shuai, TK. et al. Deletion of soluble epoxide hydrolase attenuates mice Hyperoxic acute lung injury. BMC Anesthesiol 18, 48 (2018). https://doi.org/10.1186/s12871-018-0490-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12871-018-0490-z