
Abstract
Soybean is a crucial crop for the Brazilian economy, but it faces challenges from the biotrophic fungus Phakopsora pachyrhizi, which causes Asian Soybean Rust (ASR). In this study, we aimed to identify SNPs associated with resistance within the Rpp1 locus, which is effective against Brazilian ASR populations. We employed GWAS and re-sequencing analyzes to pinpoint SNP markers capable of differentiating between soybean accessions harboring the Rpp1, Rpp1-b and other alternative alleles in the Rpp1 locus and from susceptible soybean cultivars. Seven SNP markers were found to be associated with ASR resistance through GWAS, with three of them defining haplotypes that efficiently distinguished the accessions based on their ASR resistance and source of the Rpp gene. These haplotypes were subsequently validated using a bi-parental population and a diverse set of Rpp sources, demonstrating that the GWAS markers co-segregate with ASR resistance. We then examined the presence of these haplotypes in a diverse set of soybean genomes worldwide, finding a few new potential sources of Rpp1/Rpp1-b. Further genomic sequence analysis revealed nucleotide differences within the genes present in the Rpp1 locus, including the ULP1-NBS-LRR genes, which are potential R gene candidates. These results provide valuable insights into ASR resistance in soybean, thus helping the development of resistant soybean varieties through genetic breeding programs.
Similar content being viewed by others


Background
Soybean is one of the main products of the Brazilian economy, with 155 million metric tons harvested in the 2022/2023 growing season, on a cultivated area of 44 million hectares [1]. Although Brazil has a great potential to increase its production, there are some limiting factors affecting it, such as climate, pests, and diseases. The Asian Soybean Rust (ASR), caused by the obligate biotrophic fungus Phakopsora pachyrhizi (Pp), is currently the most damaging soybean disease in Brazil, with yield losses reaching up to 80% in the absence of adequate control measures [2, 3]. Since its identification in Brazilian fields (2001/2002), economic losses due to ASR have reached billions of US dollars (USD), which includes both yield loss and the cost of chemical control [4]. Currently the main disease control applied is a chemical one, with fungicide costs to manage ASR in Brazilian fields reaching more than 2 billion USD per year [5]. Yield loss caused by ASR in a food security hotspot including South of Brazil, Paraguay, Uruguay and Argentina was estimated in 6.65%. The disease was classified as chronical, which means, that cause large crop losses in specific food security hotspots [6].
Studies focusing on the identification of sources of genetic resistance to ASR and efforts to develop resistant cultivars have been done [7,8,9,10,11]. So far, seven different P. pachyrhizi resistance (Rpp) loci have been mapped in the soybean genome. The Rpp1 locus was the first locus identified in PI 200492, and it was mapped on chromosome 18 [12]. After that, the Rpp1 locus was also identified in several other accessions such as PI 417120, PI 423958, PI 518295, PI 547875, PI 368039 as well as in the Japanese cultivar Himeshirazu (PI 594177) and the Chinese accession Xiao Jing Huang [10, 13, 14]. Recently, Rpp1 was also reported in accessions from Malaysia (WC2) [15], Uganda (UG-5) [16] and India (EC241780) [17]. Meanwhile through differential virulence profiling of Pp isolates, allelism tests and genetic mapping, other Rpp1 alleles have also been identified. Originally, the Rpp1-b allele was mapped in PI 594538A [18], and subsequently Rpp1b or other alternative alleles in the accessions PI 587880A, PI 587886, PI 594767A, PI 587905, PI 587855, PI 594723, and PI 594756 [19,20,21,22,23]. In addition, other Rpp loci were further mapped: Rpp2 on chromosome 16 in the accessions PI 230970 [7] and PI 224270 [8], and Rpp3 on chromosome 6 in PI 462312 [24]. Rpp4 from PI 459025B [7], and Rpp6 from PI 567102B [25] were identified on chromosome 18, but mapped to distinct regions when compared to the Rpp1 locus. Rpp5 was mapped on chromosome 3 in PI 200456, PI200526 and PI 471904 [8]. Lastly, Rpp7 was mapped on chromosome 19 in PI 605823 [26]. However, an ineffectiveness of most of these Rpp loci has been reported over the world widely and is thought to be due to the high variability of Pp populations and races [27,28,29]. Furthermore, Pp populations from different countries show different virulence profiles, which means that the efficacy of Rpp loci is variable and depends on the origin of the Pp population [30,31,32,33]. Therefore, knowledge about Pp populations and local pathotypes, combined with the discovery of new alleles and loci, are essential for ASR management on soybean fields.
Currently, genome-wide association studies (GWAS) are one of the major approaches to identify genomic regions associated with resistance to pathogens in soybean [34,35,36], and so far, three GWAS analysis have been conducted to identify genomic regions associated to ASR resistance [37,38,39]. The first study, using USDA data, discovered two SNP markers associated with resistance: one on chromosome 15, a novel region associated with ASR resistance, and another within the Rpp1 locus on chromosome 18 [37]. Another study screened 191 soybean accessions in the Southeastern US from 2008 to 2015, identifying eight genomic regions linked to ASR resistance, including Rpp3 and Rpp6 loci, along with new regions unrelated to major resistance genes [38]. A recent study of 3,082 soybean accessions identified several genomic regions associated with ASR resistance, with significant SNP markers near the Rpp1, Rpp2, Rpp3, and Rpp4 loci [39].. However, all three studies were performed using SNP data derived from the SoySNP50K Infinium Chip, which may limit the discovery of new SNP markers. Furthermore, all studies were performed using Pp populations and/or isolates from the US, thus limiting the discovery of potential regions associated with resistance towards different pathotypes from other countries.
The analysis of the Rpp1 locus in PI 200492 through virus-induced gene silencing (VIGS) revealed a cluster of three NBS-LRR genes, with an N-terminal ubiquitin-like protease 1 (ULP1) domain, as the best candidates for the Rpp1 gene. Interestingly, the silencing of the ULP1-NBS-LRR genes switched plants from an immune response (absence of symptoms/lesions) to a resistant reaction (RB lesions) [40]. A recent study showed that as was the case for Rpp1 from PI 200492, the Rpp1-b locus from PI 594760B also contains three ULP1-NBS-LRR genes. It was also found that resistance from the Rpp1 locus can be affected by a mechanism of dominant susceptibility (DS), when Rpp1 accessions are crossed with accession carrying a susceptibility allele. Through VIGS, yeast two-hybrid studies and in silico modelling, the study suggested the NBS-LRR proteins from resistant and susceptible lines interacted with each other to lead to the DS phenome [41]. Importantly, those two studies highlighted the complexity of the Rpp1 locus in soybean and indicate that further studies are necessary to gain a better understanding of its resistance mechanisms. It was recently demonstrated that Rpp1-b allele is still effective against current Pp populations on Brazilian soybean fields, especially if combined with other Rpp genes [42, 43]. Despite the potential of this locus in providing robust resistance to soybean cultivars, there are few studies identifying and validating SNP markers tightly associated with these alleles, hindering its use in marker-assisted selection (MAS) programs.
In this present study, we aimed to explore the allelic variability at the Rpp1 locus using a diverse set of soybean accessions bearing Rpp1, Rpp1-b or other alternative alleles, to identify SNP markers tightly associated with this locus by GWAS. We found haplotypes common to a group of Rpp1-b donors and validated them in a biparental population. We also checked the distribution of the haplotypes in a diverse set of more than one thousand accessions of soybean worldwide. Overall, our study brings insights about the genomic composition of the Rpp1 locus, contributing for future cloning approaches. These data will be helpful in identifying the specific genes conferring Rpp resistance and will provide useful data for ASR management and breeding programs, for example in the pyramiding of multiple Rpp genes.
Methods
Plant materials
The GWAS panel was composed of 100 soybean accessions: 35 Brazilian cultivars, 3 American cultivars, 42 ASR-resistant advanced breeding lines (BL) from the Embrapa Soybean breeding program that were selected as they contained sources of Rpp1 in their pedigree, 12 varieties from China, five from Japan, two from Taiwan, and one from US, all previously described harboring Rpp1/Rpp1-b (Supplementary Table 1) and here identified by their GRIN PI (Plant Introduction) codes. The advanced breeding lines were developed to harbor the Rpp1 locus from different Rpp1 donors, such as PI 587880A, PI 561356 and PI 594766. Seeds for each soybean accession were sown under greenhouse conditions (temperature between 20 °C and 34 °C). Leaf tissue for each accession was harvested individually and frozen in liquid nitrogen for DNA extraction. For the fine sequence analysis of the Rpp1 locus, leaf material of seven soybean accessions bearing either Rpp1 or Rpp1-b were also collected for DNA extraction and short-read re-sequencing.
For haplotype validation, a biparental population derived from the susceptible accession PI 594774 (used as female) and the resistant accession PI 587880A (used as male) was developed. F1 plants from the crosses were self-pollinated, producing F2 seeds used for ASR phenotyping and SNP genotyping. A set of accessions harboring different Rpp loci was also sown, and leaf tissue was used for DNA extraction and genotyping. All accessions were grown in a greenhouse under controlled conditions for leaf collection and evaluation of resistance to P. pachyrhizi. All seeds used were obtained from the Embrapa Soybean Active Germplasm Bank, Londrina, Brazil.
ASR resistance evaluation
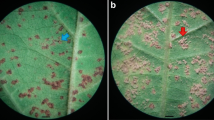
The 100 accessions of the GWAS panel and the 106 F2 progeny from the cross between PI 587880A × PI 594774 were inoculated with spores from a Brazilian P. pachyrhizi population collected from the experimental fields of Embrapa Soja, Londrina, Brazil in 2017. Briefly, plants were sowed in 8-L pots containing heat-sterilized soil. The GWAS panel accessions were arranged following a randomized block design, with three replicates (each replicate consisting of five plants per pot), in a total of 15 plants per genotype, while the F2 individuals in completely randomized design. Plants were inoculated at the V2-V3 developmental stage [44]. ASR inoculum consisted of Pp urediniospores at the concentration of 6 × 105 spores mL−1, suspended in a solution of sterile water and 0.01% (v/v) Tween-20 (Uniqema). Inoculations were carried out at the end of the day to ensure ideal conditions for spore viability and infectivity. Following inoculation, the plants were kept bagged for 24 h to ensure high humidity and ideal conditions for spore germination [36]. After that period, the bags were removed, and the plants remained in the greenhouse (80% humidity maintained by water spray) until symptoms appeared. Symptom assessment was performed approximately 10 days after inoculation. The second trifolium of each plant was evaluated qualitatively for lesion type as susceptible, characterized by tan coloured lesions with sporulating uredinia; and resistant, characterized by reddish-brown coloured lesions, with few or absent uredinia and spores [45]. Three evaluations were carried out at 10, 14, and 18 days after inoculation (DAI), to confirm the disease reaction. The phenotypic results were the same on all three evaluations.
DNA extraction, GBS approach and SNP calling
DNA extraction was performed using the DNeasy Plant Mini Kit (Qiagen, Inc., Valencia, CA) from 100 mg of young leaf tissue (14-day-old seedlings), following the manufacturer’s instructions. DNA concentration was determined using a NanoDrop ND-1000 UV–Vis spectrophotometer (Thermo Fisher Scientific) and diluted to 10 ng/μL. Sample integrity was confirmed by electrophoresis (120 V) on 1% agarose gel using 1X SB buffer (sodium borate).
Briefly, for the GBS library preparation, DNA from all the 100 accessions was digested by the enzyme ApeKI, linked to compatible adapters containing barcode sequences and primers for Ion Torrent sequencing performed at the Institut of Biologie Intégrative et des Systèmes at the Université Laval, Quebec, Canada (as per Sonah et al. [46]). Raw data (50–135 bp) were analyzed using the Fast-GBS pipeline [47]. In summary, raw paired-end reads were demultiplexed using Sabre (https://github.com/najoshi/sabre), cleaned and trimmed using Cutadapt [48]. Filtered paired-end reads were mapped to the soybean reference genome (W82.a2.v1) using BWA v0.7.17 [49] and variant calling was performed by Platypus [50]. After that, variants showing ≥ 80% of missing data were removed, and the resulting SNP catalogue as then filtered out to remove InDels. SNP markers with MAF (minor allele frequency) > 1% and heterozygosity < 10% were tehn submitted to imputation of missing data using Beagle v.4.1 [51].
Short-read data obtained from WGRS sequencing of Rpp1/Rpp1-b sources were trimmed to remove low-quality reads and adapters using Trimmomatic v0.39 [52]; filtered paired-end reads were mapped to the soybean reference genome (W82.a2.v1) with BWA, (BWA-MEM algorithm with default parameters). The resulting SAM files were converted to BAM format with SAMtools v1.9. BAM; files were then sorted, and PCR duplicates were marked with Picard Toolkit (https://github.com/broadinstitute/picard) and the variant calling was performed using GATK v4.1.4.1 with HaplotypeCaller (GATK) and GenotypeGVCFs functions [53].
Associative mapping analysis and haplotype analysis
GWAS was conducted using a compressed linear mixed model (cMLM—Compressed Mixed Linear Model) [54], implemented in the GAPIT (Genome Association and Prediction Integrated Tool) software package in R environment [55]. Population structure (three principal components) and genetic relatedness among the accessions (VanRaden kinship matrix (K)) were used to reduce confounding in the cMLM model. Since kinship is derived from all the markers, incorporating with the kinship for testing markers in a MLM model causes the confounding between the testing markers and the individuals’ genetic effects with variance structure defined by the kinship. To reduce the confounding problem, individuals are replaced by their corresponding groups in the cMLM model (https://zzlab.net/GAPIT/gapit_help_document.pdf). Only SNP-trait associations with an FDR-adjusted p-value (FDR—false discovery rate) ≤ 0.001 were considered significant. SNP markers highly associated with the ASR resistance were used to identify haplotypes to distinguish Rpp1, Rpp1-b and other Rpp1 alleles from susceptible alleles. We retrieved whole genome re-sequencing (WGRS) data from Brazilian soybean cultivars [34, 56] and combined it with our WGRS data from Rpp1/Rpp1-b donors, extracted all SNP markers in the genomic interval associated with resistance to ASR in order to capture additional SNP markers not detected by GBS, but useful for haplotype analysis. To define linkage disequilibrium (LD) patterns and candidate genes, the correlation coefficient of alleles values (r2) between the GBS-derived SNP markers and WGRS SNP markers were calculated using PLINK 1.9 [57]. LD blocks were visualized as heatmaps using the “LDheatmap” R package [58]. Predictions of SNP effects were done by SnpEff version 4.3i [59]. Candidate gene annotation was obtained from the Phytozome database (https://phytozome-next.jgi.doe.gov/info/Gmax_Wm82_a2_v1).
Phylogenetic trees were constructed using 9,555 WGRS-derived SNP markers that were polymorphic among the 28 re-sequenced soybean accessions genomes and 67 GBS-derived SNPs (from 100 soybean accessions) identified within the 568-Kb Rpp1 interval using the neighbor-joining method, with 1,000 bootstraps and bootstrap values over 50 were used as threshold (MEGA X) [60]. Phylogenetic trees were visualized with the online tool iTOL [61]. The level of identity between sequences was calculated after alignment in the Geneious program v.11.1.5 (https://assets.geneious.com/documentation/geneious/GeneiousPrimeManual) and candidate gene sequences were extracted using the Integrative Genomics Viewer (IGV) [62]. Genomic sequences of the eight NLR genes identified in the known accessions harboring the Rpp1 and Rpp1-b locus were converted to protein sequences using the ExPASy online translation tool (https://web.expasy.org/translate/), and conserved domains were predicted for PI 200492, PI 587880A, PI 587886, PI 587905, PI594754, PI 561356, and PI 594538A with the NCBI Conserved Domains Database (CDD) [63].
Validating of SNP markers associated with Rpp1 locus
To validate SNP markers associated with ASR resistance conferred by the Rpp1-b locus, a population derived from the resistant accession PI 587880A, and the susceptible accession PI 594774 was developed. A total of 106 F2 plants were generated and used for ASR disease screening and genotyping. Seven SNP markers associated with ASR resistance identified in the GWAS analysis were used (Chr18:55,976,566; Chr18:56,207,185; Chr18:56,378,428; Chr18:56,378,436; Chr18:56,412,205; Chr18:56,544,134; Chr18:56,544,813). DNA extraction was performed following the methodology previously described. Genotyping was performed by PlexSeq™ sequencing by AgriPlex Genomics in the entire F2 population. Briefly, fasta sequences containing 100 bp up- and downstream of each SNP marker were extracted from the reference genome (W82.a2.v1) masked and submitted to the company to primers designing and further amplicon sequencing. Data from the PI 587880A F2 population was analyzed by performing a goodnes-of-fit χ2 (chi-square) test to compare expected and observed resistance segregation rates and genotyping results with selected markers.
We also validated the haplotypes using PlexSeq™ sequencing in a comprehensive set of 75 soybean accessions harboring different Rpp genes and/or alleles previously describe in the literature: six Rpp1/Rpp1-b, six Rpp2, 45 Rpp3, four Rpp4, four Rpp5, six Rpp6, one Rpp7, and the three pyramids [Rpp3 + Rpp5 (cv. Hyuuga), No 6–12 1 (Rpp2 + Rpp4 + Rpp5), and An76-1 (Rpp2 + Rpp4)]. To analyze the distribution of the Rpp1-b haplotype worldwide, we downloaded the variant calling data from 1,511 diverse wild and cultivated soybean publicly available from Zhang et al. [64] and deposited in Soybase database (https://soybase.org/data/v2/Glycine/max/diversity/Wm82.gnm2.div.Zhang_Jiang_2020/). SNP marker positions were extracted by VCFtools and histogram plots made with ggplot2 package [65].
Results
Soybean resistance to Phakopsora pachyrhizi
The GWAS panel, comprising 100 soybean accessions, was inoculated with a recent Brazilian population of P. pachyrhizi collected from soybean fields and subsequently assessed for their disease reaction (Supplementary Table 1). All soybean cultivars lacking Rpp genes consistently showed tan-type lesions, indicating a susceptibility reaction, across all replicates at three evaluation time points (10, 14, and 18 DAI). Similarly, cultivars previously described as susceptible to ASR, such as cv. BRS 232, cv. BRSGO Chapadões, and cv. Bragg, also exhibited susceptibility reaction. Interestingly, among the 20 accessions harboring a known Rpp allele at the Rpp1 locus, 11 accessions (65%) showed resistance symptoms (RB-type lesions): PI 368039, PI 518295, PI 561356, PI 587880A, PI 587886, PI 587905, PI 594754, PI 594766, PI 594767A, PI594760B and PI 594538A (Rpp1-b original source). The remaining nine accessions showed susceptibility symptoms: PI 423958, PI 547875, PI 368038, PI 587866, PI 594177, PI 587855, PI 417120, PI 587916A, including PI 200492 (Rpp1 original source) (Fig. 1a). Out of the 42 advanced breeding lines harboring the Rpp1 locus in their pedigree, 30 (70%) exhibited a resistance reaction, while the remaining 12 (30%) showed susceptibility, similar to the susceptible Brazilian cultivars. For instance, among the 19 breeding lines described as having PI 587880A in their pedigree, 13 were resistant and six were susceptible. Similar results were observed in the eight breeding lines derived from PI 561356, in which, six lines showed resistance reaction and two showed susceptibility reaction.

GWAS results identified a region on chromosome 18 associated with ASR resistance. a Representation of known Rpp1 sources with resistance (green) and susceptibility (brown) against a Brazilian Pp population. b Visualization of population structure among 100 soybean accessions through the first two principal components. c GWAS results depicted in a Manhattan plot, showing SNP markers along with their negative log10 p-values across the 20 soybean chromosomes, with the quantile–quantile plot (upper left). Red line indicating the significance threshold. d Phylogenetic tree constructed based on 67 GBS-derived SNP markers, highlighting the clustering of accessions, and their corresponding haplotypes. BR 36, Vmax RR, and Davis exhibited unique haplotypes G/T-T-G/T, TGT, and TAC, respectively
GWAS results and haplotype analysis
Sequencing of the GBS library yielded a total of 55,481,894 raw paired-end reads. Variant calling performed by the Fast-GBS pipeline and further filtering steps generated a total of 49,271 high-quality SNPs. We first investigated the population structure of our GWAS panel though a principal component analysis (PCA) (Fig. 1b, Supplementary Fig. 1), with the three first PCs explaining approximately 25% of the genetic variance (PC1 explained about 12% of genetic variance, PC2 8% and PC3 about 5%). In the scatter plot of the first two PCs, it is clear that soybean accessions clustered according to their geographic origin and the nature of the accessions, with three major clusters identified. The first cluster primarily consists of old Brazilian cultivars, the second cluster was composed mainly by RILs developed to harbor ASR resistance and derived from modern cultivars, and the third cluster consisting of PIs Rpp1 sources, all of them from China, Japan, and Taiwan.
For the GWAS analysis using cMLM model, we observed a low basal level of association throughout the entire genome (P-value < 10–2), except for the genomic location of Rpp1 on chromosome 18, as expected (Fig. 1c). We identified seven SNP markers significantly associated with ASR resistance (FDR ≤ 0.001). Their FDR-adjusted p-values ranged from 1.34E-05 to 0.001, with they explained up to 68% of the phenotypic variation (Table 1). SNP markers identified on chromosome 18 delimited a genomic interval of 568.25 Kbp (55,976,566 to 56,544,813). Among the seven significant SNP markers, only the Chr18:56,412,205, which is the least significant associated, was not able to correctly distinguish between resistant (R) from the susceptible (S) accessions. In its position, both alleles (A and G) are observed in similar proportions in both resistant and susceptible accessions (Supplementary Table 2).
Neither the combination of all seven SNPs nor each SNP by itself was sufficient to assign all the resistant and susceptible accessions to a specific haplotype (Supplementary Table 2). However, two SNP markers (Chr18:56,207,185 and Chr18:56,544,134) were present in most resistant accessions, and the combination of both markers with a third SNP marker (Chr18:55,976,566) defined two common haplotypes (GTC and TTC) found exclusively among resistant accessions. For instance, the GTC haplotype was exclusively found in five sources: PI 561356, PI 587880A, PI 587886, PI 587905, and PI 594538A, and their respective derived breeding lines. The TTC haplotype was exclusively shared by the resistant sources PI 544766, PI 594767A and PI 594754. On the other hand, nine Rpp1 sources were identified as susceptible in our study. Three of them shared the TAT haplotype, which was the haplotype found in almost all of the susceptible Brazilian cultivars (33 out of 37 cultivars shared the TAT haplotype). Notably, we found the TTT haplotype shared among eight Rpp1 sources (two resistant and six susceptible). Although the TTT haplotype was not associated with ASR resistance, it seems to be common among the Rpp1 sources. We selected the breeding lines based on their pedigree from the Embrapa Soybean breeding program; consequently, some breeding lines may not harbor the Rpp1 haplotype shared by their Rpp1 donors, as those lines were developed and selected based solely on phenotypic screening. In most cases, lines derived from a specific source also exhibited the expected haplotype. For instance, we obtained 16 lines derived from PI 587880A (lines showing heterozygous SNP markers were not take in account here). Among these, 11 resistant lines shared the GTC haplotype with PI 587880A, whereas the remaining five susceptible lines showed the TAT haplotype (susceptible haplotype). Similar results were observed for the other lines derived from PI 561356 and PI 594754 donors (Supplementary Table 2).
To gain further insights into the genomic interval surrounding the Rpp1 locus, we extracted all 67 GBS-derived SNP markers from the 568-kbp interval and used them to construct a phylogenetic tree, observing how the accessions were grouped. As expected, accessions that shared the same haplotype (based on the three markers described above) were grouped together, with few exceptions, resulting in four major clusters (Fig. 1d). The first cluster grouped the sources presenting the GTC haplotype and was formed by the sources harboring the Rpp1-b alleles from the original source PI.
594538A, along with accessions that likely harbor the same resistance allele, including PI 587880A, PI 587886, PI 561356, and PI 587905. These accessions showed resistance reactions. The second cluster was composed of the Rpp1 locus from the original source PI 200492, as well as susceptible Rpp1 sources like PI 594177, PI368038, PI 423958, PI 547875, all sharing the TTT haplotype with PI 200492. Interestingly, within this cluster, PI 36803, and PI 518295, described as resistant in our study, also showed the TTT haplotype, suggesting that although the TTT haplotype was shared among Rpp1 sources, it may not be associated with the resistance. The third and largest cluster is composed primarily of Brazilian and American cultivars with susceptibility phenotypes, all sharing the TAT haplotype. Finally, the fourth cluster included resistant accessions PI 594754, PI 594767A and PI 594766, sharing the TTC haplotype and showing resistance reactions (Fig. 1d).
Identification of additional SNPs variants in the 568-kbp interval retrieved from WGRS data
To obtain more information about the genomic interval including the Rpp1 locus (Fig. 2a), we utilized WGRS data to identify additional SNP markers not captured by GBS. In total, we identified 9,555 SNP markers that were polymorphic among the 28 re-sequenced accessions within the 568-Kbp region, with 57.9% (5,536 SNP markers) located within gene models and 42.1% (4,019 SNPs) situated in intergenic regions. Subsequently, all 9,555 SNP markers were used to construct a new phylogenetic tree to observe how these 28 accessions would cluster using such a large set of SNP markers (Fig. 2b). Notably, susceptible accessions formed two major clusters, while the Rpp1 sources grouped similarly to the phylogenetic tree constructed with GBS-derived SNP markers; namely, accessions with the GTC haplotype clustered together, while PI 200492 (haplotype TTT) and PI 594754 (haplotype TTC) located close to the GTC haplotype cluster. Thus, whether using the large set of WGRS-derived SNPs or the GBS-derived SNPs, the phylogenetic analysis produced similar genetic relationships, distinguishing the known Rpp1 sources from the susceptible cultivars. We then examined the LD patterns between the three SNP markers used in the haplotype analysis using WGRS-derived SNP markers in order to define an interval to identify candidate genes. We observed that these SNP markers showed r2 values ranging from 0.26 up to 0.38 to each other (Fig. 2c). We also investigated the distribution of unique SNP markers for each source and its distribution in the interval. This information can be valuable, for instance, for the introgression of specific resistance from particular Rpp1 donor (Supplementary Fig. 2). Among the WGRS-derived SNP markers, 542 SNPs (178 in 36 gene models and 364 in intergenic regions) are PI 200492-exclusive SNPs (haplotype TTT) (Supplementary Table 3), and 390 SNPs (147 across 27 gene models and 243 in intergenic regions) are unique to PI 594754 (haplotype TTC). Finally, 217 SNPs across 41 gene models are exclusive to the GTC haplotype group: PI 587880A, PI 594538A, PI 587886, PI 587905, and PI 561356 (Supplementary Table 4).

Genomic region of Rpp1/Rpp1-b on chromosome 18, and additional WGRS-SNP markers within candidate genes.a GWAS-SNP markers and gene models situated within the Rpp1/Rpp1-b locus, spanning a 568 Kbp interval (highlighted in grey). Resistance regions associated with pathogens previously mapped within the GWAS region, such as Phytophthora root rot (PRR) caused by P. sojae [66], Soybean Cyst Nematode (SCN) resistance to Heterodera glycines [67], and ASR resistance-associated SNP markers [37] are also shown. Gene models with defense-related functions, annotated based on the Williams 82 genome, are represented by arrows on the right. b Phylogenetic tree constructed based on WGRS-SNP markers. c Heatmap representing the squared correlation coefficient (r2) among the 9,555 WGRS-SNP markers within the GWAS interval. d Gene models within the GWAS interval displaying WGRS-SNP markers that form haplotypes distinguishing between resistant and susceptible soybean accessions. NBS-LRR genes are highlighted in bold
We successfully identified 36 WGRS-derived SNP markers that differentiate six ASR-resistant accessions (PI 594538A, PI 587880A, PI 587886, PI 587905, PI 561356, and PI 594754) from 21 Brazilian susceptible cultivars, including the susceptible genotype PI 200492 (the original source of the Rpp1 locus) (Fig. 2d). Among these accessions, PI 594538A, PI 587880A, PI 561356, PI 587886, and PI 587905 exhibited higher identity based on all WGRS-SNP markers found in the GWAS interval,with values ranging from 96.3% to 96.6% in individual comparisons and 97.5% when all five accessions were considered together. They also showed values around 70% when compared to different susceptible accessions group (Supplementary Fig. 3a). As expected, PI 200492 (Rpp1-susceptible) shared around 70% identity with the other resistant Rpp1 accessions. However, PI 594754 (haplotype TTC) exhibited identity values of around 70% when compared to the GTC haplotype group, suggesting that this accession might harbor another distinct Rpp1 allele. This hypothesis is supported by the fact that we also found additional 83 WGRS-derived SNP markers (in genic regions) uniquely shared by the GTC haplotype group, but not by PI 594754 (Supplementary Fig. 3b). In summary, these results reinforce the GWAS findings, indicating that this group (haplotype GTC) is indeed distinct from other soybean accessions, potentially due to the presence of the Rpp1-b locus.
Identification of resistance gene candidates in the GWAS interval of 568 kbp
The GWAS interval comprised 67 annotated soybean gene models (Wm82.a2.v1) (Supplementary Table 5). This interval overlaps with the 93.6-Kbp region (56,218,250 to 56,383,864) on chromosome 18 previously identified by Kim et al. [68] as associated with the Rpp1/Rpp1-b locus in PI 594538A, PI 561356, PI 587880A, and PI 587886. Furthermore, the GWAS region also encompasses the Rpp1 region from PI 200492, including its candidate genes validated as the genes (ULP1-NBS-LRR genes) conferring ASR immunity in PI 200492 [40] (Fig. 2a). Among the 67 gene models, 25 showed potential molecular functions related to plant defense and/or were previously reported to be involved in plant-pathogen interactions (Table 2). Furthermore, 16 gene models had unknown function or had not been annotated yet. As for the remaining gene models, five were associated with metabolic processes, while eight were associated with plant development and growth stages, among other functions not directly related to plant defenses against pathogens (Supplementary Table 5). Among the 25 gene models annotated with defense related functions, eight with similarities to classical known R genes were identified: two encoding leucine-rich repeat (LRR) proteins, four encoding ULP1-NBS-LRR proteins, one as an LRR-NBS protein (without the ULP1 domain), and a serine/threonine kinase with an LRR domain (Table 2). Other defense-related genes found within the interval include protein-coding genes such as chitinase, ring-zinc-finger protein, and AP2 transcription factor.
We initially investigated whether the three SNP markers from the Rpp1/Rpp1-b haplotypes are located within potential resistance candidate genes. The SNP located on Chr18:55,976,566 is an intergenic SNP located 3,985 bp upstream of the Glyma.18G278200 gene model, which is predicted to encode an ATP binding protein serine/threonine kinase with a putative LRR N-terminal domain (Supplementary Table 5). The SNP on Chr18:56,207,185 was located in the 2nd exon of Glyma.18G281100, which encodes two dimers (Rpb3 and Rpb1) of the larger subunit of RNA polymerase. The third SNP marker on Chr18:56,544,134 (synonymous mutation) was located in the 5th exon of Glyma.18G284700 gene model, which is predicted to encode a tRNA nucleotidyltransferase/poly(A) polymerase. Neither of the three SNP markers were located within the 25 candidate genes associated with resistance. Subsequently, we predicted the SNP effects for all the WGRS-derived SNP markers located in the 25 candidate genes (Supplementary Table 6). As expected, several SNP markers with missense effects were identified in the NBS-LRR candidate genes compared to the few missense SNPs found in all the remaining candidate genes. Within the NBS-LRR genes, Glyma.18G280300 (ULP1-NBS-LRR) and Glyma.18G281700 (ULP1-NBS-LRR), described as R1 and R5 genes according to Pedley et al. [40] exhibited the highest number of missense SNPs. Additionally, an aspartyl protease (Glyma.18G283100) present in the locus also exhibited several missense SNPs.
To uncover and identify potential candidate genes, we conducted genomic sequence comparisons and phylogenetic trees among the NBS-LRR genes from PI 200492 (Rpp1, haplotype TTT), PI 587880A, PI 587886, PI 587905, PI 561356, and PI 594538A (Rpp1-b, haplotype GTC), as well as PI594754 (haplotype TTC), and the susceptible accessions. As a result, the sequences of Glyma.18G280300 (R1) and Glyma.18G280400 (R2) from PI 200492 showed reduced sequence conservation when compared to the other Rpp1 accessions. Specifically, Glyma.18G280300 from PI 200492 showed low similarity when compared to sequences from Williams 82 (44.2%) and Rpp1-b accessions (30.7% to 34.6%). This distinction is further supported by the distinct clustering of Rpp1, Rpp1-b, and susceptible accession groups in the Glyma.18G280300 phylogenetic tree (Supplementary Fig. 4). The other genes with LRR domains, such as Glyma.18G278200 and Glyma.18G281500 (R3), were conserved and showed high similarity between PI 200492 and Rpp1-b accessions, not distinguishing Rpp1 from Rpp1-b. Furthermore, Glyma.18G284100 was the only one that exhibited high similarity between resistant and susceptible accessions, ranging from 99.9% to 100% (Supplementary Fig. 4). Notably, through our manual selection of WGRS-derived SNP markers in the GWAS interval that could efficiently distinguish Rpp1-b accessions from susceptible accessions (including PI 200492) and PI 594754 (haplotype TTC), we found the highest number of markers inside Glyma.18G280300 (R1), totaling 36 SNPs, but only two and one SNP in Glyma.18G280400 (R2) and Glyma.18G283200 (R6) (Supplementary Fig. 3, Supplementary Table 4). Furthermore, we provided to breeders, a large set of new SNP markers for MAS selection for selection of ASR resistance conferred by Rpp1-b allele.
Haplotype characterization in a diverse set of Rpp sources and a worldwide collection of cultivated and wild soybeans
We also investigated which haplotypes were present in other known Rpp sources to determine whether the Rpp1/Rpp1-b haplotypes identified by GWAS were unique to those sources. To achieve this, we first analyzed WGRS data from the original sources of Rpp2, Rpp3, Rpp4, Rpp5, Rpp6, and Rpp7. Four Rpp2 sources (PI 230970, PI 224270, PI 197182, and PI 417125) and one Rpp3 source (PI 462312) showed the susceptible TAT haplotype. Rpp4 sources (PI 459025B and PI 635027) showed the TAC haplotype, while three Rpp5 sources (PI 200487, PI 200526, PI 471904) showed the TAT haplotype, and one Rpp5 source (PI 200456) showed the TTT haplotype. Both Rpp6 and Rpp7 sources exhibited the TTC haplotype. WGRS data from PI 587880A (haplotype GTC) and PI 200492 (haplotype TTT) confirmed their previously identified haplotypes from GBS.
To validate the Rpp1/Rpp1-b haplotypes and assess their applicability, particularly in distinguishing Rpp1-b from other Rpp loci for breeding purposes, we genotyped a large collection of soybean accessions previously described as carrying a Rpp locus through amplicon sequencing (Table 3, Fig. 3a). We then confirmed the specificity of the Rpp1-b haplotype (GTC) as no other Rpp source showed this haplotype. In summary, we observed three haplotypes in Rpp1/Rpp1-b accessions, PI 200492-type haplotype (TTT), PI 594538A-type haplotype (GTC) in three accessions including the original source (PI 594538A) and the PI 594754-type haplotype (TTC). Rpp2 accessions shared the TAT haplotype, as most of the susceptible cultivars used in our GWAS panel. Rpp3 accessions were split in several haplotypes, with the main haplotypes being (TAT) and (TTT). The remaining Rpp4 accessions shared the TAC haplotype and most of Rpp5 accessions shared TAT haplotype. Finally, Rpp6 accessions shared TAT haplotype, and Rpp7 accession showed the TTC haplotype. Therefore, we found two Rpp1/Rpp1-b haplotypes highly specific for ASR resistance, which select only for those sources. We then asked if we could find potential unreported soybean accessions harboring these Rpp1/Rpp1-b haplotypes and analyze the distribution of these loci in a worldwide collection of soybean and wild soybean. Though WGRS data from 1,511 accessions, as expected, most of the soybean accessions showed the TAT haplotype, which was associated with susceptible accessions in our study. Surprisingly, the GTC haplotype was only found in 10 soybean accessions (genotypes showing heterozygous SNPs were not considered): PI 398593, PI 398595, PI 407729, PI 437725, PI 548415, PI 89772, Nan Guan Xiao PI Qing, PI 8388, Huang Dou 1, and Mao Dou Zi. Furthermore, six wild soybean (Glycine soja) accessions (Fig. 3b, Supplementary Table 7) also showed the GTC haplotype: PI 479752, PI 479752, PI 483460B, PI 549048, PI 639635, and PI 507830B. These results suggest that these 16 accessions potently carry the Rpp1-b allele.

Rpp1/Rpp1-b haplotypes validation on other Rpp sources, biparental population and in wide set of cultivated and wild soybeans. a Distribution of GWAS-haplotypes in other Rpp sources demonstrates the exclusivity of the GTC-haplotype in Rpp1-b sources. b Distribution of GWAS-haplotypes in 1,511 cultivated and wild soybeans. c Haplotype validation using a biparental population derived from a cross between PI 594774 and PI 587880A. d Virulence profiles of Rpp sources, highlighting the distinct profiles of Rpp1-b and Rpp1 sources. Disease classification adapted from Yamanaka et al. [45]: IF: immune with flecks. S: susceptible; SR: slightly resistant; R: resistant; HR: highly resistant; I: immune. Phenotypic data sourced from Barros et al. [23]
Finally, to assess the degree of co-segregation between the SNP markers identified via GWAS and ASR resistance, we developed an F2 population derived from the cross between the ASR resistant accession PI 587880A (haplotype GTC) and the susceptible PI 594774 accession (haplotype TAT). We first analyzed the segregation of ASR resistance for both parents and for the whole F2 population. As expected, while PI 594774 plants showed only susceptible lesions (TAN lesions) in all disease evaluations, PI 587880A showed resistance reactions (RB lesions). The observed segregation in the F2 population was 82 resistant and 22 susceptible individuals, showing a goodness of fit 3 resistant: 1 susceptible (χ2 = 1.01), fitting one dominant gene model (Table 4). We then genotyped the F2 individuals to observe if the SNP markers would co-segregate with ASR resistance. Our results showed that almost all resistant plants were either homozygous or heterozygous for the GTC haplotype. Only three plants were classified as resistant but showed the susceptible haplotype (TAT). Similar patters were observed for the susceptible group, in which, 16 susceptible plants showed the TAT haplotype, with only one susceptible plant showing the GTC haplotype. (Fig. 3c). We also observed the association between all seven GWAS markers and the resistance to the Brazilian ASR population. The peak SNP marker (Chr18:56,544,134) showed the highest association with resistance, as indicated by its p-value and a goodness of fit at the 3:1 segregation ratio in the chi-square test. Therefore, we confirmed that applicability of the haplotype defined by the three SNP markers for the introgression of the Rpp1-b resistance from PI 587880A, which will be useful as this allele is still effective against different Pp isolates while the original Rpp1 is not (Fig. 3d).
Discussion
Characterization of ASR resistance reveals variability in resistance sources of Rpp1
Currently, the major pathogen associated with soybean in Brazil and tropical regions is P. pachyrhizi (Pp), which causes Asian Soybean Rust (ASR). Although seven major Rpp loci have been described in soybean germplasm, their effectiveness against recent Pp populations in Brazilian soybean fields is limited. Additionally, studies examining the virulence profile of Pp isolates from different regions of the world, including Mexico [29], Japan [69], Bangladesh [32, 70], Argentina [69], Brazil [28], Uruguay [71], Kenya [33], Malawi [72], and the United States [10], have revealed the presence of diverse Pp pathotypes associated with their geographical origin. Hence, the emergence of GWAS employing distinct isolates, is highly desirable for uncovering the genetic components in soybean that underlie resistance against the diverse pathotypes of this pathogen. In our study, we utilized a panel of soybean accessions enriched with breeding lines derived from Rpp1 donors, enabling us to identify a genomic region on chromosome 18 and SNPs associated with ASR resistance against a recent Brazilian Pp population.
In our phenotypic screening, commercial soybean varieties composed of historical (ancient) soybean cultivars before ASR emergence in Brazil (2001), were predominantly susceptible, while variability in resistance was observed among Rpp1 sources and derived breeding lines. Despite ASR's significance in Brazilian soybean production, the availability of ASR-resistant cultivars in the market remains limited, with only a few resistant cultivars from Tropical Melhoramento Genético (TMG) (cv. TMG 7062 IPRO and TMG 7363 RR), and from Embrapa-Soybean (cv. BRS 539, BRS 531, and BRS 511) available. In our panel, primarily composed of historical (ancient) soybean cultivars, we did not expect to detect any ASR resistance, as most of these cultivars were developed before the emergence of ASR in Brazil in 2001.
Among the 20 Rpp1 sources in our panel, 11 showed resistance reactions, including the original source of Rpp1-b, PI 594538A, while nine showed susceptibility, including the Rpp1 source, PI 200492. Our screening revealed varying disease reactions between Rpp1 and Rpp1-b alleles, consistent with prior findings of Chakraborty et al. [18] and Hossain et al. [20]. The weak resistance group comprised PI 200492, PI 368039, and PI 587886, while the strong resistance group included PI 594767A, PI 587905, and PI 587880A. These findings suggest the presence of different resistant alleles of Rpp1 or tightly linked loci within the same genomic region. In our study, we used a 2017 Brazilian Pp population for phenotypic screening, which suggests the efficacy of locus against different Pp populations and isolates from different countries and years. We subsequently analyzed the group of resistant Rpp1 sources to determine whether these accessions had previously been described as traditional Rpp1 or potential Rpp1-b based on their haplotypes and virulence profiles. Remarkably, within the Rpp1 resistant group, most accessions were previously described as carrying either Rpp1-b allele or a different Rpp1 allele, including PI 594538A, PI 587880A, PI 587886, PI 587905, and PI 594767 [13, 19,20,21]. In the susceptible Rpp1 group, both PI 547875 (originally L85-2378) and PI 368038 (Tainung 3) are lines derived from PI 200492 [9, 73] and were therefore expected to exhibit the same susceptibility reaction. Notably, the accession potentially carrying the Rpp1-b allele, PI 587855, was classified as susceptible in our study.
The variability in resistance responses among different Rpp1/Rpp1-b sources was expected, mainly due to the high diversity of Pp pathotypes reported in previous studies. This reinforces the importance of identifying genetic resistance against local Pp populations and pathotypes, as not all the genetic resistance identified using foreign isolates may be efficient across regions. When we compared the Rpp1-resistant accessions found in our study to a Brazilian Pp population with the same Rpp1 accessions used in GWAS with field screening data in the southeastern USA between 2008 and 2015, we observed remarkable phenotypic differences. While the Rpp1 from PI 200492 was one of the most resistant sources identified in USA fields, in our study, this accession was completely susceptible. Furthermore, the Rpp1 allele in PI 561356, the Rpp1-b allele from PI 594538A, and the Rpp1 alleles from PI 594760B and PI 594767A, all classified as resistant in our study, did not show high levels of protection as observed in PI 200492 in southeastern USA fields. It’s notable the discrepancy between Rpp1 and Rpp1-b against pathotypes from different countries. Murithi et al. [72], comparing the virulence profile of Pp isolates, also found these discrepancies. Despite conferring immunity against an American isolate from Florida (FL-07–01), Rpp1 (PI 200492) exhibited susceptibility when challenged with isolates from Tanzania, Malawi, Australia, and Argentina, indicating an ability of these isolates to overcome the Rpp1 resistance.
In summary, our phenotypic results highlight the importance of local pathotypes in screening for ASR resistance. For breeding purposes in countries like Brazil, the largest soybean producer, we were able to identify variability in resistance provided by Rpp1 sources against a Brazilian Pp population. We then used this data to perform the GWAS approach and the subsequent analysis.
SNP markers and different haplotypes associated with the Rpp1/Rpp1-b region on chromosome 18 revealed by GWAS
We genotyped 100 soybean accessions, including commercial soybean cultivars, Rpp1 sources, and derived breeding lines, using GBS-derived SNP markers, and identified a region on chromosome 18 corresponding to the Rpp1/Rpp1-b locus, associated with resistance to Brazilian Pp population. The strategic inclusion of breeding lines derived from different Rpp1 donors proved to be valuable, as it allowed us to increase the frequency of Rpp1/Rpp1-b alleles in the GWAS panel, thereby facilitating the mapping process. A recent GWAS conducted under field conditions used an even larger panel (256 accessions) compared to our study [38]. However, in this study, the frequency of Rpp1 donors showing resistance was low, and they were unable to identify the Rpp1 region. Therefore, for future GWAS targeting Rpp loci, the inclusion of breeding lines in panels is beneficial, as only a few soybean accessions harboring those Rpp genes have been identified. The combination of breeding lines and the Rpp1 donors was interesting, as we could validate the haplotypes segregating with the ASR resistance in breeding lines, which potentially will become future cultivars, demonstrating the useability of our haplotypes for MAS programs.
Seven SNP markers significantly associated with ASR resistance were mapped via GWAS in the Rpp1/Rpp1-b region, as described by several genetic mapping studies with different Rpp1 sources (Fig. 2a). Although the Rpp1/Rpp1-b physical interval defined by markers varies across different Rpp1 genetic mapping studies, our 568.25-kbp GWAS interval (55,976,566—56,544,813 bp) overlapped with all of them. Compared to the 1.6-Mbp Rpp1-b interval mapped from the original source PI 594538A, delimited between the SNP markers BARC-010495–00656 (55,011,589) and BARC-014379–01337 (56,611,810), we narrow down the region. We then observed that the GWAS-SNP markers formed haplotypes able to assign the Rpp1/Rpp1-b sources into different groups. For instance, the GTC haplotype was shared among Rpp1-b (PI 594538A) and the resistant accessions: PI 587880A, PI 587886, PI 561356, and PI 587905. This clustering was also observed in the phylogenetic trees, whether using GBS- or WGRS-derived SNP markers. In both cases, the sources with haplotype GTC formed one cluster, while the sources with haplotype TTT formed another cluster. Notably, the clustering of PI 200492, PI 547875, PI 594177, and PI 368089 (all with haplotype TTT) into a unique cluster was also reported in a previous phylogenetic analysis [17].
We validate the Rpp1/Rpp1-b haplotypes against other Rpp sources, in a biparental population and in a wide set of cultivated and wild soybeans. The distribution of GWAS-haplotypes among other Rpp sources not only point out the exclusivity of the GTC-haplotype within Rpp1-b sources but also its successful discrimination of these sources from soybean accessions carrying Rpp2, Rpp3, Rpp4, Rpp5, Rpp6, and Rpp7, and from the historical cultivars (Table 3). The haplotypes were validated using a biparental population derived from the cross between PI 594774 (susceptible) and PI 587880A (resistant, Rpp1-b), enabling the discrimination of plants with the susceptible phenotype (nearly all possessing the TAT haplotype) from those with the resistance phenotype (nearly all harboring the GTC haplotype) (Fig. 3c). Finally, we investigated the occurrence of these novel haplotypes across a comprehensive dataset of soybeans, encompassing WGRS data from 1,511 accessions. Our analysis revealed that, associated with the GTC haplotype there are 16 previously unidentified potential sources of Rpp1-b (Supplementary Table 7). These results highlight the potential and the significance of utilizing the newly identified haplotype to accurately classify soybean accessions based on both phenotype and their corresponding resistance gene (Rpp1-b), thus offering a crucial tool (to identify new sources) for precise breeding strategies tailored towards enhancing Asian soybean rust resistance.
Attempts to distinguish Rpp1 sources were previously made due to the clear differences in ASR resistance among these sources. Kim et al. [68] used 21 markers and several Rpp1 accessions to identify 21 distinct haplotypes within the Rpp1 interval. Although they are distinct among the five sources, indicating that there are indeed differences between them in the region, the Rpp1-b haplotype (PI 594538A) was shared with seven susceptible North American soybean ancestors. They were unable to identify a unique haplotype selecting solely all the potential Rpp1-b. Subsequently, Harris et al. [10] successfully distinguished Rpp1-b from PI 594538A and Rpp1 (PI 200492) as well as the Rpp loci using nine SNP markers. However, the Rpp1-b haplotype was also shared with the susceptible accessions. Furthermore, this study did not include additional Rpp1 and Rpp7 accessions, and only a few susceptible soybean accessions were tested. The challenge in identifying haplotypes that differentiate the Rpp1 sources is likely attributed to the limited saturation of SNP markers within the Rpp1 interval. Both studies relied on SNP markers derived from the SoySNP50K Infinium Chip data. Furthermore, using American isolates and/or Pp populations alongside SNP markers from the SoySNP50K data has been a common approach in previous GWAS. Nevertheless, this approach may not be applicable for ASR breeding in countries like Brazil, mainly due to differences in Pp pathotypes. In our study, we opted to screen the accessions using a recent Pp Brazilian population, which better represents the pathogen’s variability in Brazilian soybean fields and combined that if high density markers in the interval.
The concept of utilizing haplotypes to distinguish alleles at specific loci has long been successfully employed in MAS. In soybean, differences in alleles and their associated haplotypes have been identified in various resistance genes, including those related to Southern Stem Canker [74], soybean cyst nematodes [75], Fusarium graminearum [76] and soybean mosaic virus [77]. Knowledge of allelic variation in resistance loci has proven valuable not only for cloning R genes and their alleles, but also for identifying the corresponding AVR genes. For instance, in wheat, the largest allelic series of R genes is formed by the Pm3 gene, which provides resistance against powdery mildew and includes 17 functional alleles that have been validated and cloned [78, 79]. Recently, the fine mapping and the haplotype analysis of different sources of soybean aphid resistance demonstrated that the RagFMD (Fangzheng Moshidou) gene shared a unique haplotype distinct from the Rag2 haplotype (PI 587972 and PI 594879) and the Rag5 haplotype (PI 567301B) [80], within the same interval, similarly to our haplotype analysis, with distinct unique haplotypes for Rpp1 and Rpp1-b. Therefore, we cannot exclude the possibility that our GWAS is revealing the presence of multiple genes rather than different alleles of the same gene. To validate this hypothesis, allelism tests with Pp isolates capable of distinguishing between Rpp1 and the potential Rpp1-b, along with fine-mapping approaches, will be essential for clarification. These efforts will provide the groundwork for cloning the genes responsible for ASR resistance on chromosome 18.
Hence, our findings regarding the variability in virulence profiles among Rpp1 sources, the identification of distinct haplotypes, and the highly associated SNP markers with ASR resistance will be valuable for breeding purposes. These results can accelerate the introgression of Rpp1-b resistance, particularly because the pyramiding of different Rpp genes mainly depends on SSR markers. [81, 82]. Moreover, they provide valuable insights for future studies aimed at identifying the candidate genes responsible for the Rpp1 and Rpp1-b alleles.
Identification of candidate genes for Rpp1/Rpp1-b allele pointed out for the ULP1-NBS-LRR genes
Efforts have been made to identify and clone the gene responsible for Rpp1 resistance. BAC sequencing of the Rpp1 interval from PI 200492 (56,182,230–56,333,803), combined with VIGS validation and gene expression profiling, has identified ULP1-NBS-LRR genes as potential candidates for the Rpp1 gene [40]. Unfortunately, the high similarity among the ULP1-NBS-LRR genes (R3-Glyma.18G281500, R4-Glyma.18G281600, and R5-Glyma.18G281700), hampered individual gene silencing, making it challenging to distinguish which of the three genes is the causal gene. Moreover, silencing these three genes resulted in the loss of immunity. While the original PI 200492 plants exhibited an immune reaction, the silenced plants displayed RB-type resistance rather than susceptibility (TAN lesions), as expected for silencing a potential R gene. Notably, the R4 candidate (Glyma.18G281600) exhibited high expression both in the absence and presence of the Pp isolate. Recently, the Rpp1-b genomic region from PI 594760B was elucidated through long-read sequencing, revealing the presence of three ULP1-NBS-LRR genes [41]. Specifically, B-R1 shared similarity with Glyma.18G281500, while B-R2 and B-R3 are similar to Glyma.18G281600. The authors demonstrated that a ULP1-NBS-LRR gene (similar to Glyma.18G281600), found in a susceptible soybean line (TMG06_0011), interacts with the three ULP1-NBS-LRR genes from PI 594760B, resulting in a partial suppression of resistance of the F1 plants. This phenomenon was referred to as dominant susceptibility. We also observed that the NBS-LRR genes had the highest number of SNPs with non-synonymous mutations compared to the other genes in the interval. Unfortunately, we worked with WGRS data, and structural variation underlying differences between R and S accessions in the interval could not be verified with confidence. However, it is worth mentioning that both PI 200492 (susceptible) and PI 594760B (resistant) shared the TTT haplotype in our results. To date, candidate genes from other Rpp1/Rpp1-b group with different haplotypes have not been validated. Therefore, we cannot exclude the possibility that other candidate genes may still play important roles in resistance in these Rpp1 sources.
Our GWAS results, along with the haplotype analysis, identified a broader interval and revealed a diverse set of genes that could potentially contribute to Rpp1/Rpp1-b resistance in soybean. Interestingly, the three SNP markers that differentiated the accessions are not located within any ULP1-NBS-LRR gene, despite the highest number of WGRS-derived SNP markers being identified in an NBS-LRR gene (Glyma.18g280300). Previous studies have suggested that could be the causative genes underlying Rpp1 in PI 200492 and Rpp1-b in PI 594760B. Additionally, earlier fine-mapping studies on other Rpp loci have also emphasized NBS-LRR genes as potential candidates for the Rpp genes. For example, the fine mapping of Rpp2 from PI 230970 delineated a 188.1-kbp interval containing 12 candidate genes, of which 10 are TIR-NBS-LRR genes [83]. These findings align with prior research demonstrating that the silencing of EDS1 and PAD4 genes in PI 2130970 plants, which are well-known components of the TIR-NBS-LRR immunity signaling pathway [84], led to the loss of Rpp2 resistance [85]. Another example comes from BAC sequencing, VIGS, and expression profiling, where a CC-NBS-LRR gene (Rpp4C4) was identified as the likely Rpp4 gene responsible for resistance in PI 459025B [86]. Intracellular nucleotide-binding domain and leucine-rich repeat (NB-LRR) receptors, commonly referred to as NLRs, represent the largest group of intracellular immune receptors in plants. These receptors recognize pathogen effectors, triggering programmed cell death, which is known as the hypersensitive response (HR) [87, 88]. Based on their N-terminal domains, canonical NLRs are categorized into three subfamilies: coiled-coil (CC)-NLRs (CNLs), Toll/Interleukin-1 receptor/Resistance (TIR)-NLRs (TNLs) and Resistance to Powdery Mildew 8 (RPW8)-like CC domain-NLRs (RNLs) [89, 90]. While TNLs and CNLs are likely candidate genes for Rpp2 and Rpp4, respectively, all evidence suggests that Rpp1/Rpp1-b genes are the noncanonical NBS-LRR genes with an integrated ULP1 protease domain at their N-terminal, without TIR or CC domains. There are a few examples of validated NLRs with integrated domains (IDs), such as CC-NLRs Pik-1 and Pia-2 (also known as RGA5) in rice, and RRS1 TIR-NLR with a WRKY transcription factor-like domain in Arabidopsis [91].
Few resistance genes have been cloned in soybean, with most being non-canonical NLRs, including Rsc4, a canonical CC-NBS-LRR gene (resistance against soybean mosaic virus) [92], Rps11, a giant NBS-LRR gene (broad-spectrum resistance to Phytophthora sojae) [93], GmRmd1, a TIR-NBS-BSP gene with a basic secretory domain (resistance to Microsphaera diffusa) [94]. Additionally, non-NLR genes, such as Rhg1 and Rhg4, confer resistance to soybean cyst nematode [95, 96], and the recently identified through map-based cloning C2H2-type zinc finger transcription factor RpsYD29 (resistance to P. sojae) [97].
Therefore, we cannot exclude the possibility that other candidate genes identified in the GWAS interval might also play important roles in Rpp1/Rpp1-b resistance. Our WGRS data and haplotype analysis revealed that SNP markers capable of distinguishing between resistant and susceptible accessions are not exclusively located within the NBS-LRR genes but are also found in other genes.
The fact that both the Pedley et al. [40] and Wei et al. [41] studies only explored the ULP1-NBS-LRR genes, and neither of them demonstrated that silencing these ULP1-NBS-LRR genes leads to complete susceptibility, raises the possibility that other genes in the interval may be involved in the Rpp1/Rpp1-b resistance. Within our GWAS interval, we identified a few LRR receptor-like kinase (RLK) subfamilies, which have been implicated in plant immunity against rust species in other crops, including coffee [98], wheat [99,100,101], and barley [102]. For instance, Glyma.18G278200 encodes a Protein NSP-Interacting Kinase 1, which has been associated with plant defence against Geminivirus [103]. Another example is Glyma.18G280200, encoding a Proline-Rich Receptor-Like Protein Kinase (PERK8), primarily associated with plant development [104], yet it has also been implicated in plant defense, as demonstrated by BnPERK1, which is rapidly induced in response to wounding and in the presence of Sclerotinia sclerotiorum in Brassica napus [105]. The Glyma.18G282100 predicts a non-specific serine/threonine protein kinase, which has been linked to defence responses against herbivory in Arabidopsis [106]. Additionally, Glyma.18G282200 encodes a serine protease belonging to the S10 serine carboxypeptidase family, which has been demonstrated to play important roles in disease resistance in oats [107] and Arabidopsis [108]. Interestingly, we identified several SNPs with non-synonymous effects within Glyma.18G283100, which encodes an aspartyl protease. In Arabidopsis, the Bcl-2-associated athanogene (BAG) protein serves as a co-chaperone essential for basal immunity against the fungal pathogen Botrytis cinerea, with aspartyl protease-mediated cleavage of BAG6 playing a crucial role in this process; specifically, inactivation of the aspartyl protease results in the prevention of BAG6 processing and subsequent loss of resistance [67]. Remarkably, a recent study identified a Pp effector (PpEC15) that functions as an aspartyl protease, cleaving 3-deoxy-7-phosphoheptulonate synthase in soybean and suppressing host immunity [109], indicating significant roles for this class of proteases in both soybean and P. pachyrhizi pathogen and their interactions.
In summary, definitively determining whether a single NBS-LRR gene, multiple NBS-LRR genes, or another gene within the interval is responsible for resistance at this locus remains challenging. However, the identification of several SNP markers associated with ASR resistance in this locus provides valuable insights. These markers can guide the selection of different resistant accessions (with different haplotypes) for use in de novo assembly approaches with long-read sequencing, facilitating the positional cloning of the gene conferring Rpp1/Rpp1-b resistance.
Conclusions
The investigation into ASR resistance variation of Rpp1/Rpp1-b sources highlights the significance of local pathotypes in ASR resistance screening. Our study, focusing on a recent Brazilian field Pp population, revealed distinct reactions among Rpp1/Rpp1-b sources and derived breeding lines, shedding light on the variability in resistance and differences in the genomic region of these alleles.
Through GWAS, we identified SNP markers significantly associated with ASR resistance in the Rpp1/Rpp1-b region. Our study revealed distinct haplotypes segregating with resistance, particularly the GTC haplotype shared among Rpp1-b sources and resistant accessions. These haplotypes facilitated the differentiation of Rpp1 and Rpp1-b accessions, providing a valuable tool for breeding strategies aimed at enhancing ASR resistance using each of these sources. Regarding candidate genes, our analysis highlighted the potential roles of NBS-LRR genes in Rpp1/Rpp1-b resistance, although other genes within the interval may also contribute. In conclusion, our study contributes valuable information on ASR resistance variability, SNP markers and haplotypes associated with resistance, and candidate genes within the Rpp1/Rpp1-b region. These findings have significant implications for breeding programs aiming to enhance ASR resistance in soybean, offering insights into resistance and guiding marker-assisted selection strategies.
Availability of data and materials
The datasets (fastq files) for whole genome sequencing data of soybean Brazilian cultivars analyzed during the current study are available at SoyBase (https://soybase.org/projects/SoyBase.C2020.01.php). All phenotypic data are provided in supplementary files as well genotypic data, SNPs and haplotypes on chromosome 18, for all 100 soybean accessions.
Abbreviations
- ASR:
-
Asian Soybean Rust
- Rpp:
-
Resistance to Phakopsora pachyrhizi
- SNP:
-
Single nucleotide polymorphism
- GWAS:
-
Genome-wide association study
- Pp:
-
Phakopsora pachyrhizi
- PI:
-
Plant Introduction
- GBS :
-
Genotyping by sequencing
- MAF:
-
Minor allele frequency
- FDR :
-
False discovery rate
- WGRS:
-
Whole genome Re-sequencing
- CV:
-
Cultivars
- MAS:
-
Marker-assisted selection
References
CONAB. Acompanhamento da Safra Brasileira de Grãos, décimo primeiro levantamento, agosto 2023. 2023. https://www.conab.gov.br/info-agro/safras/graos/boletim-da-safra-de-graos. Accessed 15 Sep 2023.
Godoy CV, Seixas CDS, Soares RM, Marcelino-Guimarães FC, Meyer MC, Costamilan LM. Asian soybean rust in Brazil: past, present, and future. Pesq Agrop Brasileira. 2016;51:407–21.
Gupta YK, Marcelino-Guimarães FC, Lorrain C, Farmer A, Haridas S, Ferreira EGC, et al. Major proliferation of transposable elements shaped the genome of the soybean rust pathogen Phakopsora pachyrhizi. Nat Commun. 2023;14:1835.
Yorinori J, Paiva W, Frederick R, Costamilan LM, Bertagnolli PF, Hartman GE, et al. Epidemics of soybean rust (Phakopsora pachyrhizi) in Brazil and Paraguay from 2001 to 2003. Plant Dis. 2005;89:675–7.
Scherm H, Christiano R, Esker P, Del Ponte EM, Godoy CV. Quantitative review of fungicide efficacy trials for managing soybean rust in Brazil. Crop Prot. 2009;28:774–82.
Savary S, Willocquet L, Pethybridge SJ, Esker P, McRoberts N, Nelson A. The global burden of pathogens and pests on major food crops. Nat Ecol Evol. 2019;3:430–9.
Silva DCG, Yamanaka N, Brogin RL, Arias CAA, Nepomuceno AL, Di Mauro AO, et al. Molecular mapping of two loci that confer resistance to Asian rust in soybean. Theor Appl Genet. 2008;117:57–63.
Garcia A, Calvo ÉS, de Souza Kiihl RA, Harada A, Hiromoto DM, Vieira LGE. Molecular mapping of soybean rust (Phakopsora pachyrhizi) resistance genes: discovery of a novel locus and alleles. Theor Appl Genet. 2008;117:545–53.
Boerma HR, Monteros MJ, Ha B-K, Wood ED, Phillips DV, Walker DR, et al. Registration of Asian soybean rust-resistant soybean germplasm G01-PR16. J Plant Regist. 2011;5:118–22.
Harris DK, Kendrick MD, King ZR, Pedley KF, Walker DR, Cregan PB, et al. Identification of unique genetic sources of soybean rust resistance from the USDA soybean germplasm collection. Crop Sci. 2015;55:2161–76.
Chander S, Ortega-Beltran A, Bandyopadhyay R, Sheoran P, Ige GO, Vasconcelos MW, et al. Prospects for durable resistance against an old soybean enemy: a four-decade journey from Rpp1 (Resistance to Phakopsora pachyrhizi) to Rpp7. Agronomy. 2019;9: 348.
Hyten DL, Hartman GL, Nelson RL, Frederick RD, Concibido VC, Narvel JM, et al. Map location of the Rpp1 locus that confers resistance to soybean rust in soybean. Crop Sci. 2007;47:837–8.
Yamanaka N, Hossain MM, Yamaoka Y. Molecular mapping of Asian soybean rust resistance in Chinese and Japanese soybean lines, Xiao Jing Huang, Himeshirazu, and Iyodaizu B. Euphytica. 2015;205:311–24.
Kato M. Effectiveness of resistance genes to the soybean rust pathogen Phakopsora pachyrhizi. JARQ-Jpn Agr Res Q. 2017;51:199–207.
Aoyagi LN, Muraki Y, Yamanaka N. Characterization of three soybean landraces resistant to Asian soybean rust disease. Mol Breed. 2020;40:1–12.
Paul C, Hartman GL, Diers BW, Walker DR. Mapping and confirmation of two genes conferring resistance to soybean rust (Phakopsora pachyrhizi) in the soybean line UG-5 (Glycine max). Plant Breeding. 2020;139:932–42.
Ratnaparkhe MB, Marmat N, Kumawat G, Shivakumar M, Kamble VG, Nataraj V, et al. Whole genome re-sequencing of soybean accession EC241780 providing genomic landscape of candidate genes involved in rust resistance. Curr Genomics. 2020;21:504–11.
Chakraborty N, Curley J, Frederick RD, Hyten DL, Nelson RL, Hartman GL, et al. Mapping and confirmation of a new allele at Rpp1 from soybean PI 594538A conferring RB lesion-type resistance to soybean rust. Crop Sci. 2009;49:783–90.
Ray JD, Morel W, Smith JR, Frederick RD, Miles MR. Genetics and mapping of adult plant rust resistance in soybean PI 587886 and PI 587880A. Theor Appl Genet. 2009;119:271–80.
Hossain MdM, Akamatsu H, Morishita M, Mori T, Yamaoka Y, Suenaga K, et al. Molecular mapping of Asian soybean rust resistance in soybean landraces PI 594767A, PI 587905 and PI 416764. Plant Pathol. 2014;64:147–56.
Yamanaka N, Morishita M, Mori T, Muraki Y, Hasegawa M, Hossain MdM, et al. The locus for resistance to Asian soybean rust in PI 587855. Plant Breeding. 2016;135:621–6.
Meira D, Batti V de BB, Woyann LG, Miliolli AS, Bozi AH, Beche E, et al. Confirmation of Rpp genes conferring resistance to Asian soybean rust and mapping of Rpp1 Allele from PI 594723. 2021. https://doi.org/10.21203/rs.3.rs-267310/v1. Accessed 20 Sep 2023.
Barros LG, Avelino BB, da Silva DCG, Ferreira EGC, Castanho FM, Ferreira ME, et al. Mapping of a soybean rust resistance in PI 594756 at the Rpp1 locus. Mol Breed. 2023;43:12.
Hyten DL, Smith JR, Frederick RD, Tucker ML, Song Q, Cregan PB. Bulked segregant analysis using the GoldenGate assay to locate the Rpp3 locus that confers resistance to soybean rust in soybean. Crop Sci. 2009;49:265–71.
Li S, Smith JR, Ray JD, Frederick RD. Identification of a new soybean rust resistance gene in PI 567102B. Theor Appl Genet. 2012;125:133–42.
Childs SP, King ZR, Walker DR, Harris DK, Pedley KF, Buck JW, et al. Discovery of a seventh Rpp soybean rust resistance locus in soybean accession PI 605823. Theo Appl Genet. 2018;131:27–41.
Paul C, Hartman GL. Sources of soybean rust resistance challenged with single-spored isolates of Phakopsora pachyrhizi. Crop Sci. 2009;49:1781–5.
Darben LM, Yokoyama A, Castanho FM, Lopes-Caitar VS, de Carvalho MCCG, Godoy CV, et al. Characterization of genetic diversity and pathogenicity of Phakopsora pachyrhizi mono-uredinial isolates collected in Brazil. Eur J Plant Pathol. 2020;156:355–72.
García-Rodríguez JC, Vicente-Hernández Z, Grajales-Solís M, Yamanaka N. Virulence diversity of Phakopsora pachyrhizi in Mexico. PhytoFront. 2022;2022(2):52–9.
Bonde MR, Nester SE, Austin CN, Stone CL, Frederick RD, Hartman GL, et al. Evaluation of virulence of Phakopsora pachyrhizi and P. meibomiae isolates. Plant Dis. 2006;90:708–16.
Paul C, Frederick RD, Hill CB, Hartman GL, Walker DR. Comparison of pathogenic variation among Phakopsora pachyrhizi isolates collected from the United States and international locations, and identification of soybean genotypes resistant to the US isolates. Plant Dis. 2015;99:1059–69.
Hossain MdM, Yamanaka N. Pathogenic variation of Asian soybean rust pathogen in Bangladesh. J Gen Plant Pathol. 2019;85:90–100.
Murithi HM, Soares RM, Mahuku G, van Esse HP, Joosten MHAJ. Diversity and distribution of pathotypes of the soybean rust fungus Phakopsora pachyrhizi in East Africa. Plant Pathol. 2021;70:655–66.
Maldonado dos Santos JV, Valliyodan B, Joshi T, Khan SM, Liu Y, Wang J, et al. Evaluation of genetic variation among Brazilian soybean cultivars through genome resequencing. BMC genomics. 2016;17:1–18.
Alekcevetch JC, de Lima Passianotto AL, Ferreira EGC, Dos Santos AB, da Silva DCG, Dias WP, et al. Genome-wide association study for resistance to the Meloidogyne javanica causing root-knot nematode in soybean. Theor Appl Genet. 2021;134:777–92.
Ferreira EGC, Marcelino-Guimarães FC. Mapping major disease resistance genes in soybean by genome-wide association studies. Methods Mol Biol. 2022;2481:313–40.
Chang H-X, Lipka AE, Domier LL, Hartman GL. Characterization of disease resistance loci in the USDA soybean germplasm collection using genome-wide association studies. Phytopathology. 2016;106:1139–51.
Walker DR, McDonald SC, Harris DK, Boerma HR, Buck JW, Sikora EJ, et al. Genomic regions associated with resistance to soybean rust (Phakopsora pachyrhizi) under field conditions in soybean germplasm accessions from Japan, Indonesia and Vietnam. Theor Appl Genet. 2022;135:3073–86.
Xiong H, Chen Y, Pan Y-B, Wang J, Lu W, Shi A. A genome-wide association study and genomic prediction for Phakopsora pachyrhizi resistance in soybean. Front Plant Sci. 2023;14: 1179357.
Pedley KF, Pandey AK, Ruck A, Lincoln LM, Whitham SA, Graham MA. Rpp1 encodes a ULP1-NBS-LRR protein that controls immunity to Phakopsora pachyrhizi in soybean. Mol Plant Microbe Interact. 2019;32:120–33.
Wei W, Wu X, Garcia A, McCoppin N, Viana JPG, Murad PS, et al. An NBS-LRR protein in the Rpp1 locus negates the dominance of Rpp1-mediated resistance against Phakopsora pachyrhizi in soybean. Plant J. 2023;113:915–33.
Meira D, Panho MC, Beche E, Woyann LG, Madella LA, Milioli AS, et al. Gene pyramiding combinations confer resistance of Asian soybean rust. Crop Sci. 2022;62:792–801.
Panho MC, Fernandes RAT, Menegazzi CP, Campagnolli OR, de Quadra FC, Madella LA, et al. Rpp-gene pyramiding confers higher resistance level to Asian soybean rust. Euphytica. 2022;218:172.
Fehr WR, Caviness CE, Burmood DT, Pennington JS. Stage of development descriptions for soybeans, Glycine max (L.) Merrill. Crop Sci. 1971;11:929–31.
Yamanaka N, Kato M, Akamatsu H, Yamaoka Y. Laboratory manual for studies on soybean rust resistance. Japan International Research Center for Agricultural Sciences; 2023. https://www.jircas.go.jp/en/publication/manual_guideline/30. Accessed 30 Sep 2023.
Sonah H, Bastien M, Iquira E, Tardivel A, Légaré G, Boyle B, et al. An improved genotyping by sequencing (GBS) approach offering increased versatility and efficiency of SNP discovery and genotyping. PLoS ONE. 2013;8: e54603.
Torkamaneh D, Laroche J, Bastien M, Abed A, Belzile F. Fast-GBS: a new pipeline for the efficient and highly accurate calling of SNPs from genotyping-by-sequencing data. BMC Bioinformatics. 2017;18:1–7.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–2.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95.
Rimmer A, Phan H, Mathieson I, Iqbal Z, Twigg SRF, WGS500 Consortium, et al. Integrating mapping-, assembly-and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet. 2014;46:912–8.
Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81:1084–97.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinform. 2014;30:2114–20.
Van der Auwera GA, O’Connor BD (O’Reilly). Genomics in the Cloud: Using Docker, GATK, and WDL in Terra. Copyright 2020 The Broad Institute, Inc and Brian O´Connor. 978-1-491-97519-0.
Zhang Z, Ersoz E, Lai C-Q, Todhunter RJ, Tiwari HK, Gore MA, et al. Mixed linear model approach adapted for genome-wide association studies. Nat Genet. 2010;42:355–60.
Lipka AE, Tian F, Wang Q, Peiffer J, Li M, Bradbury PJ, et al. GAPIT: genome association and prediction integrated tool. Bioinformatics. 2012;28:2397–9.
Torkamaneh D, Laroche J, Valliyodan B, O’Donoughue L, Cober E, Rajcan I, et al. Soybean (Glycine max) Haplotype Map (GmHapMap): a universal resource for soybean translational and functional genomics. Plant Biotechnol J. 2021;19:324–34.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Shin J-H, Blay S, McNeney B, Graham J. LDheatmap: an R function for graphical display of pairwise linkage disequilibria between single nucleotide polymorphisms. J Stat Softw. 2006;16:1–9.
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9.
Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49:W293–6.
Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings Bioinform. 2013;14:178–92.
Wang J, Chitsaz F, Derbyshire MK, Gonzales NR, Gwadz M, Lu S, et al. The conserved domain database in 2023. Nucleic Acids Res. 2023;51:D384–8.
Zhang H, Jiang H, Hu Z, Song Q, An YGC. A versatile resource of 1500 diverse wild and cultivated soybean genomes for post-genomics research. BioRixv. 2020. https://doi.org/10.1101/2020.11.16.383950. Accessed 20 Sep 2023.
Wickham H. ggplot2: elegant graphics for data analysis. 2nd ed. New York: Springer-Verlag; 2016.
Sahoo DK, Abeysekara NS, Cianzio SR, Robertson AE, Bhattacharyya MK. A novel Phytophthora sojae resistance Rps12 gene mapped to a genomic region that contains several Rps genes. PlosOne. 2017;12: e0169950.
Li Y, Kabbage M, Liu W, Dickman MB. Aspartyl protease-mediated cleavage of BAG6 is necessary for autophagy and fungal resistance in plants. Plant Cell. 2016;28:233–47.
Kim K-S, Unfried JR, Hyten DL, Frederick RD, Hartman GL, Nelson RL, Song Q, Diers BW. Molecular mapping of soybean rust resistance in soybean accession PI 561356 and SNP haplotype analysis of the Rpp1 region in diverse germplasm. Theor Appl Genet. 2012;125:1339–52.
Akamatsu H, Yamanaka N, Soares RM, Ivancovich AJG, Lavilla MA, Bogado AN, et al. Pathogenic variation of South American Phakopsora pachyrhizi populations isolated from soybeans from 2010 to 2015. JARQ-Jpn Agr Res Q. 2017;51:221–32.
Hossain MdM, Yasmin L, Rubayet MT, Akamatsu H, Yamanaka N. A major variation in the virulence of the Asian soybean rust pathogen (Phakopsora pachyrhizi) in Bangladesh. Plant Pathol. 2022;71:1355–68.
Larzábal J, Rodríguez M, Yamanaka N, Stewart S. Pathogenic variability of Asian soybean rust fungus within fields in Uruguay. Trop Plant Pathol. 2022;47:574–82.
Murithi HM, Haudenshield JS, Beed F, Mahuku G, Joosten MHAJ, Hartman GL. Virulence diversity of Phakopsora pachyrhizi isolates from East Africa compared to a geographically diverse collection. Plant Dis. 2017;101:1194–200.
Walker DR, Boerma HR, Philips DV, Schneider RW, Buckley JB, Shipe ER, et al. Evaluation of USDA soybean germplasm accessions for resistance to soybean rust in the southern United States. Crop Sci. 2011;51:678–93.
Maldonado dos Santos JV, Fereira EGC, Passianoto ALL, Brumer BB, Dos Santos AB, Soares RM, et al. Association mapping of a locus that confers southern stem canker resistance in soybean and SNP marker development. BMC Genet. 2019;20:798.
Patil GB, Lakhssassi N, Wan J, Song L, Zhou Z, Klepadlo M, et al. Whole-genome re-sequencing reveals the impact of the interaction of copy number variants of the rhg1 and Rhg4 genes on broad-based resistance to soybean cyst nematode. Plant Biotechnol J. 2019;17:1595–611.
Cheng P, Gedling CR, Patil G, Vuong TD, Shannon JG, Dorrance AE, et al. Genetic mapping and haplotype analysis of a locus for quantitative resistance to Fusarium graminearum in soybean accession PI 567516C. Theor Appl Genet. 2017;130:999–1010.
Ilut DC, Lipka AE, Jeong N, Bae DN, Kim DH, Kim JH, et al. Identification of haplotypes at the Rsv 4 genomic region in soybean associated with durable resistance to soybean mosaic virus. Theor Appl Genet. 2016;129:453–68.
Bhullar NK, Mackay M, Keller B. Genetic diversity of the Pm3 powdery mildew resistance alleles in wheat gene bank accessions as assessed by molecular markers. Diversity. 2010;2:768–86.
Bourras S, Kunz L, Xue M, Praz CR, Müller MC, Kälin C, et al. The AvrPm3-Pm3 effector-NLR interactions control both race-specific resistance and host-specificity of cereal mildews on wheat. Nat commun. 2009;10:2292.
Yang J, Liu G, Tang J, Wang X, Diao Y, Su Y, et al. Fine mapping and characterization of an aphid-resistance gene in the soybean landrace Fangzheng Moshidou. Front Plant Sci. 2022;13: 899212.
Yamanaka N, Morishita M, Mori T, Lemos NG, Hossain MdM, Akamatsu H, et al. Multiple Rpp-gene pyramiding confers resistance to Asian soybean rust isolates that are virulent on each of the pyramided genes. Trop Plant Pathol. 2015;40:283–90.
Parhe SD, Chimote VP, Deshmukh MP, Chandra K, Akash M. Marker-assisted pyramiding of four QTL/genes for Asian rust (Phakopsora pachyrhizi) resistance in soybean. J Crop Improv. 2017;31:689–711.
Yu N, Kim M, King ZR, Harris DK, Buck JW, Li Z, et al. Fine mapping of the Asian soybean rust resistance gene Rpp2 from soybean PI 230970. Theor Appl Genet. 2015;128:387–96.
Ngou BPM, Ding P, Jones JD. Thirty years of resistance: zig-zag through the plant immune system. Plant Cell. 2022;34:1447–78.
Pandey AK, Yang C, Zhang C, Grahan MA, Horstman HD, Lee Y, et al. Functional analysis of the Asian soybean rust resistance pathway mediated by Rpp2. Mol Plant Microbe Interact. 2011;24:194–206.
Meyer JD, Silva DC, Yang C, Pedley KF, Zhang C, van de Mortel M, et al. Identification and analyses of candidate genes for Rpp4-mediated resistance to Asian soybean rust in soybean. Plant Physiol. 2009;150:295–307.
Chiang YH, Coaker G. Effector triggered immunity: NLR immune perception and downstream defense responses. Arabidopsis Book: Am Soc Plant Biol. 2015(13):0183. https://doi.org/10.1199/tab.0183.
Förderer A, Kourelis J. NLR immune receptors: structure and function in plant disease resistance. Biochem Soc Trans. 2023;51:1473–83.
Kourelis J, Van Der Hoorn RA. Defended to the nines: 25 years of resistance gene cloning identifies nine mechanisms for R protein function. Plant Cell. 2018;30:285–99.
Kourelis J, Sakai T, Adachi H, Kamoun S. RefPlantNLR is a comprehensive collection of experimentally validated plant disease resistance proteins from the NLR family. PLoS Biol. 2021;19: e3001124.
Marchal C, Michalopoulou VA, Zou Z, Cevik V, Sarris PF. Show me your ID: NLR immune receptors with integrated domains in plants. Essays Biochem. 2022;66:527–39.
Yin J, Wang L, Jin T, Nie Y, Liu H, Qiu Y, et al. A cell wall-localized NLR confers resistance to Soybean mosaic virus by recognizing viral-encoded cylindrical inclusion protein. Mol Plant. 2021;14:1881–900.
Wang W, Chen L, Fengler K, Bolar J, Llaca V, Wang X, et al. A giant NLR gene confers broad-spectrum resistance to Phytophthora sojae in soybean. Nat Comm. 2021;12:6263.
Xian P, Cai Z, Jiang B, Xia Q, Cheng Y, Yang Y, et al. GmRmd1 encodes a TIR-NBS-BSP protein and confers resistance to powdery mildew in soybean. Plant Commun. 2022;3: 100418.
Cook DE, Lee TG, Guo X, Melito S, Wang K, Bayless AM, et al. Copy number variation of multiple genes at Rhg1 mediates nematode resistance in soybean. Science. 2012;338:1206–9.
Liu S, Kandoth PK, Warren SD, Yeckel G, Heinz R, Alden J, et al. A soybean cyst nematode resistance gene points to a new mechanism of plant resistance to pathogens. Nature. 2012;492:256–60.
Li W, Zheng X, Cheng R, Zhong C, Zhao J, Liu TH, et al. Soybean ZINC FINGER PROTEIN03 targets two SUPEROXIDE DISMUTASE1s and confers resistance to Phytophthora sojae. Plant Physiol. 2023;192:633–47.
de Almeida DP, Castro ISL, Mendes TAO, Alves DR, Barka GD, Barreiros PRR, et al. Receptor-Like Kinase (RLK) as a candidate gene conferring resistance to Hemileia vastatrix in coffee. Sci Agric. 2020;78:6.
Feuillet C, Schachermayr G, Keller B. Molecular cloning of a new receptor-like kinase gene encoded at the Lr10 disease resistance locus of wheat. Plant J. 1997;11:45–52.
Gu J, Sun J, Liu N, Sun X, Liu C, Wu L, et al. A novel cysteine-rich receptor-like kinase gene, TaCRK2, contributes to leaf rust resistance in wheat. Mol Plant pathol. 2020;21:732–46.
Wang J, Wang J, Li J, Shang H, Chen X, Hu X. The RLK protein TaCRK10 activates wheat high-temperature seedling-plant resistance to stripe rust through interacting with TaH2A.1. Plant J. 2021;108:1241–55.
Brueggeman R, Rostoks N, Kudrna D, Kleinhofs A. The barley stem rust-resistance gene Rpg1 is a novel disease-resistance gene with homology to receptor kinases. Proc Natl Acad Sci. 2002;99:9328–33.
Santos AA, Lopes KV, Apfata JA, Fontes EP. NSP-interacting kinase, NIK: a transducer of plant defence signalling. J Exp Bot. 2010;61:3839–45.
Invernizzi M, Hanemian M, Keller J, Libourel C, Roby D. PERKing up our understanding of the proline-rich extensin-like receptor kinases, a forgotten plant receptor kinase family. New Phytol. 2022;235:875–84.
Silva NF, Goring DR. The proline-rich, extensin-like receptor kinase-1 (PERK1) gene is rapidly induced by wounding. Plant Mol Biol. 2002;50:667–85.
Miyamoto T, Uemura T, Nemoto K, Daito M, Nozawa A, Sawasaki T, et al. Tyrosine kinase-dependent defense responses against herbivory in Arabidopsis. Front Plant Sci. 2019;10: 776.
Mugford ST, Qi X, Bakht S, Hill L, Wegel E, Hughes RK, et al. A serine carboxypeptidase-like acyltransferase is required for synthesis of antimicrobial compounds and disease resistance in oats. Plant Cell. 2009;21:2473–84.
Liu H, Wang X, Zhang H, Yang Y, Ge X, Song F. A rice serine carboxypeptidase-like gene OsBISCPL1 is involved in regulation of defense responses against biotic and oxidative stress. Gene. 2008;420:57–65.
Chicowski AS, Qi M, Variz H, Bredow M, Montes C, Caiazza F, Dong H, et al. A soybean rust effector protease suppresses host immunity and cleaves a 3-deoxy-7-phosphoheptulonate synthase. bioRxiv. 2023. https://doi.org/10.1101/2023.09.07.556260.
Acknowledgements
Special thanks to the professionals in the Plant Biotechnology Laboratory and Marker-Assisted and Molecular Genetics Laboratory at Embrapa-Soja, Londrina, Brazil.
Funding
This work was supported by Project 440782/2022–8 grant from National Council for Scientific and Technological Development – CNPq and Ministry of Science, Technology and Innovation—(MCTI) #440782/2022–8, and EMBRAPA Soybean. The scholarship was supported by the National Council for the Improvement of Higher Education (CAPES)—Finance code 001.
Author information
Authors and Affiliations
Contributions
LNA drafted the manuscript and figures, developed the experiments and GWAS and WGRS analysis. EGCF, FCMG and DGGS wrote the final version of the manuscript. EGCP conducted WGRS analysis and figures. ABS and DCGS provided phenotypic and genotypic data collection, including SNPs assays and Agriplex analysis, and upervision; BA provided phenotypic data; FB and VSLC provided input on GBS sequencing step and analysis, WGRS genome assembly and data postprocessing. CAA and MFO developed the soybean cross and provided soybean germplasm. RVA, ERS and DCGS aided with manuscript preparation. FCMG conceptualized and supervised the study, provide resources, reviewed, and edited the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
12870_2024_5454_MOESM1_ESM.xlsx
Supplementary Table S1: Summary of the soybean varieties and Rpp sources and their respective phenotypes used in the GWAS.
12870_2024_5454_MOESM7_ESM.xlsx
Supplementary Table S7: Distribution of haplotypes across a diverse set of 1,511 cultivated and wild soybean accessions.
12870_2024_5454_MOESM11_ESM.pdf
Supplementary Figure 3: Phylogenetic tree and genetic relationships between accessions based on WGRS-data. a Phylogenetic tree constructed using WGRS-data. b Nucleotide identity between soybean accessions from different clusters on the phylogenetic tree. c Identification of WGRS-SNP markers located within candidate genes.
12870_2024_5454_MOESM12_ESM.pdf
Supplementary Figure 4: Phylogenetic trees constructed based on WGRS-SNP markers located within NBS-LRR and RLK genes, as well as using all SNP markers from the interval.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Aoyagi, L.N., Ferreira, E.G.C., da Silva, D.C.G. et al. Allelic variability in the Rpp1 locus conferring resistance to Asian soybean rust revealed by genome-wide association. BMC Plant Biol 24, 743 (2024). https://doi.org/10.1186/s12870-024-05454-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-024-05454-1