
Abstract
Background
Amylose, a prebiotic found in yams is known to be beneficial for the gut microflora and is particularly advantageous for diabetic patients’ diet. However, the genetic machinery underlying amylose production remains elusive. A comprehensive characterization of the genetic basis of amylose content in yam tubers is a prerequisite for accelerating the genetic engineering of yams with respect to amylose content variation.
Results
To uncover the genetic variants underlying variation in amylose content, we evaluated amylose content in freshly harvested tubers from 150 accessions of Dioscorea zingibensis. With 30,000 high-quality single nucleotide polymorphisms (SNP), we performed a genome-wide association analysis (GWAS). The population structure analysis classified the D. zingiberensis accessions into three groups. A total of 115 significant loci were detected on four chromosomes. Of these, 112 significant SNPs (log10(p) = 5, q-value < 0.004) were clustered in a narrow window on the chromosome 6 (chr6). The peak SNP at the position 75,609,202 on chr6 could explain 63.15% of amylose variation in the population and fell into the first exon of the ADP-glucose pyrophosphorylase (AGPase) small subunit gene, causing a non-synonymous modification of the resulting protein sequence. Allele segregation analysis showed that accessions with the rare G allele had a higher amylose content than those harboring the common A allele. However, AGPase, a key enzyme precursor of amylose biosynthesis, was not expressed differentially between accessions with A and G alleles. Overexpression of the two variants of AGPase in Arabidopsis thaliana resulted in a significantly higher amylose content in lines transformed with the AGPase-G allele.
Conclusions
Overall, this study showed that a major genetic variant in AGPase probably enhances the enzyme activity leading to high amylose content in D. zingiberensis tuber. The results provide valuable insights for the development of amylose-enriched genotypes.
Similar content being viewed by others


Background
Dioscorea species encompass over 600 species and represent one of the key tuber crops regarding their economic, pharmaceutical, and sociocultural importance [1]. Yam is positioned as the fourth most cultivated tuber crop behind potato, cassava, and sweet potato [2]. It serves as a fundamental dietary constituent and a significant economic revenue generator for an estimated 300 million people worldwide, mostly in tropical and subtropical regions [3].
Yam species, like other dietary fiber plants, contain high-amylose (resistant starch type 2), a useful prebiotic, which has a positive effect on gut microbiome, and can help with weight loss and obesity prevention [4,5,6,7]. For instance, high-amylose varieties of rice [8] and wheat [9] have a much lower glycemic load, which could be beneficial for diabetics. Additionally, high-amylose starch has been associated with various health benefits, including improved gut health, weight management, diabetes control, and potential cholesterol reduction, as indicated by studies involving animal models [10, 11]. Therefore, the genetic regulation of amylose content in crops such as rice [12], wheat [13], potato [14] and sweet potato [15] has sparked significant attention in the scientific community. Besides, amylose is widely utilized in pharmaceutical, food product, textile, and paper industries [12, 16].
The amylose content properties of yams have been studied for industrial applications. A study by Freitas et al. [17] found that defatted yam starch from D. alata cultivars had a significantly higher amylose content of 36.2% compared to 24.2% for cassava starch. This difference in amylose content is reflected in the different thermal and rheological properties of the two starches. Oscillatory rheometry revealed an initial gelatinisation temperature of 71 °C for yam starch, significantly higher than the 62 °C observed for cassava starch [17]. Furthermore, the gelatinisation process of yam starch was shown to require a higher activation energy, indicating a more energetically demanding process. Interestingly, the higher amylose content in yam starch also confers a slower gelatinisation rate at elevated temperatures and facilitates the formation of stronger gel structures upon cooling and retrogradation compared to cassava starch gels [17].
Furthermore, a study of starch granules isolated from five different yam species (D. cayenensis, D. polygonoides, D. alata, D. rotundata, and D. esculenta) revealed different characteristics in terms of amylose content, granule size, crystallinity and susceptibility to enzymatic digestion [18]. Amylose content varied significantly, with D. cayenensis having the highest amylose content at 26.5%. Notably, pronounced differences were observed in the digestibility of crude starches by porcine pancreatic α-amylase, with D. esculenta and D. polygonoides being the most susceptible to enzymatic degradation, while D. cayenensis, D. rotundata and D. alata starches exhibited greater resistance to digestion [18]. The amylose content of another yam species, D. opposita Thunb. cultivars, showed lower amylose content ranging from 20.74 to 25.94% compared to D. alata species [19].
Due to its unique properties, high-amylose starch is a valuable ingredient that offers a wide range of benefits in the food industry [20]. It has a higher melting temperature, limited granule swelling, lower water holding capacity, and a superior ability to form a gel during gelatinization [21]. Food products made from high-amylose starch, such as bread wheat and maize starch, can exhibit improved cooking quality [22, 23]. High-amylose starch possesses low enzymatic digestibility, which offers several nutritional and physiological benefits to humans such as improved glycemic control, increased dietary fiber intake, and reduced caloric value [20, 24]. High-amylose starch is also utilized in the encapsulation of probiotics and drugs, and in the formulation of oral rehydration treatments [25, 26].
As for low-amylose or amylose-free starches, they are used as thickening agents in food processing and papermaking industries [27]. In South East Asian countries such as Japan, South Korea and China, amylose-free cereals are widely consumed as part of the daily diet [28]. Low-amylose starches are also used to improve the shelf-life of products such as baked goods and snacks because they are less likely to become firm and grainy over time [29]. Low-amylose starches exhibit adhesive properties, making it a valuable alternative to petroleum-based adhesives production [30].
Despite these numerous advantages, the genetic architecture of amylose production in yam is not well understood. However, tremendous works have been done to unlock the genetic determinants of amylose biosynthesis using the plant model Arabidopsis thaliana [31], cereals including maize [32], wheat [13, 33, 34], barley [35], and rice [12, 16], and tubers such as sweet potato [15] and potato [14, 36].
Amylose biosynthesis in yam involves a series of enzyme-mediated steps and regulatory factors (Fig. 1). Sucrose from the phloem is cleaved into glucose and fructose, which are then converted to glucose-1-phosphate (G1P) [37]. G1P is activated by the enzyme ADP-glucose pyrophosphorylase (AGPase) to form ADP-glucose (ADPG), the immediate precursor of starch. AGPase catalyses this reaction, converting ATP to inorganic pyrophosphate (PPi) [38]. The catalytic activity of AGPase is inhibited by inorganic phosphate (Pi) and 3-phosphoglyceric acid (3-PGA) [39]. AGPase consists of two large (AGPLS) and two small (AGPSS) subunits, each with different functions. Granule-bound starch synthase (GBSS) elongates the glucan chains of amylose.
Genome wide association study (GWAS) has become very popular and is one of the main approaches to unlock the genetic basis of biological traits. In biomedical sector, GWAS helps scientists to identify genes associated with human diseases, enabling the development of suitable therapeutics [40]. In livestock, GWAS mainly aim to identify candidate genes related to important economic traits [41,42,43]. GWAS has also led to the discovery of large quantitative genetic loci associated with phenotypes of interest in several crops such as rice [44, 45], maize [46, 47], and peanut [48, 49].
In the past decade, the implementation of GWAS in yam breeding programs has emerged as a promising tool for accelerating genetic gains and enhancing the efficiency of breeding efforts. As for D. alata, GWAS has been employed to decipher the genetic architecture of tuber dry matter, oxidative browning [50], sex determination, cross-compatibility [51], flowering control [52], anthracnose, tuber size, tuber shape [53], and tuber flesh color [54]. Using an elite population of D. rotundata, loci associated with mosaic virus tolerance and yield tuber were identified using GWAS strategy [55]. While the greater yam, D. alata, has received considerable attention in genome-wide association studies (GWAS), interest has been growing recently in other species, including the bush yam (D. praehensilis) and white guinea yam (D. rotundata) [56, 57]. The first gene discovery efforts related to key traits in the bush yam, such as dry matter content, tuber flesh oxidation, and tuber flesh hardness have been performed [57]. As tuber quality is one of the key determinant of consumers preferences [58, 59], attention has been drawn to elucidating the genetic basis of yam food quality traits. Employing sensory quality evaluation and textural profile analysis, Asfaw et al. [56] identified putative genes underlying the textural properties of boiled and pounded yam food products derived from D. rotundata. Recently, taking advantage of whole genome sequencing of 127 genotypes of the greater yam, D. alata, Mota et al. discovered several genes involved some key tuber quality related pathways including starch and sucrose metabolism, pentose and glucuronate interconversions, and flavonoid biosynthesis [60]. Although the investigated traits are more likely related to tuber quality and agronomic performance, little is known about the genetic determinants of amylose content in yams.
Several genome-wide association studies (GWAS) have been conducted to understand the genetic regulation of starch quality traits, including starch content and pasting properties, in maize and barley [61, 62]. These studies have identified significant single nucleotide polymorphisms (SNPs) and candidate genes associated with starch traits, providing valuable insights into the genetic architecture of these traits. For example, in maize, GWAS has revealed the genetic control of starch content by multiple small effect quantitative trait loci (QTLs), and identified candidate genes related to starch pasting properties [61]. Similarly, in barley, GWAS has been used to identify novel putative alleles associated with total starch, amylose, and amylopectin content in grain [62].
D. zingiberensis is a dioecious perennial plant indigenous to southern China [63]. It has been discovered to contain over 70 bioactive compounds, exhibiting a diverse range of biological activities [64]. These include, but are not limited to, cardiovascular protection, anti-inflammatory responses, and anti-cancer properties [64, 65]. In the present study, we focused on the detection of genetic variants associated with amylose content, a key bioactive component with a wide range of applications. Molecular breeding techniques alongside CRISPR/Cas9-mediated gene knockout have been utilized to modulate the amylose content in major crops such as wheat [13] and rice [16]. Therefore, knowing the genomic regions and candidate genes underlying the biosynthesis of amylose in D. zingiberensis could pave the way for amylose-oriented genetic engineering.

A simplified schematic view of the potential routes of amylose biosynthesis in D. zingiberensis. The diagram includes two compartments: the cytosol and the amymoplast. The stepwise reactions of sucrose synthase (SuSy), UGP-glucose pyrophosphorilase (UGPase) and plastidial phosphoglucomutase (pPGM) take place in the cytosol to convert sucrose to glucose-6-phosphate, which enters the amyloplast to be used for amylose biosynthesis. The enzymes involved in amylose biosynthesis in yam tubers include the precursor ADP-glucose pyrophosphorylase (AGPase) and granule-bound starch synthase (GBSS). Abbreviations are defined as follows Fructokinase (FRK); Glucose 1-phosphate (G1P); Glucose 6-phosphate/phosphate transporter (GPT/G6PPT); Fructose 6-phosphate (F6P); Cytosolic phosphoglucomutase (cPGM); cytosolic phosphoglucose isomerase (cPGI); inorganic phosphate (Pi); inorganic pyrophosphate (PPi); 3-phosphoglyceric acid (3-PGA); AGPase large subunit (AGPLS); AGPase small subunit (AGPSS).
Results
Analysis of amylose content variation in D. Zingiberensis panel
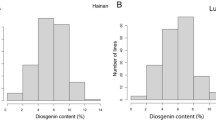
A panel of 150 D. zingiberensis accessions was screened for their amylose content. The results indicate an approximatively normal distribution of the amylose content at both Luohe (p-value = 0.125) (Fig. 2a) and Hainan (p-value = 0.181) (Fig. 2b) environments in China.

Distribution of amylose content data of D. zingiberensis accessions at Luohe (a) and Hainan (b)
The amylose content ranged from 9.11 to 32.25%, and from 10.23 to 30.78% at Luohe and Hainan, respectively (Table 1). The average amylose content among the accessions was significantly (p-value ≤ 0.001) higher in Luohe (20.41%) compared to Hainan (17.33%).
The analysis of variance also showed a highly significant (p-value ≤ 0.001) effect of environment, and genotype by environment factors on the amylose content. Besides, the broad-sense heritability was 74%, suggesting that variation in amylose content in D. zingiberensis tuber is substantially attributable to genetic causes.
Population structure and principal component analysis
Prior to conducting the genome-wide association analysis, we assessed the presence of putative sub-populations based on 30,000 high-quality SNPs. Population structure analysis revealed that the studied population can be divided into three sub-groups, with the majority of accessions being admixed (Fig. 3a; Supplementary Fig. 1). Principal component analysis (PCA) showed that the first two PC (PC1 and PC2) explained a total of 26.43% of the genetic variation in the population, with PC1 and PC2 explaining 18.91% and 7.52%, respectively. PC3 explained 4,61% of the genetic variation in the population (Supplementary Fig. 2). The low proportion of explained genetic variation suggests that the population of D. zingiberensis used in this study has low levels of population differentiation or substructure. Both STRUCTURE analysis (Fig. 3a), and PCA (Fig. 3b) confirmed the grouping of accessions into three sub-groups. Overall, the population exhibited a moderately structured pattern.

The results of the population structure (a) and principal component (b) analyses of the 150 D. zingiberensis accessions
Genome wide association study and identification loci controlling amylose variation
To identify the genetic loci accountable for the amylose content variation in D. zingiberensis, we performed a GWAS using a panel of 150 accessions. The resulting Manhattan plot highlighted at least four genomic regions containing 115 significant SNPs on the chromosomes chr1 (1), chr3 (1), chr6 (112), and chr7 (1) (Fig. 4a, Supplementary Table 1). The results of the quantile-quantile plot analysis (Fig. 4b) showed that the observed distribution did not deviate from the expected values to some extent. This result indicates a relative reduction of false positive (deviation from the expected values of the SNP markers) by the GWAS model.

Genome-wide association mapping for amylose content in D. zingiberensis. Manhattan plot for amylose content (a). Quantile-quantile plot for amylose content (b)
The highest peak was observed on chr6 at the position 75609202 and could explain 63.15% of amylose variation in the population (Table 2). This result suggests that the variant Chr6_75609202 is a major locus controlling amylose content in D. zingiberensis. Other minor SNPs detected on chr1, chr3 and chr6 had very weak contributions to amylose content variation and did not fall into genic regions. Therefore, we focused our efforts on characterizing Chr6_75609202.
The SNP Chr6_75609202 was exactly located in the ADP-glucose pyrophosphorylase (AGPase) small subunit gene which is known to play a critical role in the starch biosynthesis [66, 67]. In-depth analysis the D. zingiberensis AGPase (Dzin_AGPase) gene revealed that the SNP is a non-synonymous (A/G) variant affecting the resulting protein sequence from glycine to aspartic acid (Fig. 5a). Moreover, the SNP Chr6_75609202 is located in the first exon of the gene (Fig. 5a).

Characterization of D. zingiberensis AGPase (Dzin_AGPase) gene structure showing the location of the SNP Chr6_75609202 in the first exon. A single nucleotide polymorphism (SNP) was detected within this exon, leading to a non-synonymous alteration in the resulting protein sequence (a). Comparative amylose quantification for accessions exhibiting A and G alleles (b). Relative expression of the both versions of the gene via qRT-PCR experiment (c)
Through mining the genotypic data, a total of 102 accessions presented the allele A while 27 harbored the allele G (Fig. 5b), indicating that A is the common allele. A comparative analysis of the amylose content in both groups showed a highly significant (p-value < 0.001) difference in the G allele group compared to the A allele group. Thus, the G allele could be considered the favorable allele for higher amount of amylose production in D. zingiberensis tuber.
Since the genetic variation fell into a genic region, we tested whether it impacts on the gene expression level. We performed a qRT-PCR experiment with five accessions harboring the G allele and five accessions with the A allele. The results (Fig. 5c) revealed a similar relative expression level of Dzin_AGPase for both alleles, indicating that the discovered variant did not impact the gene transcription.
Functional analysis of the Dzin_AGPase gene
To validate the predicted function of the candidate Dzin_AGPase gene, an Arabidopsis thaliana-based transformation was conducted. The full-length cDNA of the two variants of Dzin_AGPase were ligated into different pROK II-35 S vector promoted by the CAMV35S promoter (Fig. 6a). From the positive T3 overexpressing plants, leaves from four lines of each allele type with seven replicates each, were harvested for amylose and gene expression profile assessment.
The relative expression profile (Fig. 6b) of the transgenic lines indicates a highly similar expression level among the lines for both alleles of the Dzin_AGPase gene. However, the amylose content (Fig. 6c) was significantly higher in the G allele that in the A allele overexpressing lines. From the relative expression and amylose quantification results, we deduced that the variant did not impact on the transcription and function of the protein but seemingly the enzyme activity is increased when it comes to the G allele, resulting in higher amylose content.

Generation of Arabidopsis thaliana transgenic plants for the overexpression of the DZin_AGPase gene (a). The construct of plasmid contains the Dzin_AGPase gene, the CaMV35S promoter, and NOS terminator. The NPT II was employed as a selective marker. RB, right border; LB, left border; NOS-P, nopaline synthase promoter; NOS-T, nopaline synthase terminator. Relative expression (b) and amylose content quantification (c) from the T3 generation transgenic plants. Four lines for each allele were selected. WT-VC is vector control (transformed with empty vector). Mean comparison significance of the amylose content was depicted with the letter A, B, and C
Discussion
Amylose is a valuable resource with many applications covering food, paper, textiles, adhesives, and health care industries [68, 69]. The genetic basis of amylose content, a component of starch, has been investigated in tuberous crops, such as potato [70], sweet potato [15], cassava [71] and non-tuber crops, including maize [32], wheat [33], and rice [16]. The improvement of amylose content in tubers for both consumption and industrial purposes has been a major objective for breeders. Taking advantage of a large panel combined with a high-quality genotyping dataset, we were able to dive into the genetic variants modulating amylose biosynthesis in D. zingiberensis tuber using a genome-wide association study (GWAS) approach.
In the present study, not only the genotype effect but also the genotype-by-environment effect were highlighted, influencing the amylose content in the studied D. zingiberensis panel. This implies that the environmental component is also a key determinant of amylose variation and should be considered when developing stable and high-yield amylose content genotypes. Similarly, a significant environmental effect was also found for greater yam (Dioscorea alata) on tuber quality traits including flesh colour, tuber dry matter, oxidative browning, skin texture and shape regularity [50, 72], highlighting the importance of considering environmental effect for future research and breeding programs.
From GWAS, we identified four putative loci associated with amylose content variation in D. zingiberensis. Among these, three minor SNPs had very weak contribution and were not linked to any candidate gene. Interestingly, one SNP was located in the first exon of the ADP-glucose pyrophosphorylase gene. The AGPase is well-known to be one of the key precursors at the upstream step of starch production [37]. Its role is to catalyze the conversion of 1-P-glucose into ADP-glucose to enable the biosynthesis of both amylose and amylopectin [66, 67, 73]. The regulatory properties of the ADPase gene in amylose production was firstly demonstrated in potato through overexpression approach [74]. This approach has gained popularity within some major cereals crops, with a specific focus on maize [75, 76], wheat [77, 78], and rice [79, 80]. Overexpression of AGPase in maize resulted in an increase in starch (amylose and amylopectin) production and seed weight [75]. An increase in seed yield was also observed in overexpressed AGPase wheat [77] and rice [79, 80] lines. In addition to the regulatory role of AGPase with regard to amylose production, it has also been shown to have an impact on photosynthesis and carbon metabolism. By increasing the availability of ADPglucose for starch biosynthesis, AGPase enhances the capacity of the leaf to store carbon, which in turn, stimulates photosynthesis by allowing more efficient use of light energy [78].
We also uncovered that allelic variation in the DZin_AGPase is associated with amylose content level in the studied D. zingiberensis panel. Genotypes with the G allele exhibited approximately two-fold higher amylose content than the A version of the gene. Surprisingly, the expression levels of both alleles are in the same range, indicating that both versions of the genes are equitably expressed but somehow, the amylose content in genotypes with G allele is higher. The presence of non-synonymous changes in the protein sequence between the A and G alleles of AGPase, suggests that this change may lead to functional differences. The absence of evidence for differential expression of the two alleles in transcriptome data may indicate that post-transcriptional or post-translational modifications may be responsible for observed differences in amylose content. Further investigation is required to elucidate the mechanisms underlying these differences and their potential implications for breeding programs aimed at developing cultivars with higher amylose content.
The allelic variation associated with the amylose content in the panel and transgenic lines comforts the rate-limiting enzyme function of the AGPase. In fact, the AGPase subunits interaction [81,82,83,84], and specifically, amino acid motifs [85, 86] responsible for allosteric regulation are thought to confer to AGPase, the capabilities of starch level regulation. Therefore, AGPase has become a prime target for enzyme engineering, to increase starch content in some food crops including common wheat [87], maize [75], rice [79], and potato [86].
To functionally test the role of the DZin_AGPase, we proceeded to a transgenic experiment using Arabidopsis thaliana as plant model. DZin_AGPase overexpression showed a relatively high proportion of amylose content for the G allele compared the A allele. Therefore, the G version will likely be valuable to boost the production of amylose in D. zingiberensis. In-depth genetic improvement of amylose content through CRISPR-Cas9 for example, might be a promising avenue to explore. Meanwhile, for the tetraploid potato, amylose-free genotype has been recently developed by CRISPR/Cas9-mediated mutagenesis [88]. Besides, the control of the amylose content in sweet potato has also been successfully conducted [15]. It is worth noting that these two success stories exclusively relied on the granule-bound starch synthase gene editing, which is the amylose encoding enzyme. Noticeably, the granule-bound starch synthase gene has not been found in the present study. Knowing that the accumulation of amylose and the expression of key enzymes follows a diurnal pattern [89, 90], timeline transcriptome profiling of the tuber following expansion of the tuber might potentially enlighten others key enzyme master players.
Methods
Plant materials and field experiments
To identify the genomic regions associated with amylose content in D. zingiberensis, a panel of 150 accessions was utilized in a field experiment conducted in two locations in China: Hainan (18° 56’ 22’’ North and 109° 29’ 3’’ East) and Luohe (33° 34’ 18’’ North and 114° 2’ 7’’ East). The experiment was designed as a randomized complete block with three replicates. Within each block, five replicates of each accession were sown on ridges, and all recommended in-field cultural practices were applied at both sites until tuber harvest, which occurred upon leaf senescence. The plant materials were formally identified by Prof Yunpeng Luan and all germplasms are conserved as vitro-plant at the Genebank of Southwest Forestry University. No permission is required to work on this species. The accessions originated from Southern China within relatively similar agroecological zones.
Starch isolation
Freshly harvested yam tubers were processed for starch isolation following a modified protocol of Farhat et al. [91]. Briefly, the rhizomes were washed, peeled, and cut into small pieces. A total of 100 g of the slices were ground with 900 mL of 1% sodium chloride solution in a commercial blender (Waring Commercial, Stamford, Connecticut, USA) for two minutes. The resulting slurry was then passed through a 106 μm pore diameter sieve (Fisher Scientific, Waltham, Massachusetts, USA) to remove any solid particles. The obtained suspension was left to settle overnight (12 h) at room temperature to allow for starch precipitation. The supernatant was decanted, and the starch pellet was centrifuged at 3000 g for 10 min. The top brown layer was carefully removed, and the starch was subsequently resuspended in solutions of 1% w/v sodium chloride and de-ionized water, respectively. For each washing step, the solution was re-centrifuged two to three times. Finally, the freshly isolated starch was then dried at 60 °C in an oven (Precision Scientific, GCA Equipment Corporation, Madison, Wisconsin, USA), ground, and stored at room temperature in a glass container prior to usage.
Amylose Content quantification
The amylose content was quantified according to the iodine binding colorimetric methodology outlined in Jiang et al. [92] study with modifications. Firstly, a quantity of 10 mg of the previously prepared starch was mixed with 2 mL of Dimethyl sulfoxide and subjected to heating at 85 °C for 15 min. The dissolved starch was then diluted with deionized water to attain a final volume of 25 mL. A volume of 1 mL of the starch solution was transferred into a 50 mL flask followed by the addition of 5 mL of iodine. Lastly, the optical absorbance was recorded at 620 nm using a Cary 60 UV-Vis spectrophotometer (Agilent, Santa Clara, California, USA). Triplicate apparent amylose content was carried out for each accession.
Amylose Content Data Evaluation and Statistical Analysis
The collected data were checked for normality through frequency distribution histogram plot, and Shapiro-Wilk test using R program v.4.2.2 [93]. The mean, range, and coefficient of variation were also computed in R program. Subsequently, an analysis of variance was performed following the model:
Where \({\text{y}}_{\text{ijk}}\) is the observed value of the amylose content in the ith location, jth block for the kth genotype, \(\mu\) is the overall general mean, \({\text{Loc}}_{\text{i}}\) is the effect of the ith location, \({\varepsilon _{{\text{ijk}}}}\) is the experimental pooled error effect, \({\text{Rep}}_{\text{j}}\left({\text{Loc}}_{\text{i}}\right)\) is the effect of jth block within ith location,\({\text{Gen}}_{\text{k}}\) is the effect of kth genotype, \({\text{Lo}}{{\text{c}}_{\text{i}}} \times {\text{ Ge}}{{\text{n}}_{\text{k}}}\) is the effect of the interaction between the ith location and the kth genotype, and \({\varepsilon _{{\text{ijk}}}}\) is the experimental pooled error.
The variance components were computed by fitting the mixed linear model with genotype, location, and genotype by location factors as random effect using lme4 package [94]. Furthermore, the heritability (\({H}^{2}\)) was calculated as:
where \({{\sigma }}_{\text{g}}^{2}\) is the genotype variance component, \({{\sigma }}_{\text{g}\text{e}}^{2}\) is the genotype by environment interaction variance component, nLoc is the number of environments and the nRep the number of replicates.
The best linear and unbiased predictors (BLUPs) values calculated from the model, served for the downstream GWAS analysis. Normality test was conducted with shapiro.test() in R program v.4.2.2.
SNP Genotyping
From 100 g young leaves tissues, we extracted the genomic DNA for each accession with ImaSpin® Genomic DNA Kit (Imagene Bioscience, China) following the manufacturer’s protocol. The quality of the DNA was checked using a Nanodrop 8000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). A volume of 30 mL of DNA was pipetted into 96-well PCR plates, and genotyping-by-sequencing (GBS) was conducted following a 96-plex Pst I GBS protocol [95]. Briefly, the DNA of each accession was digested with the restriction enzyme PstI (New England Biolabs, Beijing, China). Restriction cutting sites were ligated with adapters (barcodes) with the T4 ligase. The ligated products were then pooled together. Single-end sequencing was performed using an Illumina HiSeq2500 instrument (Illumina Inc. San Diego, CA, USA).
The generated raw reads were processed (sorting, demultiplexing and trimming) using the TASSEL GBS v2 pipeline [96] (Supplementary Table 2). The mapping onto the reference genome [97] was performed using the Burrows–Wheeler alignment (BWA) v0.7.17 (Li and Durbin, 2009), and the SNPs were called with DiscoverySNPCallerPluginV2 of the TASSEL GBS v2 pipeline yielding 2.3 M SNPs. A minimum locus coverage (mnLov) was set to 0.1, while other parameters were maintained to default settings. Monomorphic sites, SNPs with missing data > 20%, and with minor allele frequency (MAF) < 0.01, were excluded using vcftools v0.1.16 [98]. The resulting data was imputed with Beagle v4.1 [99] yielding (842,000 SNPs), and a second round of SNPs with MAF < 0.01 were filtered out prior to the downstream analyses. Out of 54,000 SNPs, 30,000 high-quality SNPs were retained for downstream analyses.
Population structure analysis
The population genetic structure of the 150 accessions was inferred by using a Bayesian model-based method embedded in STRUCTURE v2.3.4 [100]. The number of population clusters was predetermined as k ranging from 1 to 10. We applied five independent runs for each k. Each run involved a total of 100,000 Markov chain Monte Carlo iterations after a burn-in period of 100,000 iterations. We determined the best k population following the Evanno ΔK method. Besides, the principal component analysis (PCA) was performed using the Genomic Association and Prediction Integrated Tool (GAPIT) [101] in the R program v.4.2.2 [93].
Genome-wide Association analyses
To find out putative candidate genomic regions related to the amylose content accumulation in tuber, the association phenotype-genotype analysis was performed using the GAPIT package following the mixed linear model (MLM) option [102]. Both kinship matrix and PCA matrix were employed as random and fixed effects, respectively. Since we retained a set of 30,000 high-quality SNPs meeting the stringent filtering criteria, the genome wide significant threshold was set to 5 following the calculation -log10(p) with p = 1/30,000. The Manhattan and qq plots were rendered using the qqman package [103].
Quantitative RealTime PCR (qRTPCR) analysis
To evaluate the expression of the candidate gene, a qRT-PCR experiment was conducted in an Applied Biosystems™ 7500 Real-Time PCR machine (Thermo Fisher Scientific, Waltham, Massachusetts, USA) with a SYBR Green PCR Master Mix (Tiangen Biotech, Beijing, China). Total RNA was extracted with RNAprep Pure Plant Kit (Tiangen Biotech, Beijing, China), and the RNA was transcribed with the help of a Quantscript Reverse Transcriptase Kit (Tiangen Biotech, Beijing, China). A primer pair (5’-AGAATCTAGACCACTTAC-3’; 5’-CTCTAGGTACAGTCTCA-3’) was designed, and the PCR experiment was conducted with the following conditions: denaturation step at 95 °C for 10 min, annealing step with 40 cycles at 95 °C for 15 s, and the extension step at 60 °C for 1 min. The relative expression of the candidate genes was quantified following the comparative CT method [104]. Three replicates were applied for each gene variant, and the expression data were normalized against those of D. zingiberensis actin gene sequence (NCBI GenBank accession: JN693499).
Arabidopsis transgenics experiment
To functionally characterize the candidate gene Dzin_AGPase, we extracted the protein coding region from two genotypes, each having different alleles. The construct design and cloning were performed using the pROK II-35 S vector. The Arabidopsis transformation was performed following the floral dip method as outlined by Clough and Bent [105] using Agrobacterium tumefaciens strain LBA4404. The overexpressing plants (T3 homozygous lines) were then transferred into larger pots and maintained in greenhouse. The gene expression and the amylose content quantification were executed in accordance with the aforementioned methods.
Conclusions
In the present study, we report for the first time, a major locus associated with amylose content in a non-model plant D. zingiberenisis. DZin_AGPase, a starch rate-limiting enzyme, exhibited allelic variation with the G allele associated to higher amylose content. Overexpression of the two DZin_AGPase alleles using Arabidopsis transgenic plants corroborated the higher amylose content for the G allele. Our findings provide a valuable foundation for developing new varieties with desired amylose content levels. This study can also contribute to improving the nutritional quality of yam-based foods, as amylose content affects their digestibility and glycemic index. Future research can expand on our findings by examining the functional roles of the identified genetic variants using CRISPR-Cas9.
Data availability
The raw sequencing data are available at NCBI SRA under the project number: 716093 (https://www.ncbi.nlm.nih.gov/bioproject/716093). Codes and scripts used for data analysis are available in Supplementary file 1. Any intermediary file and all phenotypic data can be provided upon request to the corresponding author.
References
Adomėnienė A, Venskutonis PR. Dioscorea spp.: comprehensive review of antioxidant properties and their relation to Phytochemicals and Health benefits. Molecules. 2022;27. https://doi.org/10.3390/molecules27082530.
Padhan B, Panda D. Potential of neglected and underutilized yams (Dioscorea spp.) for improving Nutritional Security and Health benefits. Front Pharmacol. 2020;11:1–13. https://doi.org/10.3389/fphar.2020.00496.
Mignouna HD, Abang MM, Asiedu R. Genomics of Yams, a Common Source of Food and Medicine in the Tropics. In Genomics of Tropical Crop Plants; Springer New York: New York, NY, 2008; pp. 549–570.
Fuentes-Zaragoza E, Riquelme-Navarrete MJ, Sánchez-Zapata E, Pérez-Álvarez JA. Resistant starch as functional ingredient: a review. Food Res Int. 2010;43:931–42. https://doi.org/10.1016/j.foodres.2010.02.004.
Obidiegwu JE, Lyons JB, Chilaka CA. The Dioscorea Genus (Yam)—An Appraisal of Nutritional and therapeutic potentials. Foods. 2020;9:1304. https://doi.org/10.3390/foods9091304.
Wang Z, Zhao S, Tao S, Hou G, Zhao F, Tan S, Meng Q. Dioscorea spp.: Bioactive compounds and potential for the treatment of inflammatory and metabolic diseases. Molecules. 2023;28:1–18. https://doi.org/10.3390/molecules28062878.
Venter C, Meyer RW, Greenhawt M, Pali-Schöll I, Nwaru B, Roduit C, Untersmayr E, Adel-Patient K, Agache I, Agostoni C, et al. Role of dietary fiber in promoting immune health—An EAACI position paper. Allergy Eur J Allergy Clin Immunol. 2022;77:3185–98. https://doi.org/10.1111/all.15430.
Zenel AM, Stewart ML. High amylose white rice reduces post-prandial glycemic response but not appetite in humans. Nutrients. 2015;7:5362–74. https://doi.org/10.3390/nu7075225.
Di Rosa C, De Arcangelis E, Vitelli V, Crucillà S, Angelicola M, Trivisonno MC, Sestili F, Blasi E, Cicatiello C, Lafiandra D, et al. Effect of three Bakery products formulated with high-amylose wheat flour on post-prandial glycaemia in healthy volunteers. Foods. 2023;12:1–14. https://doi.org/10.3390/foods12020319.
Rahim MS, Kumar V, Mishra A, Fandade V, Kumar V, Kiran kondepudi K, Bishnoi M, Roy J. High resistant starch mutant wheat ‘TAC 35’ reduced glycemia and ameliorated high fat diet induced metabolic dysregulation in mice. J Cereal Sci. 2022;105:103459. https://doi.org/10.1016/j.jcs.2022.103459.
Lim SM, Choo JM, Li H, O’rielly R, Carragher J, Rogers GB, Searle I, Robertson SA, Page AJ, Muhlhausler B. A high amylose wheat diet improves gastrointestinal health parameters and gut microbiota in male and female mice. Foods. 2021;10:1–18. https://doi.org/10.3390/foods10020220.
Liu X, Ding Q, Wang W, Pan Y, Tan C, Qiu Y, Chen Y, Li H, Li Y, Ye N, et al. Targeted deletion of the First Intron of the Wxb Allele via CRISPR/Cas9 significantly increases Grain Amylose Content in Rice. Rice. 2022;15:1–12. https://doi.org/10.1186/s12284-021-00548-y.
Regina A, Berbezy P, Kosar-Hashemi B, Li S, Cmiel M, Larroque O, Bird AR, Swain SM, Cavanagh C, Jobling SA, et al. A genetic strategy generating wheat with very high amylose content. Plant Biotechnol J. 2015;13:1276–86. https://doi.org/10.1111/pbi.12345.
Veillet F, Chauvin L, Kermarrec MP, Sevestre F, Merrer M, Terret Z, Szydlowski N, Devaux P, Gallois JL, Chauvin JE. The Solanum tuberosum GBSSI gene: a target for assessing gene and base editing in tetraploid potato. Plant Cell Rep. 2019;38:1065–80. https://doi.org/10.1007/s00299-019-02426-w.
Wang H, Wu Y, Zhang Y, Yang J, Fan W, Zhang H, Zhao S, Yuan L, Zhang P. CRISPR/Cas9-Based mutagenesis of Starch Biosynthetic genes in Sweet Potato (Ipomoea batatas) for the improvement of Starch Quality. Int J Mol Sci. 2019;20. https://doi.org/10.3390/ijms20194702.
Xu Y, Lin Q, Li X, Wang F, Chen Z, Wang J, Li W, Fan F, Tao Y, Jiang Y, et al. Fine-tuning the amylose content of rice by precise base editing of the wx gene. Plant Biotechnol J. 2021;19:11–3. https://doi.org/10.1111/pbi.13433.
Freitas R, Paula R, Feitosa JP, Rocha S, Sierakowski M-R. Amylose contents, rheological properties and gelatinization kinetics of yam (Dioscorea alata) and cassava (Manihot utilissima) starches. Carbohydr Polym. 2004;55:3–8. https://doi.org/10.1016/S0144-8617(03)00142-5.
Riley CK, Wheatley AO, Hassan I, Ahmad MH, Morrison EYSA, Asemota HN. In vitro digestibility of raw starches extracted from five Yam (Dioscorea spp.) species grown in Jamaica. Starch - Stärke. 2004;56:69–73. https://doi.org/10.1002/star.200300195.
Shujun W, Hongyan L, Wenyuan G, Haixia C, Jiugao Y, Peigen X. Characterization of new starches separated from different Chinese yam (Dioscorea Opposita Thunb.) Cultivars. Food Chem. 2006;99:30–7. https://doi.org/10.1016/j.foodchem.2005.07.008.
Li HT, Zhang W, Zhu H, Chao C, Guo Q. Unlocking the potential of high-amylose starch for Gut Health: not all function the same. Fermentation. 2023;9:1–17. https://doi.org/10.3390/fermentation9020134.
Zhong Y, Zhu H, Liang W, Li X, Liu L, Zhang X, Yue H, Xue J, Liu X, Guo D. High-amylose starch as a new ingredient to balance nutrition and texture of food. J Cereal Sci. 2018;81:8–14. https://doi.org/10.1016/j.jcs.2018.02.009.
Rahim MS, Chauhan A, Madhawan A, Mishra A, Parveen A, Garg M, Bishnoi M, Kumar V, Oak MD, Singhal NK et al. Development and evaluation of chapatti quality of high amylose wheat mutants on the basis of physicochemical, textural and sensory characteristics. Lwt 2020, 133.
De Arcangelis E, Angelicola M, Trivisonno MC, Iacovino S, Falasca L, Lafiandra D, Sestili F, Messia MC, Marconi E. High-amylose bread wheat and its effects on cooking quality and nutritional properties of pasta. Int J Food Sci Technol. 2022;57:6785–94. https://doi.org/10.1111/ijfs.16028.
Obadi M, Qi Y, Xu B. High-amylose maize starch: structure, properties, modifications and industrial applications. Carbohydr Polym 2023, 299.
Montoya-Yepes DF, Jiménez-Rodríguez AA, Aldana-Porras AE, Velásquez-Holguin LF, Méndez-Arteaga JJ, Murillo-Arango W. Starches in the encapsulation of plant active ingredients: state of the art and research trends. Polym Bull. 2023. https://doi.org/10.1007/s00289-023-04724-6.
Tian S, Xue X, Wang X, Chen Z. Preparation of starch-based functional food nano-microcapsule delivery system and its controlled release characteristics. Front Nutr. 2022;9. https://doi.org/10.3389/fnut.2022.982370.
Santelia D, Zeeman SC. Progress in Arabidopsis starch research and potential biotechnological applications. Curr Opin Biotechnol. 2011;22:271–80. https://doi.org/10.1016/j.copbio.2010.11.014.
Landini A, Yu S, Gnecchi-Ruscone GA, Abondio P, Ojeda-Granados C, Sarno S, De Fanti S, Gentilini D, Di Blasio AM, Jin H, et al. Genomic adaptations to cereal-based diets contribute to mitigate metabolic risk in some human populations of east Asian ancestry. Evol Appl. 2021;14:297–313. https://doi.org/10.1111/eva.13090.
Tuberosa R, Frison E, Graner A. TILLING for Improved Starch Composition in Wheat. In Genomics of Plant Genetic Resources; Tuberosa, R., Graner, A., Frison, E., Eds.; Springer Netherlands: Dordrecht, 2014; Vol. 2, pp. 1–515 ISBN 978-94-007-7574-9.
Watcharakitti J, Win EE, Nimnuan J, Smith SM. Modified starch-based adhesives: a review. Polym (Basel). 2022;14. https://doi.org/10.3390/polym14102023.
Schneider A, Häusler RE, Kolukisaoglu Ü, Kunze R, Van der Graaff E, Schwacke R, Catoni E, Desimone M, Flügge UI. An Arabidopsis thaliana knock-out mutant of the chloroplast triose phosphate/phosphate translocator is severely compromised only when starch synthesis, but not starch mobilisation is abolished. Plant J. 2002;32:685–99. https://doi.org/10.1046/j.1365-313X.2002.01460.x.
Li C, Huang Y, Huang R, Wu Y, Wang W. The genetic architecture of amylose biosynthesis in maize kernel. Plant Biotechnol J. 2018;16:688–95. https://doi.org/10.1111/pbi.12821.
Sestili F, Janni M, Doherty A, Botticella E, D’Ovidio R, Masci S, Jones HD, Lafiandra D. Increasing the amylose content of durum wheat through silencing of the SBEIIagenes. BMC Plant Biol. 2010;10:144. https://doi.org/10.1186/1471-2229-10-144.
Gu Y, Han S, Chen L, Mu J, Duan L, Li Y, Yan Y, Li X. Expression and regulation of genes involved in the reserve starch biosynthesis pathway in hexaploid wheat (Triticum aestivum L). Crop J. 2021;9:440–55. https://doi.org/10.1016/j.cj.2020.08.002.
Islamovic E, Obert DE, Oliver RE, Harrison SA, Ibrahim A, Marshall JM, Miclaus KJ, Hu G, Jackson EW. Genetic dissection of grain beta-glucan and amylose content in barley (Hordeum vulgare L). Mol Breed. 2013;31:15–25. https://doi.org/10.1007/s11032-012-9764-1.
Blennow A, Skryhan K, Tanackovic V, Krunic SL, Shaik SS, Andersen MS, Kirk HG, Nielsen KL. Non-GMO potato lines, synthesizing increased amylose and resistant starch, are mainly deficient in isoamylase debranching enzyme. Plant Biotechnol J. 2020;18:2096–108. https://doi.org/10.1111/pbi.13367.
Salamone PR, Kavakli IH, Slattery CJ, Okita TW. Directed molecular evolution of ADP-glucose pyrophosphorylase. Proc. Natl. Acad. Sci 2002, 99, 1070–1075, https://doi.org/10.1073/pnas.012603799.
Geigenberger P. Regulation of Starch Biosynthesis in response to a fluctuating environment. Plant Physiol. 2011;155:1566–77. https://doi.org/10.1104/pp.110.170399.
Saddhe AA, Manuka R, Penna S. Plant sugars: Homeostasis and transport under abiotic stress in plants. Physiol Plant. 2021;171:739–55. https://doi.org/10.1111/ppl.13283.
Black JRM, Clark SJ. Age-related macular degeneration: genome-wide association studies to translation. Genet Med. 2016;18:283–9. https://doi.org/10.1038/gim.2015.70.
Fridaïus S, Vanvanhossou U, Scheper C, Dossa LH, Yin T, Brügemann K, König S. A multi-breed GWAS for morphometric traits in four Beninese indigenous cattle breeds reveals loci associated with conformation, carcass and adaptive traits. 2020, 1–16.
Mancin E, Tuliozi B, Pegolo S, Sartori C, Mantovani R. Genome Wide Association Study of Beef Traits in local Alpine Breed reveals the diversity of the pathways involved and the role of Time Strati fi cation. 2022, 12, 1–22, https://doi.org/10.3389/fgene.2021.746665.
Lu Z, Yue Y, Yuan C, Liu J, Chen Z. Genome-Wide Association Study of Body Weight Traits in Chinese Fine-Wool Sheep. 1–11.
Yoshida H, Hirano K, Yano K, Wang F, Mori M, Kawamura M, Koketsu E, Hattori M, Ordonio RL, Huang P, et al. Genome-wide association study identifies a gene responsible for temperature-dependent rice germination. Nat Commun. 2022;13. https://doi.org/10.1038/s41467-022-33318-5.
Yano K, Yamamoto E, Aya K, Takeuchi H, Lo PC, Hu L, Yamasaki M, Yoshida S, Kitano H, Hirano K, et al. Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat Genet. 2016;48:927–34. https://doi.org/10.1038/ng.3596.
Li H, Peng Z, Yang X, Wang W, Fu J, Wang J, Han Y, Chai Y, Guo T, Yang N, et al. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat Genet. 2013;45:43–50. https://doi.org/10.1038/ng.2484.
Lin M, Qiao P, Matschi S, Vasquez M, Ramstein GP, Bourgault R, Mohammadi M, Scanlon MJ, Molina I, Smith LG, et al. Integrating GWAS and TWAS to elucidate the genetic architecture of maize leaf cuticular conductance. Plant Physiol. 2022;189:2144–58. https://doi.org/10.1093/plphys/kiac198.
Zhang X, Zhang J, He X, Wang Y, Ma X, Yin D. Genome-wide association study of major agronomic traits related to domestication in peanut. Front Plant Sci. 2017;8:1–10. https://doi.org/10.3389/fpls.2017.01611.
Li L, Cui S, Dang P, Yang X, Wei X, Chen K, Liu L, Chen CY. GWAS and bulked segregant analysis reveal the loci controlling growth habit-related traits in cultivated peanut (Arachis hypogaea L). BMC Genomics. 2022;23:1–13. https://doi.org/10.1186/s12864-022-08640-3.
Gatarira C, Agre P, Matsumoto R, Edemodu A, Adetimirin V, Bhattacharjee R, Asiedu R, Asfaw A. Genome-wide association analysis for tuber dry matter and oxidative browning in water Yam (Dioscorea alata L). Plants. 2020;9:1–19. https://doi.org/10.3390/plants9080969.
Mondo JM, Agre PA, Asiedu R, Akoroda MO, Asfaw A. Genome-Wide Association Studies for Sex Determination and cross-compatibility in Water Yam (Dioscorea alata L). Plants. 2021;10:1412. https://doi.org/10.3390/plants10071412.
Cormier F, Martin G, Vignes H, Lachman L, Cornet D, Faure Y, Maledon E, Mournet P, Arnau G, Chaïr H. Genetic control of flowering in greater yam (Dioscorea alata L). BMC Plant Biol. 2021;21:1–12. https://doi.org/10.1186/s12870-021-02941-7.
Bredeson JV, Lyons JB, Oniyinde IO, Okereke NR, Kolade O, Nnabue I, Nwadili CO, Hřibová E, Parker M, Nwogha J, et al. Chromosome evolution and the genetic basis of agronomically important traits in greater yam. Nat Commun. 2022;13:1–16. https://doi.org/10.1038/s41467-022-29114-w.
Dossa K, Morel A, Houngbo ME, Mota AZ, Irep J, Diman J, Mournet P, Causse S, Nguyen K. Genome-wide association studies reveal novel loci controlling tuber flesh color and oxidative browning in. bioRxiv 2023, https://doi.org/10.1101/2023.03.12.532275.
Agre PA, Norman PE, Asiedu R, Asfaw A. Identification of quantitative trait nucleotides and candidate genes for tuber yield and mosaic virus tolerance in an elite population of white guinea yam (Dioscorea rotundata) using genome-wide association scan. BMC Plant Biol. 2021;21:1–16. https://doi.org/10.1186/s12870-021-03314-w.
Asfaw A, Agre P, Matsumoto R, Olatunji AA, Edemodu A, Olusola T, Odom-Kolombia OL, Adesokan M, Alamu OE, Adebola P, et al. Genome‐wide dissection of the genetic factors underlying food quality in boiled and pounded white Guinea yam. J Sci Food Agric. 2023. https://doi.org/10.1002/jsfa.12816.
Adewumi AS, Asare PA, Akintayo OT, Adejumobi II, Adu MO, Taah KJ, Afutu E, Opoku VA, Stanley AE, Akaba S, et al. Genetic architecture of post-harvest tuber quality traits in bush yam (Dioscorea praehensilis Benth.) Germplasm through association mapping. Front Hortic. 2024;3. https://doi.org/10.3389/fhort.2024.1373327.
Otegbayo B, Oluyinka O, Tanimola AR, Bisi F, Ayomide A, Tomilola B, Madu T, Okoye B, Chijioke U, Ofoeze M, et al. Food quality profile of pounded yam and implications for yam breeding. J Sci Food Agric. 2023. https://doi.org/10.1002/jsfa.12835.
Alamu EO, Adesokan M, Awoyale W, Oyedele H, Fawole S, Amele A, Maziya-Dixon B. Assessment of biochemical, cooking, sensory and textural properties of the boiled food product of white yam (D. Rotundata) genotypes grown at different locations. Heliyon. 2022;8:e11690. https://doi.org/10.1016/j.heliyon.2022.e11690.
Mota APZ, Dossa K, Lechaudel M, Cornet D, Mournet P, Santoni S, Lopez D, Chaïr H. Whole-genome sequencing and comparative genomics reveal candidate genes associated with quality traits in Dioscorea alata. BMC Genomics. 2024;25:248. https://doi.org/10.1186/s12864-024-10135-2.
Guo X, Ge Z, Wang M, Zhao M, Pei Y, Song X. Genome-wide association study of quality traits and starch pasting properties of maize kernels. BMC Genomics. 2023;24:1–15. https://doi.org/10.1186/s12864-022-09031-4.
Li M, Geng L, Xie S, Wu D, Ye L, Zhang G. Genome-wide association study on total starch, amylose and amylopectin in barley grain reveals novel putative alleles. Int J Mol Sci. 2021;22:1–13. https://doi.org/10.3390/ijms22020553.
Zhou W, Li B, Li L, Ma W, Liu Y, Feng S, Wang Z, Scoles GJ. Genome survey sequencing of Dioscorea zingiberensis. Genome. 2018;61:567–74. https://doi.org/10.1139/gen-2018-0011.
Zhang X, Jin M, Tadesse N, Dang J, Zhou T, Zhang H, Wang S, Guo Z, Ito Y. Dioscorea Zingiberensis C. H. Wright: an overview on its traditional use, phytochemistry, pharmacology, clinical applications, quality control, and toxicity. J Ethnopharmacol. 2018;220:283–93. https://doi.org/10.1016/j.jep.2018.03.017.
Epping J, Laibach N. An underutilized orphan tuber crop—Chinese yam : a review. Planta. 2020;252:1–19. https://doi.org/10.1007/s00425-020-03458-3.
Mugford ST, Fernandez O, Brinton J, Flis A, Krohn N, Encke B, Feil R, Sulpice R, Lunn JE, Stitt M, et al. Regulatory properties of ADP glucose pyrophosphorylase are required for adjustment of leaf starch synthesis in different photoperiods. Plant Physiol. 2014;166:1733–47. https://doi.org/10.1104/pp.114.247759.
Ghosh HP, Preiss J. Adenosine Diphosphate Glucose Pyrophosphorylase. J Biol Chem. 1966;241:4491–504. https://doi.org/10.1016/S0021-9258(18)99747-4.
Ogunsona E, Ojogbo E, Mekonnen T. Advanced material applications of starch and its derivatives. Eur Polym J. 2018;108:570–81. https://doi.org/10.1016/j.eurpolymj.2018.09.039.
García-Guzmán L, Cabrera-Barjas G, Soria-Hernández CG, Castaño J, Guadarrama-Lezama AY, Rodríguez, Llamazares. Progreso en materiales a base de almidón para alimentos aplicaciones de embalaje. Polysaccharides. 2022;3:136–77.
Van Harsselaar JK, Lorenz J, Senning M, Sonnewald U, Sonnewald S. Genome-wide analysis of starch metabolism genes in potato (Solanum tuberosum L). BMC Genomics. 2017;18:1–18. https://doi.org/10.1186/s12864-016-3381-z.
Saithong T, Rongsirikul O, Kalapanulak S, Chiewchankaset P, Siriwat W, Netrphan S, Suksangpanomrung M, Meechai A, Cheevadhanarak S. Starch biosynthesis in cassava: a genome-based pathway reconstruction and its exploitation in data integration. BMC Syst Biol. 2013;7. https://doi.org/10.1186/1752-0509-7-75.
Ehounou AE, Cormier F, Maledon E, Nudol E, Vignes H, Gravillon MC, N’guetta ASP, Mournet P, Chaïr H, Kouakou AM, et al. Identification and validation of QTLs for tuber quality related traits in greater yam (Dioscorea alata L). Sci Rep. 2022;12:1–14. https://doi.org/10.1038/s41598-022-12135-2.
Huang B, Hennen-Bierwagen TA, Myers AM. Functions of multiple genes encoding ADP-glucose pyrophosphorylase subunits in maize endosperm, embryo, and leaf. Plant Physiol. 2014;164:596–611. https://doi.org/10.1104/pp.113.231605.
Stark DM, Timmerman KP, Barry GF, Preiss J, Kishore GM. Regulation of the Amount of Starch in Plant Tissues by ADP glucose pyrophosphorylase. Sci (80-). 1992;258:287–92. https://doi.org/10.1126/science.258.5080.287.
Li N, Zhang S, Zhao Y, Li B, Zhang J. Over-expression of AGPase genes enhances seed weight and starch content in transgenic maize. Planta. 2011;233:241–50. https://doi.org/10.1007/s00425-010-1296-5.
Hannah LC, Futch B, Bing J, Shaw JR, Boehlein S, Stewart JD, Beiriger R, Georgelis N, Greene T. A shrunken-2 transgene increases Maize Yield by acting in maternal tissues to increase the frequency of seed development. Plant Cell. 2012;24:2352–63. https://doi.org/10.1105/tpc.112.100602.
Smidansky ED, Clancy M, Meyer FD, Lanning SP, Blake NK, Talbert LE, Giroux MJ. Enhanced ADP-glucose pyrophosphorylase activity in wheat endosperm increases seed yield. Proc Natl Acad Sci. 2002;99:1724–9. https://doi.org/10.1073/pnas.022635299.
Smidansky ED, Meyer FD, Blakeslee B, Weglarz TE, Greene TW, Giroux MJ. Expression of a modified ADP-glucose pyrophosphorylase large subunit in wheat seeds stimulates photosynthesis and carbon metabolism. Planta. 2007;225:965–76. https://doi.org/10.1007/s00425-006-0400-3.
Sakulsingharoj C, Choi SB, Hwang SK, Edwards GE, Bork J, Meyer CR, Preiss J, Okita TW. Engineering starch biosynthesis for increasing rice seed weight: the role of the cytoplasmic ADP-glucose pyrophosphorylase. Plant Sci. 2004;167:1323–33. https://doi.org/10.1016/j.plantsci.2004.06.028.
Smidansky ED, Martin JM, Hannah CL, Fischer AM, Giroux MJ. Seed yield and plant biomass increases in rice are conferred by deregulation of endosperm ADP-glucose pyrophosphorylase. Planta. 2003;216:656–64. https://doi.org/10.1007/s00425-002-0897-z.
Kavakli IH, Greene TW, Salamone PR, Choi SB, Okita TW. Investigation of subunit function in ADP-glucose pyrophosphorylase. Biochem Biophys Res Commun. 2001;281:783–7. https://doi.org/10.1006/bbrc.2001.4416.
Hwang SK, Nagai Y, Kim D, Okita TW. Direct appraisal of the potato tuber ADP-glucose pyrophosphorylase large subunit in enzyme function by study of a novel mutant form. J Biol Chem. 2008;283:6640–7. https://doi.org/10.1074/jbc.M707447200.
Tuncel A, Kavakli IH, Keskin O. Insights into subunit interactions in the heterotetrameric structure of potato ADP-glucose pyrophosphorylase. Biophys J. 2008;95:3628–39. https://doi.org/10.1529/biophysj.107.123042.
Barıs I, Tuncel A, Ozber N, Keskin O, Kavakli IH. Investigation of the Interaction between the large and small subunits of potato ADP-Glucose pyrophosphorylase. PLoS Comput Biol. 2009;5:e1000546. https://doi.org/10.1371/journal.pcbi.1000546.
Cross JM, Clancy M, Shaw JR, Boehlein SK, Greene TW, Schmidt RR, Okita TW, Hannah LC. A polymorphic motif in the small subunit of ADP-glucose pyrophosphorylase modulates interactions between the small and large subunits. Plant J. 2005;41:501–11. https://doi.org/10.1111/j.1365-313X.2004.02315.x.
Seferoglu AB, Koper K, Can FB, Cevahir G, Halil Kavakli I. Enhanced heterotetrameric assembly of potato ADP-glucose pyrophosphorylase using reverse genetics. Plant Cell Physiol. 2014;55:1473–83. https://doi.org/10.1093/pcp/pcu078.
Kang G, Liu G, Peng X, Wei L, Wang C, Zhu YJ, Ma Y, Jiang Y, Guo T. Increasing the starch content and grain weight of common wheat by overexpression of the cytosolic AGPase large subunit gene. Plant Physiol Biochem. 2013;73:93–8. https://doi.org/10.1016/j.plaphy.2013.09.003.
Toinga-Villafuerte S, Vales MI, Awika JM, Rathore KS. CRISPR/Cas9-Mediated mutagenesis of the granule-bound starch synthase gene in the Potato Variety Yukon Gold to Obtain Amylose-Free Starch in Tubers. Int J Mol Sci. 2022;23:4640. https://doi.org/10.3390/ijms23094640.
Geigenberger P, Stitt M. Diurnal changes in sucrose, nucleotides, starch synthesis and AGPS transcript in growing potato tubers that are suppressed by decreased expression of sucrose phosphate synthase. Plant J. 2000;23:795–X806. https://doi.org/10.1046/j.1365-313x.2000.00848.x.
Smith SM, Fulton DC, Chia T, Thorneycroft D, Chapple A, Dunstan H, Hylton C, Zeeman SC, Smith AM. Diurnal changes in the transcriptome encoding enzymes of starch metabolism provide evidence for both transcriptional and posttranscriptional regulation of starch metabolism in arabidopsis leaves. Plant Physiol. 2004;136:2687–99. https://doi.org/10.1104/pp.104.044347.
Farhat IA, Oguntona T, Neale RJ. Characterisation of starches from west African yams. J Sci Food Agric. 1999;79:2105–12. https://doi.org/10.1002/(SICI)1097-0010(199912)79:15%3C2105::AID-JSFA510%3E3.0.CO;2-R.
Jiang Q, Gao W, Li X, Man S, Shi Y, Yang Y, Huang L, Liu C. Comparative susceptibilities to alkali-treatment of A-, B- and C-type starches of Dioscorea zingiberensis, Dioscorea Persimilis and Dioscorea Opposita. Food Hydrocoll. 2014;39:286–94. https://doi.org/10.1016/j.foodhyd.2014.01.012.
R Core Team R. A Language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria. https://www.R-project.org/ 2022.
Bates D, Mächler M, Bolker BM, Walker SC. Fitting linear mixed-effects models using lme4. J Stat Softw. 2015;67. https://doi.org/10.18637/jss.v067.i01.
Ishizaki T, Ueda Y, Takai T, Maruyama K, Tsujimoto Y. In-frame mutation in rice TEOSINTE BRANCHED1 (OsTB1) improves productivity under phosphorus deficiency. Plant Sci. 2023;330:111627. https://doi.org/10.1016/j.plantsci.2023.111627.
Glaubitz JC, Casstevens TM, Lu F, Harriman J, Elshire RJ, Sun Q, Buckler ES. TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS ONE. 2014;9. https://doi.org/10.1371/journal.pone.0090346.
Li Y, Tan C, Li Z, Guo J, Li S, Chen X, Wang C, Dai X, Yang H, Song W, et al. The genome of Dioscorea zingiberensis sheds light on the biosynthesis, origin and evolution of the medicinally important diosgenin saponins. Hortic Res. 2022;9. https://doi.org/10.1093/hr/uhac165.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8. https://doi.org/10.1093/bioinformatics/btr330.
Browning BL, Browning SR. Genotype imputation with millions of reference samples. Am J Hum Genet. 2016;98:116–26. https://doi.org/10.1016/j.ajhg.2015.11.020.
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14:2611–20. https://doi.org/10.1111/j.1365-294X.2005.02553.x.
Lipka AE, Tian F, Wang Q, Peiffer J, Li M, Bradbury PJ, Gore MA, Buckler ES, Zhang ZGAPIT. Genome association and prediction integrated tool. Bioinformatics. 2012;28:2397–9. https://doi.org/10.1093/bioinformatics/bts444.
Yu J, Pressoir G, Briggs WH, Bi IV, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet. 2006;38:203–8. https://doi.org/10.1038/ng1702.
Turner D. Qqman: an R package for visualizing GWAS results using Q-Q and manhattan plots. J Open Source Softw. 2018;3:731. https://doi.org/10.21105/joss.00731.
Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat Protoc. 2008;3:1101–8. https://doi.org/10.1038/nprot.2008.73.
Clough SJ, Bent AF. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998;16:735–43. https://doi.org/10.1046/j.1365-313X.1998.00343.x.
Acknowledgments
Not applicable.
Funding
The present study was funded by the National Natural Science Foundation of China (NSFC) (41867027, 42167057, 32160223, 31860254), the Key Special Program of Yunnan Province’s Science and Technology Planning Project ( 202201AS070028), the Outstanding Young Talent Projects of Yunnan Ten Thousand Talents Program (80201442), the Reserve Talents Project for Young and Middle-aged Academic and technical Leaders of Department of Science and Technology of Yunnan Province (202105AC160047), and the General Program of Science and Technology Department of Yunnan Provincial (2018FG001-039).
Author information
Authors and Affiliations
Contributions
Conceptualization, S S, B G, L Z, and Y L; methodology, S S, B G, Y X, X L, L L, Y L; software, K D; validation, X L, L L, Y L, L J, S Y; formal analysis, S S, B G; investigation, S S, B G; resources, Y L, L J; data curation, K D; writing—original draft preparation, S S, B G; writing—review and ed-iting, K D, L Z, Y L; visualization, S S; supervision, L Z, Y L; project administration, L Z, Y L; funding acquisition, L Z, Y L. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All relevant institutional, national, and international guidelines and legislations were followed while conducting this experiment. All germplasms are conserved at the Genebank of Southwest Forestry University.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sun, S., Guan, B., Xing, Y. et al. Genome-wide association analysis and transgenic characterization for amylose content regulating gene in tuber of Dioscorea zingiberensis. BMC Plant Biol 24, 524 (2024). https://doi.org/10.1186/s12870-024-05122-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-024-05122-4