
Abstract
Background
Color is the major ornamental feature of the Rhododendron genus, and it is related to the contents of flavonoid in petals. However, the regulatory mechanism of flavonoid biosynthesis in Rhododendron pulchrum remains unknown. The transcriptome and metabolome analysis of Rhododendron pulchrum with white, pink and purple color in this study aimed to reveal the mechanism of flavonoid biosynthesis and to provide insight for improving the petal color.
Results
Flavonoids and flavonols are the major components of flavonoid metabolites in R.pulchrum, such as laricitrin, apigenin, tricin, luteolin, isoorientin, isoscutellarein, diosmetin and their glycosides derivatives. With transcriptome and metabolome analysis, we found CHS, FLS, F3’H, F3′5’H, DFR, ANS, GT, FNS, IFR and FAOMT genes showed significantly differential expression in cultivar ‘Zihe'. FNS and IFR were discovered to be associated with coloration in R.pulchrum for the first time. The FNS gene existed in the form of FNSI. The IFR gene and its related metabolites of medicarpin derivatives were highly expressed in purple petal. In cultivar ‘Fenhe', up-regulation of F3’H and F3′5’H and down-regulation of 4CL, DFR, ANS, and GT were associated with pink coloration. With the transcription factor analysis, a subfamily of DREBs was found to be specifically enriched in pink petals. This suggested that the DREB family play an important role in pink coloration. In cultivars ‘Baihe', flavonoid biosynthesis was inhibited by low expression of CHS, while pigment accumulation was inhibited by low expression of F3′5'H, DFR, and GT, which led to a white coloration.
Conclusions
By analyzing the transcriptome and metabolome of R.pulchrum, principal differential expression genes and metabolites of flavonoid biosynthesis pathway were identified. Many novel metabolites, genes, and transcription factors associated with coloration have been discovered. To reveal the mechanism of the coloration of different petals, a model of the flavonoid biosynthesis pathway of R.pulchrum was constructed. These results provide in depth information regarding the coloration of the petals and the flavonoid metabolism of R.pulcherum. The study of transcriptome and metabolome profiling gains insight for further genetic improvement in Rhododendron.
Similar content being viewed by others


Background
Floral color is a main ornamental feature of plants. Several factors, including Vacuole pH, metal ions, pigment composition, and the structure of the petal, influence flower color [1]. One of the most critical factors is the pigment composition, which contains flavonoids, carotenoids, betalains, and chlorophylls [2]. Flavonoids produce a variety of colors ranging from pale yellow to deep blue [3], which have been divided into flavonoids, flavonols, anthocyanins, isoflavones, flavan-3-ols, dihydroflavonols, dihydroflavones, and chalcons [4, 5]. Among these, chalcones were reported in deep yellow color, while flavones and flavonols in faintly yellow or almost colorless [6]. A variety of colors can be produced by anthocyanins [7]. To date, nearly 700 anthocyanin derivatives have been identified in plants [8].
The flavonoid biosynthesis is a complex process in which structural genes such as early and late biosynthetic genes (EBGs and LBGs) are involved [9]. These EBGs encode enzymes that provide precursors for flavonoid synthesis, including phenylalanine ammonia-lyase (PAL), cinnamate-4-hydroxylase (C4H), 4-coumarate CoA ligase 4 (4CL), chalcone synthase (CHS), and chalcone isomerase (CHI). LBGs are responsible for the synthesis of proanthocyanidins, which includes flavanone 3-hydroxylase(F3H), flavonoid 3’-hydroxylase(F3’H), flavonoid 3’,5’-hydroxylase(F3′5’H), dihydroflflavonol 4-reductase (DFR), anthocyanidin synthase (ANS), flavonol synthase (FLS), glucosyltransferase (GT), etc. It was reported CHS had the potential to affect petal coloration [10, 11]. A high expression of CHS, DFR, and ANS genes led to an accumulation of anthocyanins [12], while the insertion of ANS coding led to a reduction in anthocyanin content [13]. Yang et al. observed that the expression of CHI, C3 'H and F3 'H genes increased in red Eucommia ulmoides leaves, while the expression of F3 ′5'H and GT genes decreased [14]. In addition, flavonoid biosynthesis is also jointly controlled by a variety of transcription factors, including MYB, bHLH, TTG1 (WD40 repeat protein), TTG2 (WRKY44), TT16/AGL32 (MADS protein) and TT1 [15]. In recent years, genes and transcription factors involved in anthocyanin biosynthesis have been identified in many plants [16,17,18,19,20].
The genus Rhododendron contains brightly colored flowers and is of great use in horticulture [21]. Flavanols and anthocyanins are the main pigments in Rhododendron [22], their quantities determine the petal color ranging from light pink to violet [23,24,25,26,27,28]. At present, several studies have been done on genes and transcription factors involved in the flavonoid biosynthesis pathway in Rhododendron [29,30,31]. Rhododendron pulchrum Sweet is a species of Rhododendron belonging to the Ericaceae family and is widely cultivated in temperate Europe, Asia and North America [32]. It is highly resistant and widely cultivated in the Yangtze Delta region [33]. Based on the study of Qian et al., the main anthocyanin in purple petals is Peonidin, while Pelargonidin is the main component in pink petals, and no anthocyanin is detected in white petals of R.pulchrum [34]. There were 149 flavonoids and their derivatives identified in R.pulchrum cultivars with different colors, among them flavone and C-glycosylated flavone were the most important flavonoid metabolites [35]. In one transcriptome analysis of R.pulchrum petals, it was found that most differentially expressed genes (DEGs) were located between flower buds and early flowering, while transcripts for the genes MYC2, TIR1, CYCD3, COL-1, and EIN3 peaked at flower buds [36]. However, the regulatory mechanism of flavonoid biosynthesis in R.pulchrum remains unclear.
Recent studies have used integrated multi-omics analyses to identify and analyze traits related genes and pathways in plants [5, 6, 37,38,39,40,41]. A combined analysis of transcriptomes and metabolomes can directly and accurately reflect organismal change. We performed the analysis of metabolome and transcriptome to understand the flavonoid biosynthesis pathway in petals of R.pulchrum for the first time. The findings of this study explain the coloring mechanism and provide a theoretical basis for color breeding of R.pulchrum.
Results
Six hundred eighty-eight of differential metabolites were identified in three cultivars of R.pulchrum
Liquid chromatography–mass spectrometry (LC–MS) analysis was conducted to identify the compositions of metabolites within cultivars 'Baihe', 'Fenhe' and 'Zihe' petals. According to the results of the orthogonal projections to latent structures discriminant analysis (OPLS-DA), there is a clear separation of three colors, 'Baihe' and 'Fenhe' show a closer relationship when compared with 'Zihe' (Fig. 1). In all, 688 differential metabolites were identified in three comparison groups (VIP ≥ 1 and P < 0. 05). 176 differential metabolites were found between cultivars 'Baihe' and ‘Fenhe’. Of these, 109 were up-regulated, and 67 were down-regulated in ‘Fenhe’. There were 252 (139 up-regulated and 113 down-regulated) and 260 (155 up-regulated and 105 down-regulated) differential metabolites in cultivar 'Zihe' compared to cultivars 'Baihe' and 'Fenhe', respectively (Fig. S1).

OPLS-DA score plot of three materials and numbers of potential markers for each color petals. Note: a OPLS-DA score plot of ‘Baihe’ and ‘Fenhe’ petals; b OPLS-DA score plot of ‘Zihe’ and ‘Baihe’ petals; c OPLS-DA score plot of ‘Fenhe’ and ‘Zihe’ petals
One hundred one flavonoid-related differential metabolites were discovered in three cultivars of R. pulchrum
To understand the coloration mechanism in more detail, our study focused on the metabolite components enriched in the flavonoid biosynthesis pathway. 36 flavonoid related metabolites were identified in cultivars of 'Baihe' (BMJ) and 'Fenhe' (FMJ) group, 65 in cultivars 'Zihe' (ZMJ) and 'Baihe' (BMJ) group, and 61 in cultivars 'Fenhe' (FMJ) and 'Zihe' (ZMJ) group (Fig. 2a). The patterns of metabolites accumulation between different colors were studied by hierarchical cluster analysis (HCA) (Fig. S2). For identifying the differential expression metabolites, we defined the criterion of the absolute Log2FC ≥ 1 and VIP value ≥ 1. According to the flavonoid structure of cultivars 'Baihe' and 'Fenhe' group, we compared the expression levels of 36 metabolites including five dihydroflavones, four flavanols, five chalcones, two anthocyanidin, nine flavonoids and 11 flavonols (Fig. 2b). In them, 13 metabolites were up-regulated and five down-regulated in cultivars ‘Fenhe’. Neoeriocitrin, Hesperetin 7-glucoside, Avicularin, Viscumneoside III, and Glucodistylin (Log2 FC = 2.67–3.73) are higher in cultivar ‘Fenhe’ than in ‘Baihe’ (Table S1), indicating that these compounds played an important role in the pink coloration of R.pulchrum. Three dihydroflavones, 11 flavanols, five chalcones, five anthocyanidins, 15 flavonoids, 25 flavonols, and one isoflavone were found in cultivars ‘Zihe’ and ‘Baihe’ group (Fig. 2c). 45 flavonoid metabolites with significantly differential expressions were identified (71.88% of the total), of which 29 showed up-regulated and 16 down-regulated in 'Zihe'. The amounts of Neoeriocitrin, 5-Methyleriodictyol 7-[glucosyl-(1- > 4)-galactoside], Hesperetin 7-glucoside, Kaempferol 3-rhamnosyl-(1- > 3)(4'''-p-coumarylrhamnosyl)(1- > 6)-glucoside, Hesperetin 7-neohesperidoside (Log2 FC = 4.87–9.69) are significantly higher in cultivar ‘Zihe’ than in ‘Baihe’ (Table S1), indicating these compounds are involved in the purple coloration of R.pulchrum. For cultivars 'Fenhe' and 'Zihe' group, we identified metabolites of five dihydroflavones, eight flavanols, two chalcones, seven anthocyanidins, 15 flavonoids, 23 flavonols, and one isoflavone (Fig. 2d), and 38 metabolites were differentially expressed (63.33% of the total), including 22 of them up-regulated and 16 down-regulated in cultivar ‘Zihe’. A significantly higher enrichment of Isovitexin 2''-(6'''-feruloylglucoside) 4'-glucoside, Medicarpin 3-O-(6'-malonylglucoside), Baicalein 6-methylether 7-glucosyl-(1- > 3)-rhamnoside, Kaempferol 3-rhamnosyl-(1- > 3)(4'''-p-coumarylrhamnosyl)(1- > 6)-glucoside, and Hesperetin 7-neohesperidoside(Log2 FC = 3.18–7.47) was found in cultivar 'Zihe' than in 'Fenhe'(Table S1). This indicates these metabolites are more specific to the purple coloration rather than pink in R.pulchrum. In summary, the flavonoid metabolites are primarily flavonoids and flavonols, and there are abundant amounts of Laricitrin, Apigenin, Tricin, Luteolin, Isoorientin, Isoscutellarein, Diosmetin,Triclin, and their glycosides. Importantly, it has been found that medicarpin 3-O-(6'-malonylglucoside) classified as isoflavones were more abundant in cultivar 'Zihe' than in the other two cultivars (Fig. 3a). Additionally, a total of 8 anthocyanins were identified, including cyanidin, pelargonidin, delphinidin, and malvidin. The content of malvidin 3-O-(6-O-(4-O-malonyl-alpha-rhamnopyranosyl)-beta-glucopyranoside)-5-O-beta-glucopyranoside and Cyanidin 3-O-[b-D-Xylopyranosyl-(1- > 2)-[4-hydroxycinnamoyl-(- > 6)-b-D-glucopyranosyl-(1- > 6)]-b-D-galactopyranoside] in cultivar 'Zihe' was significantly higher than the other two cultivars (Table S1). It is thus believed the two anthocyanins are responsible for the purple coloration in R.pulchrum [42, 43]. Leucodelphinidin 3-[Galactosyl -(1- > 4)-glucoside] concentration in dark colored petals was significantly higher than in light colored petals (Table S1). On the other hand, the amount of Cyanidin 3-O-(2 "-O-Galloyl-6"-O-alpha-rhamnopyranosyl-beta-galactopyranoside in light colored petals was significantly higher than in dark colored petals (Fig. 3b, Table S2). These results showed that Leucodelphinidin 3-[Galactosyl -(1- > 4)-glucoside] and Hesperetin 7-glucoside may contribute to the coloration of petals, while Malvidin 3-O-(6-O-(4-O-malonyl-alpha-rhamnopyranosyl)-beta-glucopyranoside)-5-O-beta-glucopyranoside,Cyanidin 3-O-[b-D-Xylopyranosyl-(1- > 2)-[4-hydroxycinnamoyl-(- > 6)-b-D-glucopyranosyl-(1- > 6)]-b-D-galactopyranoside], Medicarpin 3-O-(6'-malonylglucoside) and Hesperetin 7-neohesperidoside may contribute to the purple coloration of petals.

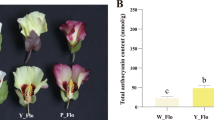
Flavonoid metabolites in R.pulchrum with different colors. Note: BMJ, cultivar ‘Baihe’; FMJ, cultivar ‘Fenhe’; ZMJ, cultivar ‘Zihe’. a Measurement of total flavonoids in R.pulchrum with different colors. b Flavonoid metabolites in ‘Baihe’ and ‘Fenhe’. c Flavonoid metabolites in ‘Zihe’ and ‘Baihe’. d Flavonoid metabolites in ‘Fenhe’ and ‘Zihe’

Fold change of the flavonoid and anthocyanin metabolites in R.pulchrum with different colors. Note: BMJ, cultivar ‘Baihe’; FMJ, cultivar ‘Fenhe’; ZMJ, cultivar ‘Zihe’. a Flod change of the flavonoid metabolites. b Fold change of the anthocyanin metabolites
Seven thousand seven hundred thirteen differentially expressed genes were found among three cultivars of R.pulchrum
To further investigate the molecular basis for flavonoid biosynthesis, transcriptome analysis was carried out to identify differentially expressed genes (DEGs) in petals of R.pulchrum. 61.84 Gb of clean data was produced from the petals of R.pulchrum (Table S3). The transcriptome data were then mapped to the genome of Rhododendron simsii and the mapping rate ranged from 84.21% to 85.27%. A total of 7713 DEGs were identified using the cut-off values of |log2 Fold change|≥ 1 and p Value < 0.05. In the comparison group between cultivars ‘Baihe’ (BMJ) and ‘Fenhe’ (FMJ), 381 DEGs were up-regulated while 1320 were down-regulated. Between the cultivars 'Zihe'(ZMJ) and 'Baihe'(BMJ) group, 1390 were up-regulated and 1278 were down-regulated. There were 2117 up-regulated genes and 1227 down-regulated genes in cultivar ‘Fenhe’ (FMJ) and ‘Zihe’ (ZMJ) group (Fig. 4).

Differently expressed genes in R.pulchrum with different colors. Note: a Statistic of differently expressed gene in cultivars 'Baihe'(BMJ),'Fenhe'(FMJ) and 'Zihe'(ZMJ). b The Venn diagram of differently expressed gene between three R.pulchrum cultivars
Comparative analysis of genes enriched by GO and KEGG in three comparison groups
To understand their biological functions and gene interactions, 5156 of 7713 DEGs were annotated to GO database. 1208 and 1739 DEGs were categorized into 50 functional groups when cultivar ‘Baihe’ was compared with ‘Fenhe’ and ‘Zihe’, respectively. The number become to 2209 DEGs and 52 functional groups when comparing cultivar ‘Fenhe’ and ‘Zihe’. Three major GO categories were ‘Biological process’, ‘Cellular component’, and ‘Molecular function’. In detail, ‘Cellular process’, ‘Single-organism process’, and ‘Metabolic process’ were the most enriched terms in ‘Biological process’, ‘Cell’, ‘Cell part’, and ‘Organelle’ were the most enriched terms in ‘Cellular component’, while ‘Binding’, ‘Catalytic activity’, and ‘Transporter activity’ were the most enriched terms in ‘Molecular function’ (Fig. 5). 22 anthocyanin biosynthetic processes and 29 flavonoid biosynthetic processes related genes were identified in the ‘Biological process’, while two anthocyanin biosynthetic processes and one flavonoid biosynthetic process related genes were identified in the ‘Molecular function’.

Transcript GO annotation classification statistics graph in three comparison groups. Note: BMJ, cultivar ‘Baihe’; FMJ, cultivar ‘Fenhe’; ZMJ, cultivar ‘Zihe’. a Transcript GO annotation classification between BMJ and FMJ; b Transcript GO annotation classification between ZMJ and BMJ; c Transcript GO annotation classification between FMJ and ZMJ
Moreover, to identify the metabolic pathways involved in the flavonoids’ biosynthesis process, 1433 DEGs were annotated with the KEGG database. 326, 472, and 635 DEGs were enriched in 19 pathways belonging to 6 categories (Fig. 6). According to KEGG analysis, the top pathways were the metabolic pathways for carbohydrates, lipids, and other secondary metabolites. The classification showed that a large number of genes were enriched in the ‘Phenylpropanoid biosynthesis’(ko00940) and ‘Flavone and flavonol biosynthesis’(ko00944), which is important to the petal coloration (Table S4). Our study found DEGs enriched in the ‘Brassinosteroid biosynthesis pathway’(ko00905) in three comparison groups, and 22 DEGs were enriched in ‘Plant hormone signal transduction pathway’(ko04075) between cultivar ‘Baihe’ and ‘Fenhe’ group. Also, 9 DEGs were enriched in ‘Photosynthesis’(ko00195) between cultivars ‘Zihe’ and ‘Baihe’ group (Table S4). It was reported that plant hormones and light signals were closely related to the synthesis of anthocyanins [44, 45]. Our results indicated that plant hormones play an important role in white to pink coloration, while light signals play an active role in white to purple coloration in R.pulchrum.

KEGG pathway classification statistics graph in three comparison groups. Note: BMJ, cultivar ‘Baihe’; FMJ, cultivar ‘Fenhe’; ZMJ, cultivar ‘Zihe’. a KEGG pathway classification statistics between BMJ and FMJ; b KEGG pathway classification statistics between ZMJ and BMJ; c KEGG pathway classification statistics between FMJ and ZMJ
Specific transcription factor families enriched in the different color of R.pulchrum
Transcription factors (TFs) play a critical role in the growth and development of plants by regulating gene expression. A total of 64 families of transcription factors were reported [46]. With transcriptome analysis, 32, 33 and 43 families of differentially expressed TFs were detected in the cultivars ‘Baihe’(BMJ) vs. ‘Fenhe’(FMJ), ‘Zihe’(ZMJ) vs. ‘Baihe’(BMJ), and ‘Fenhe’(FMJ) vs. ‘Zihe’ (ZMJ), respectively (Fig. 7). These TFs mainly belong to the tAP2/ERF, MYB, WRKY, bHLH, MyB-related, NAC and bZIP families (Fig. S3). In them, WRKY family was enriched in ‘Baihe’ and ‘Fenhe’, while MADS-M family was enriched in ‘Zihe’. ‘Fenhe’ had a high expression level of tAP2/ERF and NAC families. The HB-HD-ZIP, FAR1, and OFP families were highly expressed in ‘Zihe’. The results indicated that tAP2/ERF and NAC families were involved in the regulation of pink coloration, while HB-HD-ZIP, FAR1 and OFP families were involved in the purple coloration. The HSF family had a higher expression in ‘Baihe’. It was possible that HSF family inhibits color related gene expression. In addition, 16 genes were annotated as MYB-related genes, including MYB5, MYB6, MYB15, MYB101, MYB73, MYB61, MYB308, and other MYB-dominant proteins. 12 genes encoding bHLH. These TFs are associated with the flavonoid biosynthesis in the petals of R.pulchrum.

Distribution of transcription factor types in three comparison groups. Note: BMJ, cultivar ‘Baihe’; FMJ, cultivar ‘Fenhe’; ZMJ, cultivar ‘Zihe’. a Distribution of transcription factor types in BMJ and FMJ group; b Distribution of transcription factor types in ZMJ and BMJ group; a Distribution of transcription factor types in FMJ and ZMJ group
Modeling of a new color-related flavonoid biosynthesis pathway in R.pulchrum
Based on the expression patterns of key genes and metabolites, a model of the flavonoid biosynthesis pathway in R.pulchrum was constructed. In the modeling of cultivar ‘Baihe’, we speculated the inhibition of CHS gene led to a decrease in chalcone and naringin at the early stage (Fig. 8a). FLS, F3′5'H, and F3'H competed for the same substrate of dihydrokaempferol. Then, up-regulated of FLS and F3'H genes accompanied by down-regulated of F3′5'H gene drive the synthesis of kaempferol and quercetin derivatives in cultivar 'Baihe' (Fig. 8). Inhibition of F3′5'H, DFR and GT genes led to an inactivation of pigment accumulation, which resulted in the appearance of white petals (Fig. 9).

Biosynthesis pathway of flavonoid in three different color petals of R.pulchrum Sweet. Note: a Key structural genes and their expression level of involved in flavonoid biosynthesis pathway in R.pulchrum Sweet with different colors. b The expression levels of differential metabolites in flavonoid synthesis pathway

A model for the flavonoid biosynthesis pathway in three different color petals of R.pulchrum Sweet. Note: a cultivar ‘Baihe’. b cultivar ‘Fenhe’. c cultivar ‘Zihe’. The red color indicates genes and metabolites with high expression, and the blue color indicates genes and metabolites with low expression
In the modeling of cultivar ‘Zihe’, CHS gene showed a higher expression level in 'Zihe' than in other two cultivars. Previous studies have reported that the high expression of CHS gene is responsible for purple anthocyanins in Freesia hybrids [47] and wheat [48]. F3'H, F3′5'H, DFR, ANS, GT, and Violdelphin-GT had higher expression levels while the FLS, F3'H and LAR genes with a lower expression in 'Zihe' compared with ‘Baihe’ and ‘Fenhe’ (Fig. 8a). The differential expression of F3′5'H, FLS and F3'H genes led to synthesize many delphinidins from dihydrokaempferol. It was found that delphinidin and malvidin derivatives were accumulated in 'Zihe', such as delphinidin 3,5-di(6-O-malonylglucoside) and malvidin3-O-(6-O-(4-O-malonyl-alpha-rhamnopyranosyl)-beta-glucopyranoside)-5-O-beta-glucopyranoside (Fig. 8b). The low expression of F3'H gene and the high expression of DFR, ANS and GT genes leading to accumulation of epicatechins and cyanidins. In the meantime, cyanidin could be converted to cyanidin and pelargonidin glycosides catalyzed by GT. The metabolome analysis found pelargonidin 3-(6-p-coumaroyl) glucoside and cyanidin3-O-[b-D-Xylopyranosyl-(1- > 2)-[4-hydroxycinnamoyl-(- > 6)-b-D-glucopyranosyl-(1- > 6)]-b-D-galactopyranoside] were enriched in cultivar 'Zihe'. These DEGs are significantly associated with the production of reddish purple pigmentation in 'Zihe' (Fig. 9).
In the modeling of ‘Fenhe’, coumaric acid expression was higher, but the low expression of the 4CL gene restrained the accumulation of 4-Coumaroyl-CoA at the early stage of flavonoid synthesis pathway (Fig. 8b). The expression of FLS gene is inhibited, meanwhile the F3'H and F3′5'H genes were highly expressed, which led to overaccumulation of dihydromyricetin and dihydroquercetin (Fig. 8a). Because of the inhibition of the DFR, ANS, and GT genes, the delphinidin and cyanidin synthesis pathways are shut down leading to the formation of light purplish pink petals in cultivar 'Fenhe'. (Fig. 9).
To validate the key genes related to flavonoid biosynthesis, nine genes were randomly selected for further verification with RT-PCR (Table S6). It was found that six genes (4CL, CHS, ANS, F3 ′5' H, GT, Viodelphin-GT) were up-regulated in cultivar 'Zihe', three genes (F3H, FLS, LAR) were up-regulated in cultivars ‘Baihe’ and ‘Fenhe’. These results were consistent with what we discovered from RNA-seq (Fig. S4).
The integrated transcriptome and metabolome analysis was performed to gain a deeper understanding of the color-specific genes. 10 DEMs and 6 DEGs, 36 DEMs and 22 DEGs, and 37 DEMs and 23 DEGs enriched in flavonoid pathway were selected from the comparison groups of 'Baihe' vs. 'Fenhe', 'Zihe' vs. 'Baihe', and 'Fenhe' vs. 'Zihe', respectively. A Pearson correlation analysis of gene expression and metabolite response intensity was performed [49, 50]. CHS, F3′5'H, ANS, and Viodelphin-GT were highly expressed, and LAR had a low expression in purple petals, and the metabolites associated with these genes were also enriched in the petals (Table S5). Our integrative analysis suggested that differential expression of CHS, FLS, F3' H, F3 ′5 'H, DFR, GT, FAOMT in the flavonoid biosynthesis caused different colors between white and purple. In addition, F3' H and F3 ′5 'H up-regulated, 4CL, DFR, ANS and GT down-regulated are significantly associated with pink coloration.
Color-specific modules and hub genes were identified by using WGCNA
To further validated the flavonoid biosynthesis related key genes, a WGCNA (Weighted Correlation Network Analysis) was performed [51]. After filtering out the genes with a low expression (FPKM < 0.05), 4066 genes were obtained for the WGCNA. 12 distinct modules were identified (labeled with different colors) shown in the dendrogram in Fig. 10a. Heat map of the 12 modules are shown in Fig. 10b. The lightcyan1, mediumpurple3 and grey60 modules with 903, 41 and 658 genes, were highly associated with the cultivars 'Zihe' (r = 0.97, 0.80 and -0.98). GT, FAOMT, F3′5’H, 4CL, IFR genes are enriched in the lightcyan1 module, and DFR and Viodelphin-GT genes were enriched in mediumpurple3 module, while the grey60 module contained LAR and FNS genes. The cyan module with 238 genes was highly associated with cultivars 'Fenhe' (r = -0.92), in which ANS was enriched. The midnightblue module with 379 genes was highly associated with cultivars 'Baihe' (r = 0.90), which contained FLS gene (Fig. 11). The WGCNA results suggested that ANS, FLS, LAR, FNS, GT, 4CL, F3′5’H, IFR, DFR, FAOMT and Viodelphin-GT genes are associated with the coloration of petals in R.pulchrum, in which the genes of ANS, FLS, LAR, GT, 4CL, F3′5’H, DFR, FAOMT and Viodelphin-GT are found in the integrated analysis of transcriptome and metabolome. The results indicated these hubs genes are significantly important for the coloration of R.pulchrum.

WGCNA of genes in three different color petals of R.pulchrum Sweet. a Hierarchical clustering tree (cluster dendrogram) results showed 12 expression modules, labeled with different colors. b Module–sample association. Each row corresponds to a module, labeled with a color as in (a). The number of genes in each module is indicated on the left

Coexpression network analysis in three different color petals of R.pulchrum Sweet. (a), (c), (e), (g), and (i) Heatmaps showing genes in cyan, midnightblue, mediumpurple3, lightcyan1, and grey60 modules that were significantly over-represented in BMJ, FMJ, and ZMJ, respectively. (b), (d), (f), (h), and (j) The correlation networks in the module corresponding to (a), (c), (e), (g), and (i), respectively. Candidate hub genes are shown in red
Discussion
Multi-omics analysis can be used to study the molecular mechanisms of plant coloration [36]. Multiple metabolites are mainly accumulated in flavonoid biosynthesis, flavonoid and flavonol biosynthesis, and anthocyanin biosynthesis pathways in R.pulchrum. We provided a model illustrating the color related key genes and metabolites and their regulatory mechanism in R. pulcherum.
Floral color and key genes in flavonoid biosynthesis of R.pulchrum
At the early stage of flavonoid biosynthesis, 4CL and CHS genes showed differential expression levels among three cultivars, but not happened with CHI gene. 4-Coumarate CoA Ligase (4CL) participates in the metabolism of a variety of secondary compounds in plants, such as lignin, coumarin, flavonoids, anthocyanins, and phenols [52,53,54]. We had identified four genes that encode 4-Coumarate CoA Ligase, three of them (Rhsim10G0185800, Rhsim10G0199800, and Rhsim10G0199000) are highly expressed in cultivar ‘Zihe’, and the other one (Rhsim10G0200300) is highly expressed in cultivar ‘Fenhe’. The high expression of these genes contributed to the accumulation of more precursor compounds in cultivar 'Zihe'. Chalcone synthase (CHS), as a key initiating enzyme in the flavonoid synthesis pathway, exists in a multi-gene family in most species [55]. CHS expression was significantly lower in cultivar 'Baihe' compared with 'Fenhe' and 'Zihe', which resulted in the inhibition of flavonoid biosynthesis in 'Baihe' at the early stage. CHI gene did not show differential expressions in different color petals. As Takos et al. discovered there was no difference in CHI expression between the peels of red and non-red apple varieties, but when CHI was completely absent, anthocyanin synthesis was limited [56]. Therefore, they believed CHI was not a key enzyme for the catalytic synthesis of chalcone naringin, but rather acted as rate-limiting enzyme in the flavonoid pathway.
At the late stage of flavonoid biosynthesis, FLS, F3H, F3 'H, DFR, LAR, F3 ′5' H, ANS, GT, and violdelphin-GT genes showed differential expression in three petals. Flavonol synthase (FLS), a key enzyme in the biosynthesis of flavonols, catalyzes the production of kaempferol and myricetin from dihydrokaempferol and dihydromyricetin respectively. A deletion of FLS or an alteration of gene expression can affect the coloration of plant organs [57, 58]. As a result of high FLS expression in cultivar 'Baihe', high levels of kaempferol and quercetin derivatives were found. FLS play a more active role in catalyzing the dihydrokaempferol than F3'H [59], resulting in the accumulation of kaempferol and quercetin derivatives in 'Baihe'. It was reported that FLS and DFR compete for substrates to synthesize flavonols, and an upregulation of FLS and a downregulation of DFR may result in a reduction of anthocyanin accumulation [60], this is in line with our findings. There are MYB transcription factor binding sites and light signal response sites in most FLS gene promoters, indicating that FLS gene transcription is regulated by MYB transcription factors and light signals [61, 62]. In this study, the DEGs are enriched in the 'light signaling pathway' in 'Fenhe' and 'Zihe' group (Table S4). However, whether the FLS gene is regulated by light signal and MYB transcription factors in R.pulchrum needs further study. The flavonoid 3 'hydroxylase (F3'H) is a critical enzyme in producing Purplish Red [63]. As well as catalyzing kaempferol to form quercetin, it also catalyzes dihydrokaempferol to form dihydroquercetin. There are two genes that encode F3 'H (Rhsim01G0176800 and Rhsim08G0143600). Rhsim01G0176800 was highly expressed in cultivar 'Baihe', whereas Rhsim08G0143600 was highly expressed in cultivar 'Fenhe'. We speculate that these two genes may have distinct regulatory pathways, Rhsim01G0176800 catalyzes kaempferol to produce quercetin, while Rhsim08G0143600 catalyzes dihydrokaempferol to produce dihydroquercetin, further validations are required. F3 ′5' H is a key gene associated with the synthesis of blue color. In our study, F3 ′5' H (Rhsim13G0208100, Rhsim13G0208200) are highly expressed in cultivar 'Zihe', leading to the synthesis of dihydromyricetin, a substrate of delphinidin and malvidin glycosides. Both LAR and ANS play an important role in the synthesis of proanthocyanins and anthocyanins, which are downstream genes in the flavonoid biosynthesis pathway. It is known that leucocyanidin is the substrate of LAR and ANS genes, which could catalyze leucocyanidin into catechins and cyanins, respectively [45]. LAR gene expression is affected by temperature, light, and hormones [64], and ANS gene expression is closely linked to plant organ coloration [65, 66]. In our finding, LAR was highly expressed in cultivars ‘Fenhe’ and ‘Baihe’, resulting in the synthesis of more Catechin derivatives using leucocyanidin. ANR and GT were competing for substrate cyanins. The enzyme ANR catalyzes the production of epicatechin from cyanins, which is also affected by light, temperature, and hormones [64]. By catalyzing the glycosylation of anthocyanin, flavonoid 3-O-glucosyltransferase (GT) is an important enzyme in the last stage of anthocyanin synthesis [67]. Interestingly, there was no difference in ANR expression among the three cultivars, but cultivar 'Zihe' exhibited a higher concentration of epicatechin derivatives, suggesting that the use of cyanidin as a substrate to synthesize epicatechin derivatives resulted in accumulation of less red pigmentation.
Floral color and MYBs, AP2/ERFs transcription factors in R.pulchrum
MYB is one of the largest transcription factor families in plants. It mainly regulates the flavonoid biosynthesis via two pathways. Firstly, MYBs activate the expression of flavonoid synthesis genes by interacting with bHLHs and WD40s to form the MBW complex [68]. FaMYB5 may act as negative regulators of PA/ anthocyanin biosynthesis in strawberry [69]. MYB5 (Rhsim01G0174700) was highly expressed in cultivars 'Baihe' and 'Fenhe', suggesting that it may negatively regulate anthocyanin synthesis. Secondly, MYBs can regulate the expression of structural genes independently to influence flavonoid synthesis. As reported by Luo et al., FTMYB15 was transferred from Tartary Buckwheat to Arabidopsis thaliana, the pigment accumulation was found in the leaves and seed coat of the transgenic plant [70]. MYB15 (Rhsim04G0061700) was discovered with downregulation in cultivar ‘Zihe’. It is suggested that it may inhibit purple coloration in R. pulcherum. According to previous studies, MYB6 regulates flavonol biosynthesis through activating the promoters of F3H and FLS [71]. Based on our findings, MYB6 (Rhsim07G0002700) was highly expressed in 'Fenhe', and rutin content in 'Fenhe' was significantly higher than in 'Zihe'. It is indicated MYB6 was also involved in the pink coloration of petals through the regulation of flavonol synthesis. The MYB101 participated in pollen tube reception and leaf development [72,73,74]. According to our results, MYB101 (Rhsim06G0163100) was highly expressed in cultivar ‘Fenhe’, suggesting that MYB101 may be responsible for regulating flavonoids in R.pulchrum with pink petals.
UV-B can be used as a signal to regulate plant development, such as regulating photomorphogenesis and promoting the accumulation of flavonoids and anthocyanins [75,76,77]. MYB73 is an important transcription factor associated with plant hormones [78]. UVR8 interacts with MYB73/MYB77 (MYB DOMAIN PROTEIN 73/77) in a UV-B-dependent manner, it can affect plant growth and development through light and auxin signal [79]. When MYB73 (Rhsim05G0088000) was highly expressed in cultivar 'Zihe', MYB73 may contribute to the accumulation of flavonoids and anthocyanins in R.pulchrum by regulating the photomorphogenesis process. MYB61 (Rhsim11G0106400) was highly expressed in dark flowers, suggesting that it may regulate the coloration in R.pulchrum.
The AP2/ERF family is one of the largest transcription factors families in plants and regulate primary and secondary metabolism [80,81,82]. It has been shown that AP2/ERF can regulate the biosynthesis of anthocyanins in plants. MdMYB1 is a positive regulator of anthocyanin biosynthesis, and MdERF38 interacts with MdMYB1 to promote the binding of MdMYB1 to its target gene in the drought-induced conditions [83]. DREB subfamily, which belongs to AP2/ERF family, was only found to be highly expressed in cultivar 'Fenhe'. But further research is needed to confirm whether DREB can regulate flavonoid biosynthesis in pink petals by affecting the interactions of TFs under certain induction conditions. Rhsim07G0115900 and Rhsim03G0161100, which encode ERF5, were discovered in this study. Rhsim07G0115900 was lowly expressed in cultivar ‘Zihe’ when compared with ‘Fenhe’ and ‘Baihe’, while Rhsim03G0161100 was highly expressed in cultivar ‘Baihe’ when compared with ‘Fenhe’. It is suggested that ERF5 might be involved in regulation of flavonoid biosynthesis in R. pulcherum.
The FNS and IFR genes were identified in R.pulchrum
Flavone synthases had two different subtypes, Flavone synthase I (FNSI) and Flavone synthase II (FNSII). FNSI is a nonheme iron-dependent a-ketoglutarate dioxygenase, which is found in the cytosol [84]. As a member of the P450 family, FNSII is found in the inner membrane of cells. Both enzymes can convert naringin to apigenin, and they are not simultaneously in the same species. Researcher has reported that Medicago sativa and soybean contain only FNSII [71, 85]. Metabolome analysis revealed that the apigenin derivatives had a lower expression in cultivar 'Zihe' than in 'Baihe' and 'Fenhe’. It was found that Rhsim03G0216000, which encodes FNS, was lowly expressed in cultivar 'Zihe'. Sequence alignment results revealed that Rhsim03G0216000 had a great similarity (74%) to FNSI(AT5G24531.1). Based on these findings, it is likely that FNS is in the form of FNSI in R.pulchrum.
Isoflavone is a type of phytoestrogens, which have only been found in some species of Papilionoideae [86, 87]. The isoflavone reductase (IFR) catalyzes the conversion of isoflavones to Medicarpin, which is the first step in isoflavone decomposition [88, 89]. The IFR gene (Rhsim07G0136900) was highly expressed in cultivar 'Zihe', meanwhile metabolite of isoflavones (Medicarpin3-O-(6'-malonylglucoside)) highly enriched in cultivar 'Zihe'. Therefore, it is necessary to investigate whether the isoflavone derivatives cause the purple coloring of R.pulchrum. Medicalarpin is a natural phytoalexin of the pterocarpan subfamily. It has been found in many traditional Chinese medicines, such as Pueraria Lobata [90]and Hedysari Radix [91]. The studies have reported that it has various biologically benefits, including stimulation of bone regeneration, induction of apoptosis, and induction of lipolysis [92]. Interesting, many medicarpin derivatives have been found in cultivar 'Zihe' for the first time, suggesting that this cultivar is valuable for medicine research. It has been observed that a few purple spots are sometimes on the white petals of R.pulchrum, we discovered delphinidin and malvidin were accumulated in the petals. The blockage of glycosylation related genes in the flavonoid synthesis pathway may explain why white flowers accumulated some delphinidin and malvidin.
Conclusion
The transcriptome and metabolome analysis were used to identify genes, transcription factors, and metabolites associated with the coloration of R.pulchrum. For known color-related transcription factors, the DREB family were found to play a critical role in the formation of the pink color. Additionally, our results have shown that FNS and IFR were highly expressed in the flavonoid biosynthesis pathway, and that FNS exists in the form of FNSI in R.pulchrum. The IFR gene and its related metabolites of medicarpin derivatives were highly expressed in cultivar 'Zihe', indicating that IFR involved in the purple coloration of R.pulchrum. CHS, FLS, F3' H, F3 ′5 'H, DFR, GT and FAOMT genes were correlated with white and purple coloration, whereas F3' H and F3 ′5 'H up-regulated, 4CL, DFR, ANS and GT down-regulated were associated with pink coloration. We provided a model illustrating the color related key genes and metabolic and their regulatory mechanism of R. pulcherum. The study provided some basis for breeding new cultivars with different colors of petals.
Methods
Plant materials and treatment
The white-flowered R.pulchrum Sweet ('Baihe'), pink-flowered R.pulchrum Sweet ('Fenhe') and purple-flowered R.pulchrum Sweet ('Zihe') were grown at the Rhododendron resource center in the Shanghai Botanical Garden (31°15′ N, 121°45′ E), Shanghai, China (Fig. 12). After collecting the petals from ‘Baihe’,’Fenhe’ and’Zihe’, all materials were frozen in liquid nitrogen and stored at − 80 °C for RNA and metabolite extraction. In the transcriptome analysis of ‘Baihe’, ‘Fenhe’ and ‘Zihe’, for each color, we gathered three samples as biological replicates, then each sample randomly selected three blooms petals from the same plant and pooled into one for RNA-Seq. For metabolism analysis, we had made five biological replicates from five individual plant for each color.

Three different colors of Rhododendron pulchrum Sweet. Note: a. ‘Baihe’; b.‘Fenhe’; c.‘Zihe’
Sample preparation and metabolite extraction
Each color of petal was collected from five individual plants. 1 mL of methanol: water (7:3, v:v) was added to 80 mg samples. 2-chloro-l-phenylalanine (20μL, 0.3 mg / mL) was added as an internal standard. All samples were precooled at -20 °C for 2 min, pulverized with grinder at 60 HZ for 2 min, then ultrasonicated for 30 min and placed at -20 °C for 20 min. The samples were centrifuged for 10 min (13,000 rpm, 4 °C). The supernatants (150μL) from each tube were collected with crystal syringes, filtered through 0.22 μm microfilters, and injected into LC vials. Until the liquid chromatography–mass spectrometry (LC–MS) analysis was performed, the vials were stored at -80 °C. A pooled QC sample was prepared by mixing aliquots of all the samples. For the metabolic profiling, an ACQUITY UHPLC system (Waters Corporation, Milford, USA) in conjunction with an AB SCIEX Triple TOF 5600 System (AB SCIEX, Framingham, MA) was used. ACQUITY UPLC BEH C18 columns (1.7 μm, 2.1 × 100 mm) were used in both positive and negative mode. The binary gradient elution system consisted of (A) water (containing 0.1% formic acid, v/v) and (B) acetonitrile (containing 0.1% formic acid, v/v). The gradient program consisted of 0 min, 95% A; 2 min, 80% A; 4 min, 75% A; 9 min, 40% A; 17 min, 0% A; 19 min, 0% A; 19.1 min, 95% A; and 20.1 min, 95% A. The flow rate was 0.4 mL/min and the injection volume were 5μL. The temperature of the column was maintained at 45 °C. During the LC–MS analysis, samples were maintained in the instrument chamber at 4 °C.
Metabolite identification and quantification
The metabolome data was analyzed using Progenesis QI software (Nonlinear Dynamics, Newcastle, United Kingdom). Simca software package (version 14.0, Umetrics, Umeå, Sweden) was used to analyze the combined positive and negative data. Human Metabolome (http://www.hmdb.ca/), METLIN (http://metlin.scripps.edu) and LIPID MAPS (http://www.lipidmaps.org/) databases were used for metabolite identification. An orthogonal partial least squares discriminant analysis (OPLS-DA) was conducted to identify potential biomarker variables. Significantly different metabolites between groups were determined by variable importance in projection (VIP) ≥ 1 and an absolute Log2FC (foldchange) ≥ 1. A hierarchical clustering analysis was conducted with R (http://www.r-project.org/).
RNA-seq and annotation
For each sample, 1 g fully opened stage petals were used for extracting the RNA using the mirVana miRNA Isolation Kit (Ambion). RNA purity and quantification were assessed using a NanoDrop 2000 spectrophotometer (Thermo Scientific, USA). RNA integrity was evaluated using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Following the manufacturer's instructions, libraries were constructed using TruSeq Stranded mRNA LTSample Prep Kit (Illumina, San Diego, CA, USA). Afterwards, these libraries were sequenced on the Illumina sequencing platform (Illumina HiSeq X Ten) and 150 bp paired-end reads were obtained. Raw reads were first processed using Trimmomatic [93], and low-quality reads and adaptors were filtered out. HISAT2 [94] was used to map the remaining reads to the reference genome. The differential expression analysis was carried out using the DESeq R package [95]. According to the DESeq analysis, genes with adjusted P-values of < 0.05 were significantly differentially expressed. A hierarchical cluster analysis of DEGs was performed to analyze gene expression patterns. GO enrichment and KEGG [96] pathway enrichment analysis of DEGs were performed with R based on the hypergeometric tests. The annotation of gene functions was carried out using the following databases: Kyoto Encyclopedia of Genes and Genomes (KEGG), eukaryotic Clusters of Orthologous Groups (KOG), NCBI non-redundant (NR), Swiss PROT sequence protein, Gene Ontology (GO) and the homologous protein family (Pfam). Three biological replicates of each sample were performed.
RT-qPCR verification
A total of nine structural genes related to flavonoid biosynthesis were selected for validation by quantitative real-time PCR (RT-qPCR). Total RNA was extracted with Total RNA Kit (Ambion).The RNA was treated with DNase I(Invitrogen) to remove trace genomic DNA, then reverse-transcribed in a GeneAmp® PCR System 9700 (Applied Biosystems, USA).The specific primers were designed in the laboratory and synthesized by TsingKe Biotech (Supplementary Table S6). RT-qPCR was performed using a LightCycler® 480 II Real-time PCR with Instrument (Roche, Swiss).The PCR amplification was 94℃ for 30 s, followed by 45 cycles of 94℃ for 5 s, 60℃ for 30 s. The expression levels of mRNAs were normalized to GAPDH and were calculated using the 2−ΔΔCt method [97]. The primers of RT-qPCR are showed in Table S6.
Metabolites-transcripts correlation and weighted gene coexpression network analysis
The Pearson correlation algorithm was used to calculate the correlation between genes and metabolites. DEGs and DEMs have been mapped simultaneously to the KEGG Pathway database to obtain information on their pathways. The correlation analysis was performed by R 3.5.1 using Pearson correlation analysis. Coexpression networks was performed with WGCNA analysis [51].
Availability of data and materials
All relevant supporting data sets are included in the article and its supplemental files.
Abbreviations
- PAL:
-
Phenylalanine ammonia-lyase
- 4CL:
-
4-Coumarate:coenzyme A ligase
- CHS:
-
Chalcone synthase
- CHI:
-
Chalcone isomerase
- F3H:
-
Flavanone 3-hydroxylase
- FLS:
-
Flavonol synthase
- F3′H:
-
Flavanone 3′-hydroxylase
- F3′5’H:
-
Flavonoid 3′, 5′- hydroxylase
- DFR:
-
Dihydroflavonol 4-reductase
- ANS:
-
Anthocyanidin synthase
- LAR:
-
Leucoanthocyanidin reductase
- ANR:
-
Anthocyanidin reductase
- GT:
-
Glucosyltransferase
- Violdelphin-GT:
-
Viodelphin-glucosyltransferases
- F30GT:
-
Flavonol-3-O-glucosyl transferase
- COG:
-
Clusters of orthologous groups of proteins database
- DEG:
-
Differentially expressed gene
- DEM:
-
Differentially expressed metabolite
- FPKM:
-
Fragments per kilobase of exon model per million mapped reads
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- MYB:
-
V-myb avian myeloblastosis viral oncogene homolog
- bHLH:
-
Basic helix-loop-helix
- RNA-Seq:
-
RNA sequencing
- RT-qPCR:
-
Real-time quantitative polymerase chain reaction
- NGS:
-
Next generation sequencing
- HCA:
-
Hierarchical cluster analysis
- LC–MS:
-
Liquid chromatography–mass spectrometry
- WGCNA:
-
Weighted correlation network analysis
References
Yoshida K, Toyama-Kato Y, Kameda K, Kondo T. Sepal color variation of Hydrangea macrophylla and vacuolar pH measured with a proton-selective microelectrode. Plant Cell Physiol. 2003;44:262–8.
Liu L, Zhang LY, Wang SL, Niu XY. Analysis of anthocyanins and flavonols in petals of 10 Rhododendron species from the Sygera Mountains in Southeast Tibet. Plant Physiol Biochem. 2016;104:250–6.
Hang NTT, Miyajima I, Ureshino K, Masuda JI, Okubo H. Comparison of morphological characteristics of Rhododendron simsii. Planch distributed in Vietnam and Japan. J Faculty of Agriculture Kyushu Univ. 2010;55(2):233–7.
Dong XK, Chen W, Wang WS, Zhang HY, Liu XQ, Luo J. Comprehensive profiling and natural variation of flavonoids in rice. J Integr Plant Biol. 2014;56:876–86.
Jiang T, Zhang MD, Wen CX, Xie XL, Tian W, Wen SQ, Lu RK, Liu LD. Integrated metabolomic and transcriptomic analysis of the anthocyanin regulatory networks in Salvia miltiorrhiza Bge. flowers. BMC Plant Biol. 2020;20(1):349.
Luo XN, Sun DY, Wang S, Luo S, Fu YQ, Niu LX, Shi QQ, Zhang YL. Integrating full-length transcriptomics and metabolomics reveals the regulatory mechanisms underlying yellow pigmentation in tree peony (Paeonia suffruticosa Andr.) flowers. Horticulture Res. 2021;8:235.
Silva FM, Bailon MTE, Alonso JJP, Gonzalo JCR. Anthocyanin pigments in strawberry. LWT-Food Sci Technol. 2007;40:374–82.
Diaconeasa Z, Stirbu I, Xiao JB, Leopold N, Ayvaz Z, Danciu C, Ayvaz H, Stǎnilǎ A, Nistor M, Socaciu C. Anthocyanins, vibrant color pigments, and their role in skin cancer prevention. Biomed. 2020;8(9):336.
Yang FS, Nie S, Liu H, Shi TL, Tian XC, Zhou SS, Bao YT, Jia KH, Guo JF, Zhao W, An N, Zhang RG, Yun QZ, Wang XZ, Mannapperuma C, Porth I, El-Kassaby YA, Street NR, Wang XR, Van de Peer Y, Mao JF. Chromosome-level genome assembly of a parent species of widely cultivated azaleas. Nature commun. 2020;11(1):5269.
Chen J, Wang ZY. Progress in the study of plant MYB transcription factors. J Plant Physiol Mol Biol. 2002;28(2):81–8.
Alexander RCDK, Leon AM, Pieter DL, Joseph NMM, Antoine RS. Inhibition of flower pigmentation by antisense CHS genes: promoter and minimal sequence requirements for the antisense effect. Plant Mol Biol. 1990;14(4):457–66.
Wang YJ, Li HH, Fu WY, Gao Y, Wang BJ, Li L. Flavonoids contents and expression analysis of related genes in red cell line of Saussurea medusa. Sheng Wu Gong Cheng Xue Bao (in Chinese). 2014;30(8):1225–34.
Ben-Simhon Z, Judeinstein S, Trainin T, Harel-Beja R, Bar-Ya’akov I, Borochov-Neori H, Holland D. A “White” anthocyanin-less pomegranate (Punica granatum L.) caused by an insertion in the coding region of the leucoanthocyanidin dioxygenase (LDOX; ANS) gene. PloS one. 2015;10(11):e0142777.
Yang yun. Metabolomes and transcriptomes analysis on leaves in red color of Eucommia ulmoides ‘Huazhong No. 12.’ Henan: Henan agricultural university; 2019.
Ben-Simhon Z, Judeinstein S, Nadler-Hassar T, Trainin T, Bar-Ya’akov I, Borochov-Neori H, Holland D. A pomegranate (Punica granatum L.) WD40-repeat gene is a functional homologue of Arabidopsis TTG1 and is involved in the regulation of anthocyanin biosynthesis during pomegranate fruit development. Planta. 2011;234(5):865–81.
Li M, Cao YT, Ye SR, Irshad M, Pan TF, Qiu DL. Isolation of CHS gene from brunfelsia acuminata flowers and its regulation in anthocyanin biosysthesis. Molecules. 2016;22(1):44.
Li L, Zhai YH, Luo XN, Zhang Y, Shi QQ. Comparative transcriptome analyses reveal genes related to pigmentation in the petals of red and white Primula vulgaris Cultivars. Physiol Mol Biol Plants. 2019;25(4):1029–41.
Zhou Z, Ying Z, Wu ZG, Yang YP, Fu SB, Xu W, Yao LJ, Zeng AP, Huang J, Lan SR, Wang XL, Liu ZJ. Anthocyanin Genes Involved in the Flower Coloration Mechanisms of Cymbidium kanran. Front Plant Sci. 2021;12:737815.
Zhou H, Wang KL, Wang HL, Gu C, Dare AP, Espley RV, He HP, Allan AC, Han YP. Molecular genetics of blood-fleshed peach reveals activation of anthocyanin biosynthesis by NAC transcription factors. Plant J. 2015;82(1):105–21.
Yin XJ, Zhang YB, Zhang L, Wang BH, Zhao YD, Irfan M, Chen LJ, Feng YL. Regulation of MYB transcription factors of anthocyanin synthesis in lily Flowers. Front Plant Sci. 2021;12:761668.
Wang SZ, Zhang YJ, Huang SY, Luo YY, Jin ZQ, Liang LZ, Jin WB. Development and application of SSR markers based on buds transcriptomic data of Rhododendron pulchrum Planch. For Res. 2019;32(3):97–104.
Wang Yu, Zhang GH, Hee JS, Xu SS, Liu XY, Ma CH, Zhang JL. Research progress of Rhododendron flower color. World Forestry Research. 2020;33(5):19–24 (in Chinese).
Du H, Lai L, Wang F, Sun W, Zhang L, Li X, Wang L, Jiang L, Zheng Y. Characterisation of flower colouration in 30 Rhododendron species via anthocyanin and flavonol identification and quantitative traits. Plant Biol. 2018;20(1):121–9.
Mizuta D, Ban T, Miyajima I, Nakatsuka A, Kobayashi N. Comparison of flower color with anthocyanin composition patterns in evergreen azalea. Sci Hortic. 2009;122(4):594–602.
Miyajima I, Ureshino K, Kobayashi N, Akabane M. Flower color and pigments of intersubgeneric hybrid between white-flowered evergreen and yellow-flowered deciduous azaleas. J Japanese Soc Horticultural Sci. 2002;69:280–2.
Tanaka Y, Sasaki N, Ohmiya A. Biosynthesis of plant pigments: anthocyanins, betalains and carotenoids. Plant J. 2008;54:733–49.
Heursel JB. Diversity of flower colours in Rhododendron simsii Planch. and prospects for breeding. Euphytica. 1981;30(1):9–14.
Harborne JB. Plant polyphenols-XV, flavonols as yellow flower pigments. Phytochemistry. 1965;4(5):647–57.
Ye LJ, Mӧller M, Luo YH, Zou JY, Zheng W, Wang YH, Liu J, Zhu AD, Hu JY, Li DZ, Gao LM. Differential expressions of anthocyanin synthesis genes underlie flower color divergence in a sympatric Rhododendron sanguineum complex. BMC Plant Biol. 2021;21(1):204.
Zhou GL, Zhu P. De novo transcriptome sequencing of Rhododendron molle and identification of genes involved in the biosynthesis of secondary metabolites. BMC Plant Biol. 2020;20(1):414.
Maitre NCL, Pirie MD, Bellstedt DU. An approach to determining anthocyanin synthesis enzyme gene expression in an evolutionary context: an example from Erica plukenetii. Ann Bot. 2019;124(1):121–30.
Zhu CY, Li ZY, Bao CS, Xu YQ. Conservation and Utilization of Rich Rhododendron Germplasm Resources in China. Chinese Wild Plant Resources. 2007;26(2):28–30 (in chinese).
Tang YJ, Wu XY. Photosynthesis characteristics of Rhododendron pulchrum leaves on full flowering stage. Northern Horticulture. 2010;21:68–71.
Qian DW, Chen XM, Li M, Qiu DL. Content analysis of anthocyanin pigment and total phenols in the petals of Rhododendron pulchrum Sweet. Subtropical Agriculture Res. 2020;16(1):29–34 (in chinese).
Wang SZ, Huang SY, Yang J, Li ZL, Zhang MJ, Fang YP, Yang QF, Jin WB. Metabolite profiling of violet, white and pink flowers revealing flavonoids composition patterns in Rhododendron pulchrum Sweet. J Biosci. 2021;46:3.
Wang SZ, Li ZL, Jin WB, Fang YP, Yang QF, Xiang J. Transcriptome analysis and identification of genes associated with flower development in Rhododendron pulchrum Sweet(Ericaceae). Gene. 2018;679(30):108–18.
Wu Q, Wu J, Li SS, Zhang HJ, Feng CY, Yin DD, Wu RY, Wang LS. Transcriptome sequencing and metabolite analysis for revealing the blue flower formation in waterlily. BMC Genomics. 2016;17(1):897.
Lu JJ, Zhang Q, Lang LX, Jiang C, Wang XF, Sun HM. Integrated metabolome and transcriptome analysis of the anthocyanin biosynthetic pathway in relation to color mutation in miniature roses. BMC Plant Biol. 2021;21(1):257.
Chen WC, Zhang JQ, Zheng S, Wang ZQ, Xu CM, Zhang QX, Wu JS, Lou HQ. Metabolite profiling and transcriptome analyses reveal novel regulatory mechanisms of melatonin biosynthesis in hickory. Horticulture Research. 2021;8(1):196.
Wang J, Lv JH, Liu ZB, Liu YH, Song JS, Ma YQ, Ou LJ, Zhang XL, Liang CL, Wang F, Juntawong N, Jiao CH, Chen WC, Zou XX. Integration of Transcriptomics and Metabolomics for Pepper (Capsicum annuum L.) in Response to Heat Stress. Int J Mol Sci. 2019;20(20):5042.
Wang ZR, Song MY, Wang Z, Chen SW, Ma HQ. Metabolome and transcriptome analysis of flavor components and flavonoid biosynthesis in fig female flower tissues (Ficus carica L.) after bagging. BMC Plant Biol. 2021;21(1):396.
Fossen T, Rayyan S, Holmberg MH, Nateland HS, Andersen ØM. Acylated anthocyanins from leaves of Oxalis triangularis. Phytochemistry. 2005;66(10):1133–40.
Kawano K, Sakamura S. Cyanidin 3-[O-β-D-Xylopyranosyl(1→2)-β-D-galactopyranoside] from Aralia elata and Aralia cordata. Agric Biol Chem. 1972;36(1):27–32.
Winkel-Shirley B. Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol. 2001;126(2):485–93.
Springob K, Nakajima J, Yamazaki M, Saito K. Recent advances in the biosynthesis and accumulation of anthocyanins. Nat Prod Rep. 2003;20(3):288–303.
Liu QL, Liaquat F, He YF, Munis MFH, Zhang CY. Functional annotation of a full-length transcriptome and identification of genes associated with flower development in Rhododendron simsii (Ericaceae). Plants. 2021;10(4):649.
Sun W, Meng XY, Liang LJ, Jiang WS, Huang YF, He J, Hu HY, Almqvist J, Gao X, Wang L. Molecular and biochemical analysis of chalcone synthase from Freesia hybrid in flavonoid biosynthetic pathway. PLoS ONE. 2015;10(3):e0119054.
Trojan V, Musilová M, Vyhnánek T, Klejdus B, Hanáček P, Havel L. Chalcone synthase expression and pigments deposition in wheat with purple and blue colored caryopsis. J Cereal Sci. 2014;59:48–55.
Kong LC, Tap J, Aron-Wisnewsky J, Pelloux V, Basdevant A, Bouillot JL, Zucker JD, Doré J, Clément K. Gut microbiota after gastric bypass in human obesity: increased richness and associations of bacterial genera with adipose tissue genes. Am J Clin Nutr. 2013;98(1):16–24.
Schwab C, Berry D, Rauch I, Rennisch I, Ramesmayer J, Hainzl E, Heider S, Decker T, Kenner L, Müller M, Strobl B, Wagner M, Schleper C, Loy A, Urich T. Longitudinal study of murine microbiota activity and interactions with the host during acute inflammation and recovery. ISME J. 2014;8(5):1101–14.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559.
Hu YS. Molecular cloning and characterization of five key genes involved in lignan biosynthesis pathway from Isatis indigotica Fort. Shanghai: The Second Military Medical University; 2010. (in chinese).
Li L, Wang XB, Yang L, Kong LY. Quantitative evaluation of 4-Coumarate: CoA Ligase(4CL) activity and correlated chemical constituents in four plant materials by chromatographic analysis. Chin J Nat Med. 2010;8(4):274–9.
Ehlting J, Shin JJK, Douglas CJ. Identification of 4- coumarate: coenzyme A ligase (4CL) substrate recognition domains. Plant J. 2001;27(5):455–65.
Niesbach KU, Brazen E, Bernhardt J, Rohde W, Schwarz SZ, Reif HJ, Wienand U, Saedler H. Chalcone synthase genes in plants: a tool to study evolutionary relationships. J Mol Evol. 1987;26:213–25.
Takos AM, Jaffé FW, Jacob SR, Bogs J, Robinson SP, Walker AR. Light induced expression of a MYB gene regulates anthocyanin biosynthesis in red apples. Plant Physiol. 2006;142(3):1216–32.
Takahashi R, Githiri SM, Hatayama K, Dubouzet EG, Shimada N, Aoki T, Ayabe S, Iwashina T, Toda K, Matsumura H. A single-base deletion in soybean flavonol synthase gene is associated with magenta flower color. Plant Mol Biol. 2007;63(1):125–35.
Nielsen K, Deroles SC, Markham KR, Bradley MJ, Podivinsky E, Manson D. Antisense flavonol synthase alters copigmentation and flower color in lisianthus. Mol Breeding. 2002;9(4):217–29.
Lin QF, Liu TT, Liu JR, Cai M, Cheng TR, Wang J, Zhang QX, Pan HT. Flavonoids composition and content in petals of Lagerstroemia and Heimia species and cultivars. Acta Horticulturae Sinica. 2021;48(10):1956–68.
Lou Q, Liu YL, Qi YY, Jiao SZ, Tian FF, Jiang L, Wang YJ. Transcriptome sequencing and metabolite analysis reveals the role of delphinidin metabolism in flower colour in grape hyacinth. J Exp Bot. 2014;65(12):3157–64.
Xu F, Li LL, Zhang WW, Cheng H, Sun NN, Cheng SY, Wang Y. Isolation, characterization, and function analysis of a flavonol synthase gene from Ginkgo biloba. Mol Biol Rep. 2012;39(3):2285–96.
Henry-Kirk RA, Plunkett B, Hall M, Mcghie T, Allan AC, Wargent JJ, Espley RV. Solar UV light regulates flavonoid metabolism in apple (Malus × domestica). Plant Cell Environ. 2018;41(3):675–88.
Seitz C, Ameres S, Forkmann G. Identification of the molecular basis for the functional difference between flavonoid 3’-hydroxylase and flavonoid 3’,5’-hydroxylase. FEBS Lett. 2007;581(18):3429–34.
Song Y, Liu HD, Wang HB, Zhang HJ, Liu FZ. Molecular cloning and functional identification of proanthocyanidin synthesis related genes VcLAR and VcANR of blueberry. Jiangsu J Agricultural Sci. 2019;35(3):682–8 (in chinese).
Gong Z, Yamazaki M, Sugiyama M, Tanaka Y, Saito K. Cloning and molecular analysis of structural genes involved in anthocyanin biosynthesis and expressed in a form-specific manner in Perilla frutescens. Plant Mol Biol. 1997;35(6):915–27.
Rosati C, Cadic A, Duron M, Ingouff M, Simoneau P. Molecular characterization of the anthocyanin synthesis gene in Forsythia × intermedia reveals organ-specific expression during flower development. Plant Sci. 1999;149:73–9.
Terrier N, Torregrosa L, Ageorges A, Vialet S, Verriès C, Cheynier V, Romieu C. Ectopic expression of VvMybPA2 promotes proanthocyanidin biosynthesis in grapevine and suggests additional targets in the pathway. Plant Physiol. 2009;149(2):1028–41.
Jochen B, Jaff FW, Takos AM, Walker AR, Robinson SP. The grapevine transcription factor VvMYBPA1 regulates proanthocyanidin synthesis during fruit development. Plant Physiol. 2007;143(3):1347–61.
Schaart JG, Dubos C, Romero De La Fuente I, van Houwelingen AMML, de Vos RCH, Jonker HH, Xu W, Routaboul JM, Lepiniec L, Bovy AG. Identification and characterization of MYB-bHLH-WD40 regulatory complexes controlling proanthocyanidin biosynthesis in strawberry(Fragaria × ananassa) fruits. New Phytologist. 2013;197(2):454–67.
Luo XP, Zhao HX, Yao PF, Li QQ, Huang YJ, Li CL, Chen H, Wu Q. An R2R3-MYB Transcription Factor FtMYB15 Involved in the Synthesis of Anthocyanin and Proanthocyanidins from Tartary Buckwheat. J Plant Growth Regul. 2018;37:76–84.
Yan JH, Wang B, Jiang YN, Cheng LJ, Wu TL. GmFNSII-controlled soybean flavone metabolism responds to abiotic stresses and regulates plant salt tolerance. Plant Cell Physiol. 2014;55(1):74–86.
Liang Y, Tan ZM, Zhu L, Niu QK, Zhou JJ, Li M, Chen LQ, Zhang XQ, Ye D. MYB97, MYB101 and MYB120 Function as Male Factors That Control Pollen Tube-Synergid Interaction in Arabidopsis thaliana Fertilization. PLos Genet. 2013;9(11):e1003933.
An R, Liu XY, Wang R, Wu HC, Liang S, Shao JX, Qi YF, An LJ, Yu F. The over-expression of two transcription factors, ABS5/bHLH30 and ABS7/MYB101, leads to upwardly curly leaves. PLoS ONE. 2014;9(9):e107637.
Hou HL, Zhang CW, Hou XL. Cloning and Functional Analysis of BcMYB101 Gene Involved in Leaf Development in Pak Choi (Brassica rapa ssp. Chinensis). Int J Mol Sci. 2020;21(8):2750.
Liang T, Mei SL, Shi C, Yang Y, Peng Y, Ma LB, Wang F, Li X, Huang X, Yin YH, Liu HT. UVR8 interacts with BES1 and BIM1 to regulate transcription and photomorphogenesis in Arabidopsis. Dev Cell. 2018;44(4):512–23.
Yang Y, Liang T, Zhang LB, Shao K, Gu XX, Shang RX, Shi N, Li X, Zhang P, Liu HT. UVR8 interacts with WRKY36 to regulate HY5 transcription and hypocotyl elongation in Arabidopsis. Nature Plants. 2018;4(2):98–107.
Wan JP, Zhang P, Wang RL, Sun LL, Wang WY, Zhou HK, Xu J. UV-B radiation induces root bending through the flavonoid-mediated auxin pathway in Arabidopsis. Front Plant Sci. 2018;9:618.
Jia J, Xing JH, Dong JG, Han JM, Liu JS. Functional analysis of MYB73 of Arabidopsis thaliana against Bipolaris oryzae. Agric Sci Chin. 2011;10:721–7.
Yang Y, Zhang LB, Chen P, Liang T, Li X, Liu HT. UVR8 interacts with MYB73/MYB77 in a UV-B-dependent manner, regulating auxin responses and lateral root development. EMBO J. 2020;39(2):e101928.
Zhuang J, Chen JM, Yao QH, Xiong F, Sun CC, Zhou XR, Zhang J, Xiong AS. Discovery and expression profile analysis of AP2/ERF family genes from Triticum aestivum. Mol Biol Rep. 2011;38(2):745–53.
Okamuro JK, Caster B, Villarroel R, Van Montagu M, Jofuku KD. The AP2 domain of APETALA2 defines a large new family of DNA binding proteins in Arabidopsis. Proc Natl Acad Sci USA. 1997;94(13):7076–81.
Feng K, Hou XL, Xing GM, Liu JX, Duan AQ, Xu ZS, Li MY, Zhuang J, Xiong AS. Advances in AP2/ERF super-family transcription factors in plant. Crit Rev Biotechnol. 2020;40(6):750–76.
An JP, Zhang XW, Bi SQ, You CX, Wang XF, Hao YJ. The ERF transcription factor MdERF38 promotes drought stress-induced anthocyanin biosynthesis in apple. Plant J. 2020;101(3):573–89.
Britsch L, Heller W, Grisebach H. Conversion of flavanone to flavone, dihydroflavonol and flavonol with an enzyme-system from cell cultures of parsley. Zeitschrift Fur Naturforschung C. 1981;36:742–50.
Zhang J, Subramanian S, Zhang YS, Yu O. Flavone synthases from Medicago truncatula are flavanone-2-hydroxylases and are important for nodulation. Plant Physiol. 2007;144(2):741–51.
Xu BJ, Chang SKC. Characterization of phenolic substances and antioxidant properties of food soybeans grown in North Dakota-Minnesota Region. J Agric Food Chem. 2008;56(19):9102–13.
Tsukamoto C, Shimada S, Igita K, Kudou S, Kokubun M, Okubo K, Kitamura K. Factors affecting isoflavone content in soybean seeds: Changes in isoflavones, saponins and composition of fatty acids at different temperarure during seed development. J Agricultural Food Chem. 1995;43(5):1184–92.
Chiari L, Piovesan ND, Naoe LK, Jose IC, Viana JMS, Moreira MA, Barros EG. Genetic parameters relating isoflavone and protein content in soybean seeds. Euphytica. 2004;138(1):55–60.
Dixon RA, Steele CL. Flavonoids and isoflavonoids-a gold mine for metabolic engineering. Trends Plant Sci. 1999;4(10):394–400.
Kim JM, Lee YM, Lee GY, Jang DS, Bae KH, Kim JS. Constituents of the roots of Pueraria lobata inhibit formation of advanced glycation end products (AGEs). Arch Pharm Res. 2006;29(10):821–5.
Liu Y, Chen HB, Zhao YY, Wang B, Zhang QY, Zhang L, Tu PF. Quantification and stability studies on the flavonoids of Radix hedysari. J Agric Food Chem. 2006;54(18):6634–9.
Imran KM, Yoon D, Lee TJ, Kim YS. Medicarpin induces lipolysis via activation of Protein Kinase A in brown adipocytes. BMB Rep. 2018;51(5):249–54.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–60.
Anders S, Huber W. Differential expression of RNA-Seq data at the gene level- the DESeq package. Heidelberg, Germany: EMBL; 2013.
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:D480–4.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–8.
Acknowledgements
We thank the staff of Shanghai Oe Biotechnology Co., Ltd. (Shanghai, China), for their support during the metabolite and transcriptome data analysis.
Material statements
The white-flowered R.pulchrum Sweet('Baihe'), pink-flowered R.pulchrum Sweet('Fenhe') and purple-flowered R.pulchrum Sweet('Zihe') were grown at the Rhododendron resource center at the Shanghai Botanical Garden (31°15′ N, 121°45′ E), which is an open Rhododendron resources. The plant materials are cultivated species, not wild or endangered plants. Experiment compied with the IUCN Policy Statement on Research Involving Species at Risk of Extinction and the Convention on the Trade in Endangered Species of Wild Faunaand Flora. Experimental research, including the collection of plant materials, comply with relevant institutional, national, and international guidelines and legislation. All plant materials belong to Shanghai Botanical Garden, and the use of these materials was approved by the Resource and Safety Committee of Shanghai Botanical Garden. Binjie Ge identified the voucher specimens, which were stored at the Shanghai Chenshan Botanical Garden (CSH0080557 and CSH0080665).The samples could be available by contacting with Xi Xia (xiaxi@shbg.org).
Funding
This work was supported by the scientific and technological project of Shanghai science and technology commission [20392000700], the funder provided fees for transcriptome and metabolomics testing, test verification and paper publication fees, etc.
Author information
Authors and Affiliations
Contributions
CZ and XX designed the experiments and analyzed the results. XX prepared the manuscript. CZ and RG revised the manuscript. All authors have read and approved the manuscript for publication.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable. The experiments did not involve endangered or protected species. No specific permits were required for these activities, the Rhododendron used in this study were obtained from Shanghai botanical garden.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Volcano plot of differential metabolits in R.pulchrum Sweet.(a) volcano plot of differential metabolits between cultivars ‘Baihe’ and ‘Fenhe’.(b)volcano plot of differential metabolits between cultivars ‘Zihe’ and ‘Baihe’.(c)volcano plot of differential metabolits between cultivars ‘Fenhe’ and ‘Zihe’.
Additional file 2: Fig. S2.
Hierarchical cluster analysis of differential flavonoid metabolites in R.pulchrum Sweet. Note: White, cultivar ‘Baihe’; Pink, cultivar ‘Fenhe’; Purple, cultivar ‘Zihe’. (a)Analysis of flavonoid metabolites between cultivars ‘Baihe’ and ‘Fenhe’;(b) Analysis of flavonoid metabolites between cultivars ‘Zihe’ and ‘Baihe’; (c) Analysis of flavonoid metabolites between cultivars ‘Fenhe’ and ‘Zihe’.
Additional file 3: Fig. S3.
Transcription factor distribution between three R.pulchrum Sweet cultivars. Note: BMJ, cultivar ‘Baihe’; FMJ, cultivar ‘Fenhe’; ZMJ, cultivar ‘Zihe’. (a) Transcription factor distribution between cultivars ‘Baihe’ and ‘Fenhe’; (b) Transcription factor distribution between cultivars ‘Zihe’ and ‘Baihe’; (c) Transcription factor distribution between cultivars ‘Fenhe’ and ‘Zihe’.
Additional file 4: Fig. S4.
Transcript accumulation measurements of colour-related genes involved in the flavonoid metabolic process. Note: BMJ, cultivar ‘Baihe’; FMJ, cultivar ‘Fenhe’; ZMJ, cultiv.
Additional file 5: Table S1.
Flavonoid metabolites between three comparison groups of R.pulchrum Sweet.
Additional file 6: Table S2.
The anthocyanin derivatives between three comparison groups of R.pulchrum Sweet.
Additional file 7: Table S3.
Transcriptome sequencing results from three R.pulchrum Sweet cultivars.
Additional file 8: Table S4.
Enrichment of TOP20 pathways in R.pulchrum Sweet based on KEGG analysis.
Additional file 9: Table S5.
Association analysis of DEGs and DEMs in R.pulchrum Sweet.
Additional file 10: Table S6.
The primers used for qRT-PCR analysis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xia, X., Gong, R. & Zhang, C. Integrative analysis of transcriptome and metabolome reveals flavonoid biosynthesis regulation in Rhododendron pulchrum petals. BMC Plant Biol 22, 401 (2022). https://doi.org/10.1186/s12870-022-03762-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-022-03762-y