
Abstract
Background
Filamentation temperature-sensitive H (FtsH) is an ATP-dependent zinc metalloprotease with ATPase activity, proteolysis activity and molecular chaperone-like activity. For now, a total of nine FtsH proteins have been encoded in rice, but their functions have not revealed in detail. In order to investigate the molecular mechanism of OsFtsH2 here, several osftsh2 knockout mutants were successfully generated by the CRISPR/Cas9 gene editing technology.
Results
All the mutants exhibited a phenotype of striking albino leaf and could not survive through the stage of three leaves. OsFtsH2 was located in the chloroplast and preferentially expressed in green tissues. In addition, osftsh2 mutants could not form normal chloroplasts and had lost photosynthetic autotrophic capacity. RNA sequencing analysis indicated that many biological processes such as photosynthesis-related pathways and plant hormone signal transduction were significantly affected in osftsh2 mutants.
Conclusions
Overall, the results suggested OsFtsH2 to be essential for chloroplast development in rice.
Similar content being viewed by others

Background
As the sites of photosynthesis to take place in plants, chloroplasts are unique organelles to capture and transform light energy into chemical energy, and thus provide energy for plant growth and development [1]. Also chloroplasts are responsible for the biosynthesis of various metabolites including tetrapyrroles, amino acids, lipids, terpenoids, hormones, etc. [2]. As a semi-autonomous organelle, chloroplast has its own DNA genome and protein synthesis machinery, but only a part of the proteins can be synthesized in chloroplast, and most proteins are synthesized on ribosomes of cytoplasm [3]. The development of chloroplast is a complex process modulated by both the plastid and nuclear genes, which can be divided into three steps [4]. The first step is to activate replication and DNA synthesis of plastid. The second step is the “build up” of chloroplast, characterized by the establishment of chloroplast genetic system, in which plastid genes to encode the gene expression machineries are preferentially transcribed by NEP (nuclear encoded plastid RNA polymerase), and the transcription / translation activity in chloroplast is strikingly increased [5]. In the third step, the plastid and nuclear genes encode photosynthetic apparatus are massively expressed, the plastid genes are principally transcribed by PEP (plastid-encoded RNA polymerase) [6], and expression of these genes results in chloroplast biosynthesis and assembly. Up to now, however, the molecular mechanism to regulate the development of chloroplasts in higher plants remain largely unknown [7].
Filamentation temperature-sensitive H (FtsH) is an ATP-dependent zinc metalloprotease, which exists widely in eukaryotes (mitochondria and chloroplasts) and prokaryotes [8, 9]. It is a member of the AAA (ATPase associated with diverse cellular activities) protein family, and possesses ATPase activity, proteolysis activity and molecular chaperone-like activity [10]. In general, FtsH protein contains tow transmembrane α -helices at the N terminus, which anchor the protein to the membrance of thylakoid or mitochondria. The C terminus of FtsH protein is located in the cytoplasm, consisting of an AAA-type domain and a M41-like endoprotease domain. FtsH was firstly found in Escherichia coli as an essential gene, which mediated various processes including the exporting of proteins from cell, membrane modeling, protein quality control to resist colicin and mRNA decay [11,12,13]. While FtsH is encoded by only a single gene in most prokaryotes genomes, multiple isoforms are identified in algae, cyanobacteria and plants [14]. It has been proposed that the multiplication of FtsH genes is related to the evolution of oxygenic photosynthesis and this trend is maintained in higher plants [15]. For example, four homologous genes of FtsH have been found in the cyanobacterium, Synechocystis sp. PCC 6803 [16], and among them, slr0228 was reported to play vital roles in the degradation of the photodamaged D1 proteins and removing the unassembled PSII subunits from the thylakoid membrane [17, 18]. In the soybean genome, a total of 11 FtsH genes have been identified, in which GmFtsH9 could be involved in regulating PSII function [19].
A total of 12 FtsH proteins have been encoded in Arabidopsis thaliana. Among them, three members (AtFtsH3, AtFtsH4, and AtFtsH10) are targeted to mitochondria and eight members (AtFtsH1, AtFtsH2, AtFtsH5 to AtFtsH9, and AtFtsH12) to chloroplasts, while AtFtsH11 appears to be dual-targeted to both mitochondria and chloroplasts [20, 21]. Under normal conditions of growth, AtFtsH2 (also termed VAR2) is the most abundantly expressed gene, followed by AtFtsH5 (also termed VAR1), AtFtsH8 and AtFtsH1, and the others are expressed at very low levels [22]. In addition, chloroplastic FtsHs predominantly form a hetero-hexameric complex comprising at least two types of isomers, type A (AtFtsH1/5) and type B (AtFtsH2/8) which are functionally distinguishable from each other [23, 24]. Both AtFtsH1/AtFtsH5 and AtFtsH2/AtFtsH8 proteases have been confirmed to participate in degradation of the photodamaged PSII D1 protein [25,26,27] and unassembled thylakoid membrane proteins [28, 29]. Disruption of AtFtsH2 results in a severe leaf variegation phenotype (var2) and disruption of AtFtsH5 results in a weak leaf variegation phenotype (var1), but AtFtsH1 and AtFtsH8 mutants have no visible phenotypes [21, 30,31,32]. Meanwhile, the double mutants of Atftsh1Atftsh5 and Atftsh2Atftsh8 show an albino-like phenotype, suggesting that each subunit is required for chloroplast biogenesis [28, 33]. The mitochondrial AtFtsh3, AtFtsh4 and AtFtsh10 play crucial roles in the assembly/stabilization of the mitochondrial complexes, and the activities of mitochondrial complexes I and V are significantly reduced in these mutants [34]. Moreover, those mutants lacking of AtFtsH4 show severe abnormal development of late rosette leaves, accompanied by ultrastructural impairment in chloroplasts and mitochondria [35]; AtFtsH6 will contribute to the degradation of the light-harvesting complex of PSII under conditions of high light and senescence [36], and can also restrict the thermomemory by regulating HSP21 protein abundance [37]. Furthermore, the mutation of AtFtsH11 causes a significant decrease in photosynthetic capability when environmental temperature raise above optimal, indicating its essential role in maintaining the thermostability in Arabidopsis plants [38,39,40], and also a recent study reported that the modulation of AtFtsH12 abundance causes an altered composition of the plastid import machinery, which will affect development of functional photosynthetic chloroplast [41]. All of the above research results suggest that the FtsH gene family participates in multiple processes in Arabidopsis, but the detailed molecular mechanisms need to be further studied.
So far nine FtsH proteins have been encoded in the genome of rice (Oryza sativa) [28, 42], including three members of OsftsH3, OsftsH4 and OsftsH5 to be predictably targeted to mitochondria, and the others to chloroplasts [42]. However, there are few reports on the functions and molecular mechanism of FtsH genes in rice. In this study, the knockout mutants of OsFtsH2 with an albino seedling phenotype were generated by CRISPR/Cas9 gene editing technology, and the molecular mechanism of OsFtsH2 was explored. By combining a phenotypic and RNA sequencing analysis, it was found that OsFtsH2 could play a vital role in the chloroplast development.
Results
Identification and sequence analysis of OsFtsH2
Among the nine members of the FtsH family identified in rice genome [28], sequence analysis showed that OsFtsH2 (LOC_Os01g43150) contained a complete ORF of 2031 bp and encoded a protein of 676 amino acids. Here a phylogenetic tree including ten FtsH2 proteins in rice and other plants was constructed (Fig. 1), and phylogenetic analysis showed that OsFtsH2 was clustered more closely with ZmFtsH2A and ZmFtsH2B than other FtsH2 proteins (Fig. 1A). Besides, OsFtsH2 protein harbors the conserved domains such as transmembrane region, AAA domain and peptidase M41 region, similar to its counterparts in plant species (Fig. 1B).

Phylogenetic and sequence analyses of OsFtsH2. A Phylogenetic analysis of OsFtsH2 and its closely related FtsH proteins from other species. The accession numbers of selected FtsHs are as follows: from Arabidopsis thaliana, AtFtsH2 (NP_850156.1) and AtFtsH8 (NP_001321589.1); from Oryza sativa, OsFtsH2 (XP_015643053.1); from Zea mays, ZmFtsH2A (NP_001120720.1) and ZmFtsH2B (NP_001120721.1); from Brachypodium distachyon, Bdftsh2 (XP_010240568.1); from Nicotiana tabacum, NtFtsH2 (XP_016436682.1); from Capsicum annuum, CaFtsH2 (XP_016580880.1); from Cucumis sativus, CsFtsH2 (XP_004136837.1); from Setaria italica, SiFtsH2 (XP_004966311.1). B Multiple sequence alignment of amino acid sequences of OsFtsH2 proteins with other FtsH2 proteins from various plants
Expression pattern and subcellular localization of OsFtsH2
Expression of genes in various tissues may display functional diversity. To determine expression pattern of OsFtsH2, RNA was extracted from roots, leaves, stems and panicle of wild type plants, and then implemented its reverse transcription to cDNA. The expression levels of OsFtsH2 in these tissues were assessed by qRT-PCR. As shown in Fig. 2A, leaves revealed the highest level of OsFtsH2 expression, followed in turn by sheaths, stems, seeds, panicles and roots. Therefore, OsFtsH2 mainly functions in green tissues, just as predicted to be a chloroplast-targeted protein [42]. To verify the precise subcellular localization of OsFtsH2, the fusion vector of 35S: OsFtsH2-GFP was constructed and transiently expressed in rice protoplasts (Fig. 2B). As seen, the green fluorescent signals of OsFtsH2-GFP fusion proteins overlapped with chloroplast auto-fluorescence in transformed rice protoplasts, indicating that OsFtsH2 was localized in chloroplasts.

Expression pattern and subcellular localization of OsFtsH2. A Expression analysis of the OsFtsH2 gene in different tissues. B Subcellular localization of the OsFtsH2 proteins. GFP signals of OsFtsH2-GFP fusion protein was located in the chloroplasts by transient expression analyses in rice protoplasts. Green fluorescence shows GFP, red fluorescence shows chloroplast auto-fluorescence and yellow fluorescence shows the merged fluorescence. Scale bar, 5 μm
The albino seedling phenotype caused by knockout of OsFtsH2
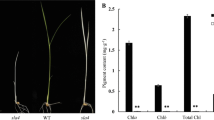
To further understand the function of OsFtsH2, a CRISPR/Cas9 vector with carrying two target sites in the first exon of OsFtsH2 was constructed (Fig. 3A, B). Then, the plasmid was integrated into the calli of wild-type rice (Donjin) by Agrobacterium-mediated transformation. For the T2 generation, sequencing analysis showed three transgene-free homozygous knockout lines of osftsh2–1, osftsh2–2 and osftsh2–3 (Fig. 3C), and all three of mutants exhibited the albino leaf phenotypes (Fig. 3D). Consistent with their phenotypes, photosynthetic pigment contents of the three osftsh2 mutants were dramatically reduced as compared with that of the wild type (Fig. 4A). In addition, the plant height and seedling fresh weight of these osftsh2 mutants were much lower than that of wild type, while there was no visible difference in their root lengths (Fig. 4B, C, D). It was also observed that the osftsh2 mutants could not survive through the stage of three leaves.

Production of OsFtsH2 knockout mutants via the CRISPR/Cas9 system. A Diagram of CRISPR/Cas9 system for editing OsFtsH2. B Schematic diagram of targets sites in OsFtsH2. Black boxes show exons, black lines show introns and white boxes show untranslated regions (UTR). C Mutation sites of OsFtsH2 knockout lines. The osftsh2–1 mutant has a 1-bp insertion; the. osftsh2–2 mutant has a 15-bp deletion; and the osftsh2–3 mutant has a 96-bp deletion. D Phenotypes of osftsh2 mutants, 7-day-old seedlings were photographed. Scale bar, 1 cm

Characteristics of osftsh2 mutants at 7-day-old seedling stage. A Pigment content of WT and osftsh2 mutants. Chlorophyll a (Chla), chlorophyll b (Chlb), total chlorophyll (Chl) and carotenoid (Car). B Plant height of wild type and osftsh2 mutants. C Root length of WT and osftsh2 mutants. D Fresh weight of WT and osftsh2 mutants. The data are mean ± SD (n = 3) and ** indicates statistical significance at p < 0.01
Photosynthetic characteristics of osftsh2 mutants
With a non-invasive feature of photosynthesis, chlorophyll fluorescence has been extensively used to monitor the changes in physiological state of photosynthetic apparatus [43]. The fluorescence analysis showed that the values of Fv/Fm in wild type rice and the osftsh2 mutant were 0.80 and 0.36, respectively, and the actual photochemical efficiency (ФPSII and ФPSI) of the osftsh2 mutant was also reduced significantly as compared with that of wild type (Fig. S1). In order to further determine the photosynthesis changes in osftsh2 mutants, the photosynthetic parameters of the wild type and its mutants were measured. Compared with wild type plants as shown in Fig. 5, the net photosynthetic rate (Pn) of wild type was about 9.14 μmol CO2 m− 2 s− 1 but those of the osftsh2 mutants dropped down to negative domain (Fig. 5A); the intercellular CO2 concentration (Ci) of each mutant was significantly higher than that of wild type (Fig. 5B); both stomatal conductance (Gs) and transpiration rate (Tr) of the osftsh2 mutants were lower than that of the wild type (Fig. 5C and D). In brief, these results indicate that the light energy harvest and transfer were seriously blocked in osftsh2 mutants, which would cause the loss of their autotrophic ability of photosynthesis.

Measurement of leaf photosynthetic parameters in WT and osftsh2 mutants at the seedling stage. A Pn, the net photosynthetic rate; B Ci, the intercellular CO2 concentration; C Gs, the stomatal conductance; D Tr, the transpiration rate. The data are mean ± SD (n = 3) and ** indicates statistical significance at p < 0.01
Impairment of chloroplast development in osftsh2 mutants
Chloroplast was developed by successive biosynthesis and assembly of chlorophyll into photosynthetic apparatus [44]. The albino leaf phenotypes (Fig. 3D) displayed the unhealthy development of chloroplast in osftsh2 mutants. For further verification, the ultrastructure of chloroplasts in both wild type and osftsh2–1 mutant leaves was analyzed by transmission electron microscopy (Hitachi H-7650). As illustrated in Fig. 6, the chloroplasts in wild type showed normal shape and contained a large number of well-structured and dense grana stacks (Fig. 6A, B), and in contrast, the osftsh2 mutant only revealed the mesophyll cells with vesicle-like structures instead of regular chloroplasts with visible grana lamellae stacks (Fig. 6C, D). These results demonstrated that the development of chloroplast in the osftsh2 mutant was severely impaired.

Transmission electron microscopic images of chloroplasts in WT (A, B) and osftsh2 mutants (C, D)
Accumulation of reactive oxygen species (ROS) in osftsh2 mutants
FtsH proteins play important roles in photo-oxidative stress, which can participate in degradation of photodamaged D1 protein [24]. ROS levels in chloroplasts were always increased under photo-oxidative stress, and function as signaling molecules to regulate chloroplast-to-nucleus signal transduction [45, 46]. Therefore, we further determined the ROS levels in osftsh2 mutants. As shown in Fig. 7, the contents of H2O2 and O2 – in osftsh2 mutants were significantly increased as compared to that in wild type, indicating that the osftsh2 mutants may suffer photo-oxidative damage due to the accumulation of excessive ROS.

Production of reactive oxygen species in WT and osftsh2 mutants. A Contents of hydrogen peroxide. B Production rate of superoxide. The data are mean ± SD (n = 3) and ** indicates statistical significance at p < 0.01
Analysis of differentially expressed genes (DEGs) in osftsh2 mutants
In order to unravel the molecular mechanism of OsFtsH2, transcriptome analysis of wild type plants and osftsh2 mutants were comparatively conducted with RNA-seq. In detail, total RNA was firstly extracted from the second leaves of wild type and osftsh2–1 mutants at three leaves stage. Then, three biological replicates were conducted for each sample and a total of six libraries were constructed. High-throughput sequencing generated 46.31–56.69 million raw reads per library, and after a stringent quality filtering process, 45.89–56.14 million clean reads were obtained for each library (Table 1). About 98% of the Q20 percentage and 95% of the Q30 percentage were observed from RNA sequencing data, and the calculated GC content of each library showed that the average GC content was in the range of 53.16–54.97% (Table 1). Moreover, about 96% of the clean reads can be mapped to the rice reference genome, and more than 89% of the reads were uniquely mapped to the genome in each sample. Overall, the quality of sequencing results was qualified for further transcriptome analyses. In addition, the level of gene expression was characterized by calculating RPKM (reads per kb per million reads) values, and a total of 38,866 expressed genes with a RPKM > 1 value were detected from the RNA-seq data. By the end, a total of 6461 DEGs were identified in wild type or osftsh2 mutants as examined with the criteria |log2(fold change) | > 1 and p-value < 0.05, and in osftsh2 mutants there were 3226 significantly up-regulated genes and 3235 down-regulated genes, respectively (Table S1).
Functional annotations and classifications of the DEGs
The DEGs were annotated and enriched in three sets of ontologies including molecular function (MF), cellular component (CC) and biological process (BP) based on GO database. The DEGs annotated in MF category included binding, catalytic activity, transporter activity, nucleic acid binding transcription factor activity and 9 other GO terms; the DEGs in CC consisted of 16 GO terms such as cell, cell part, organelle, membrane and macromolecular complex; also the DEGs in BP involved metabolic process, cellular process, single-organism process, biological regulation, response to stimulus and 16 other GO terms (Fig. S2). Furthermore, the top 20 most significantly enriched GO terms of these functional DEGs were shown in Fig. 8, notifying that no term of MF or CC categories emerged. In other words, only those terms in BP category such as branched-chain amino acid catabolic process, light harvesting in photosystem I, alpha-amino acid catabolic process and amino glycan metabolic process were enriched, suggesting that they were significantly influenced by OsFtsH2.

GO enrichment analysis of differentially expressed genes (DEGs) in osfstsh2 mutants compared with WT
To determine the effect of OsFtsH2 on the signal transduction pathways and metabolic mechanism in rice, KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis of DEGs in osftsh2 mutants was conducted. In the first, the DEGs were classified into 138 pathways (Table S2), and then KEGG pathway enrichment analysis of DEGs was performed using Fisher’s exact test and the pathway with Padjust < 0.05 was considered to be significantly enriched. As a result, the top 20 most significantly enriched KEGG pathways were identified (Fig. 9), which include glyoxylate and dicarboxylate metabolism, nitrogen metabolism, plant hormone signal transduction, glycolysis / gluconeogenesis, etc. Overall, it was found that OsFtsH2 was actively involved in regulating various pathways in rice.

KEGG enrichment analysis of DEGs in osfstsh2 mutants compared with WT
Suppression of photosynthetic genes in osftsh2 mutants
As known, FtsH could participate in the progressive degradation of chloroplast proteins along with other proteases, and a comparative microarray analysis showed that numerous photosynthetic genes were repressed strongly in the var2 white sectors [47]. According to the pathway enrichment analysis in this work, the photosynthesis-related pathways involving photosynthesis (34 genes), photosynthesis-antenna (14 genes) and carbon fixation in photosynthetic organisms (44 genes) were significantly enriched, and almost all genes of photosystem subunits and chlorophyll a-b binding protein were greatly down-regulated in osftsh2 mutants (Table S2). Consequently, suppression of these photosynthetic genes could cause chloroplast defects in osftsh2 mutants.
Changes of plant hormone signal transduction in osftsh2 mutants
Plant hormones play distinctive roles in controlling plant growth and development. It is worth noting that a total of 83 DEGs have involved in the plant hormone signal transduction pathway, which comprised the maximum number of the top 20 most significantly enriched KEGG pathways (Fig. 9). Moreover, a total of 32 DEGs were identified in auxin pathway, and most of them such as AUX/IAA and SAUR family members were significantly down-regulated, suggesting that OsFtsH2 might participate in the regulation of auxin signaling pathway (Table S3). In addition, we also found that most genes associated with cytokinine, gibberellin and brassinosteroid signals were suppressed, especially the transcription factors PIF4 and BZR1. These findings may give an explanation to the retarded growth in osftsh2 mutants. However, the expression levels of most DEGs in the ethylene signaling pathway were up-regulated, which was consistent with the senescence phenotype in mutants. Anyhow the altered expression patterns of plant hormone signals could provide an insight into the molecular mechanism of OsFtsH2 involved in the growth and development of rice seedlings.
qRT-PCR validation of DEGs identified by RNA-seq
To validate the RNA-seq transcriptome data, 12 genes associated with photosynthesis and plant hormone signals were selected for qRT-PCR analysis. The results showed that the expression tendency of these genes was basically consistent with the RNA-seq data (Fig. 10), verifying the reliability of these data.

Validation of RNA-seq by qRT-PCR
Discussion
OsFtH2 plays a vital role in early development of chloroplast
Rice (Oryza sativa) contains nine FtsH genes in its genome [28, 42], and the function mechanisms of FtsH genes in rice have not been fully understood so far. In this study, we have successfully edited the coding region of OsFtsH2 by CRISPR/Cas9 system to generate osftsh2 knockout mutants (Fig. 3). These osftsh2 mutants exhibited phenotype of albino leaves and their contents of photosynthetic pigment were significantly less than that of wild type. The evaluation of photosynthetic parameters showed that osftsh2 mutants lacked autotrophic ability for photosynthesis (Fig. 5), which was consistent with the lethal phenotype of their seedling. Subcellular localization demonstrated that the OsFtsH2 protein was targeted to chloroplast (Fig. 2), and no normal chloroplasts were formed in osftsh2 mutants (Fig. 6). In addition, transcriptome analysis showed various genes related to photosynthesis were suppressed in osftsh2 mutants (Table S2). Also OsFtH2 had the close evolutionary relationships with type B (AtFtsH2/8) chloroplastic FtsHs in Arabidopsis thaliana (Fig. 1A) and lacking of AtFtsH2 caused a severe leaf variegation phenotype and atftsh8 mutants have no visible phenotypes [21, 30,31,32]. Moreover, double mutants of AtFtsh2AtFtsh8 showed an albino-like phenotype, suggesting that they had redundant functions to some extent and each one was required for chloroplast biogenesis [28, 33]. Considering the seedling lethal trait of osftsh2 mutants, it can be concluded that OsFtsH2 performs a distinguishable function in chloroplast development and has no redundancy with other chloroplast localized FtsH genes in rice. All the results reveal that OsFtH2 plays a vital role in early development of chloroplasts in plants.
OsFtH2 is crucial to biosynthesis in photosystem
Photosystem is one of the sites for light reactions to take place, which converts solar energy to chemical energy through energy transfer, photoelectric conversion and electron transfer. Previous studies demonstrated that chloroplast FtsH metalloproteases mainly participated in degrading the proteins at the photodamaged D1 reaction center during the PSII repair cycle, as well as facilitated to remove unassembled PSII subunits, light-harvesting complex II (LHCII) proteins and cytochrome b6 f Rieske FeS proteins proteins [17, 29, 36]. Interestingly, both var1 (atftsh2) and var2 (atftsh5) mutants had lower amount of PSI complex proteins under normal conditions of growth, and also had similar levels of PSII core protein D1 as compared with the wild type. In addition, either var1 or var2 mutants showed no differences in psaA/B transcript accumulation and translation as compared with the wild type, suggesting that later stages of PsaA/B protein expression were impaired in these mutants [48]. Similarly, slr0228 encoded a chloroplast FtsH gene in Synechocystis sp. PCC 6803, and its disruption could triger a major reduction in the abundance of PSI without affecting the cellular content of PSII or phycobilisomes [16]. Moreover, the abundance of PsaA was significantly reduced in the C. reinhardtii ftsh1–1 mutant under 50 or 150 mmol photons m− 2 s− 1 [49]. In one word, FtsH gene was required for the biosynthesis of PSI, which was evolutionarily conserved in oxygenic photosynthetic organisms [48]. In this study, we found that almost all genes of PSI and PSII subunits were greatly down-regulated in osftsh2 mutants (Table S2), and also the actual photochemical efficiency (ФPSII and ФPSI) was much lower in osftsh2 mutants (Fig. S1). Thus, we can conclude that OsFtsH2 is crucial to biosynthesis of photosystem in rice. However, it is not clear whether OsFtsH2 is directly or indirectly involved in the assembly of the photosystem and what its substrates are. The specific mechanism needs further study in the future.
OsFtH2 may influence chloroplast-to-nucleus signaling
Retrograde signaling established an important regulatory mechanism for chloroplasts to regulate their metabolism and development state through chloroplast-to-nucleus signaling [50]. ROS in chloroplast, could act as signaling molecules to regulate the transduction of chloroplast-to-nucleus signal [51]. Retrograde signaling was triggered in the var2 (atftsh2) mutant through accumulation of singlet oxygen, which activated the unfolded/misfolded protein response to balance the FtsH2 deficiency [52]. Similarly, the contents of H2O2 and O2 − in osftsh2 mutants were significantly increased as compared to that in wild type (Fig. 7). In addition, some chloroplast-localized nuclear genes such as RbcS genes and Lhcb genes were substantially suppressed in osftsh2 mutants (Table S2). These results suggested that OsFtH2 would influence chloroplast-to-nucleus signaling in rice.
Conclusions
In conclusion, three transgene-free homozygous and OsFtsH2 knockout mutants were generated by CRISPR/Cas9 genome editing system. Phenotypic analysis revealed that these osftsh2 mutants were albino seedlings and eventually died at three leaves stage. OsFtsH2 targeted to chloroplast and remained much higher levels of expression in green tissues. The observation by transmission electron microscopy showed that the ultrastructure of chloroplasts was severely impaired in osftsh2 mutants, and the measurement of photosynthetic parameters verified that the net rate of photosynthesis in the mutants had negative values. Moreover, RNA sequencing analysis indicated that the expression of genes related photosynthetic pathways were seriously inhibited in osftsh2 mutants. Overall, OsFtsH2 would play a vital role in early development of chloroplasts in rice.
Materials and methods
Plant materials and growth conditions
A japonica rice named as ‘Donjin’ was used for genetic transformation and physiological experiments in this study. The seeds of Donjin were provided by Zhejiang University, China. The germinated seeds of rice were grown in hydroponic solution according to the recommendation of the International Rice Research Institute. Rice seedlings were grown in growth chambers under a 12-h-light (30 °C)/12-h-dark (22 °C) photoperiod and the photon flux density of about 500 μmol m− 2 s− 1 as previously described [53].
Plasmid construction and plant transformation
The CRISPR/Cas9 system was adopted to generate osftsh2 mutants according to the previous procedure [54]. In brief, two sites of CRISPR/Cas9 target were selected from the first coding exon of OsFtsH2 in the CRISPR-PLANT database (www.genome.arizona.edu/crispr/), and then two cassettes of sgRNA expression were inserted into the CRSPR/Cas9 binary vector (pYLCRISPR/Cas9-MH) by Golden Gate cloning. Callus were derived from mature seeds of wild type rice and then transformed with the constructed CRISPR/Cas9 vector using the Agrobacterium tumiefaciens strain EHA105 in accordance with a conventional method [55]. All of the primers are listed in Table S4.
Analysis of protein sequence
Homologous protein sequences of OsFtsH2 were obtained by the BLASTP program (www.ncbi.nlm.nih.gov) and they were aligned using the DNAMAN software. The neighbor-joining phylogenetic tree was generated using the software MEGA 5.1. Bootstrap analysis with1000 replicates was applied to assess the significances of the nodes. Conserved motif analysis of FtsH2 proteins was performed by the MEME (http://meme-suite.org/) and SMART (http://smart.embl-heidelberg.de/) online program.
Measurement of photosynthetic pigment and photosynthetic parameters
Photosynthetic pigment contents of leaves were determined according to the previously described method [56]. Fresh second top leaves (0.2 g) collected at 7-day-old seedling stage were cut into small pieces and then immersed in 5 mL of 95% ethanol for 48 h at room temperature under dark conditions. After residual debris was discarded by centrifugation, the supernatants were analysed with a spectrophotometric scanning (DU800, Beckman, Fullerton, USA) to detect absorption values at 663, 645 and 470 nm. Three biological replicates were analyzed for each sample.
Photosynthetic parameters of leaves in wild type plants and osftsh2 mutants, including net photosynthetic rate (Pn), transpiration rate (Tr), stomatal conductance (Gs) and intercellular CO2 concentration (Ci), were measured using a portable photosynthesis system (Licor-6400, LI-COR, Lincoln, NE, USA) according to the manufacturer’s instructions. In the sample chamber, all measurements were performed at a photon flux density of 1000 μmol m− 2 s− 1, a leaf temperature of 30 °C and CO2 concentration of 400 μmol mol− 1. Each measurement was repeated three times and its value was averaged.
Transmission electron microscopy
Analysis of transmission electron microscopy was conducted according to our previous procedure [57]. Briefly, a sample of the leaves was fixed in a solution of 2.5% glutaraldehyde and then in 1% osmium tetroxide at 4 °C. After fixation, the tissues were further dehydrated in gradient ethanol series and finally embedded in resin. Ultrathin sections (50 nm) were performed using a Leica EM UC7 ultra-microtome, and stained with uranyl acetate. Samples were observed under a Hitachi H-7650 transmission electron microscope.
Subcellular localization of OsFtsH2
The full-length CDS sequence of OsFtsH2 without the termination codon was amplified, and then inserted into the modified pCambia1300-GFP vector with the CaMV35S promoter. The as-obtained OsFtsH2-GFP fusion vector was transformed into rice protoplasts as described previously [58], and the GFP fluorescence was determined by a florescence microscope (Zeiss LSM710). The OsFtsH2-GFP vector construction primers are listed in Table S4.
Determination of H2O2 and O2 − production
Leaf samples of wild type plants and osftsh2 mutants at 7-day-old seedling stage were collected for ROS content measurement. H2O2 and O2− production in leaves were measured using reagent kits (Beijing Solarbio Science and Technology, China) according to the manufacturer’s instructions.
RNA sequencing (RNA-seq) analysis
The total RNA of wild type plants and osftsh2–1 mutants was extracted from their second leaves of 7-day-old seedlings. Each sample needed more than 1 μg of RNA to construct the RNA-seq libraries. The mRNA for sequencing was purified using poly(T) oligonucleotide-attached magnetic beads (Illumina, Inc., San Diego, CA, USA) and then was fragmented to small pieces about 300 bp. Random hexamer primers were used to synthesize the first-strand cDNA, and the second-strand cDNA was synthesized over DNA polymerase I and RNase H. Six RNA libraries were constructed and sequenced on an Illumina Novaseq 6000 sequencing platform (Majorbio, Shanghai). The sequencing data were analyzed according to a previous study [59]. RPKM (reads per kb per million reads) was used to describe the expression levels of genes, and genes with RPKM > 1 were considered to be expressed. The differentially expressed genes (DEGs) were assigned as |Log2(fold change) | > 1.0 and p values < 0.05. GO (Gene Ontology) enrichment analysis of DEGs was conducted with using agriGO web-based tools (http://bioinfo.cau.edu.cn/agriGO/analysis.php). KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway enrichment analysis of DEGs was performed by the online KEGG web server (https://www.kegg.jp/).
RNA extraction and qRT-PCR
Total RNA was extracted using an RNA Prep Pure Plant kit (TIANGEN, Beijing, China) according to the manufacturer’s instructions. First-strand cDNA was synthesized from 1 μg total RNA using Trans® Script One-Step gDNA Removal and cDNA Synthesis SuperMix (TransGen Biotech, Beijing, China). The qRT-PCR was performed with TransStart® Top Green qPCR SuperMix (TransGen Biotech, Beijing, China) using a LightCycler480 instrument (Roche, Sweden). The qRT-PCR reaction was carried out by incubation at 94 °C for 30 s followed by 40 cycles for 5 s and lasted at 60 °C for 30 s. The rice housekeeping gene (LOC_Os03g50885) was chosen as normalization control, and comparative expression levels were calculated by the 2-ΔΔCT method. Three technical replicates on each of three biological replicates were conducted. All qRT-PCR primers are listed in Table S4.
Availability of data and materials
The RNA-seq data has been submitted to the NCBI Sequence Read Archive (BioProject ID PRJNA735323, https://submit.ncbi.nlm.nih.gov/subs/bioproject/SUB9800680/overview). The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- BP:
-
Biological process
- CC:
-
Cellular component
- Ci:
-
Intercellular CO2 concentration
- DEGs:
-
Differentially expressed genes
- FtsH:
-
Filamentation temperature-sensitive H
- GO:
-
Gene ontology
- Gs:
-
Stomatal conductance
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- LHCII:
-
Light-harvesting complex II
- MF:
-
Molecular function
- NEP:
-
Nuclear encoded plastid RNA polymerase
- PEP:
-
Plastid-encoded RNA polymerase
- Pn:
-
Photosynthetic rate
- PSII:
-
Photosystem II
- PSI:
-
Photosystem I
- RPKM:
-
Reads per kb per million reads
- ROS:
-
Reactive oxygen species
- Tr:
-
Transpiration rate
- UTR:
-
Untranslated regions
- WT:
-
Wild type
References
Mirkovic T, Ostroumov EE, Anna JM, van Grondelle R. Govindjee, Scholes GD. Light absorption and energy transfer in the antenna complexes of photosynthetic organisms. Chem Rev. 2017;117:249–93.
Li Y, Zhang J, Li L, Gao L, Xu J, Yang M. Structural and Comparative Analysis of the Complete Chloroplast Genome of Pyrus hopeiensis-“Wild Plants with a Tiny Population”-and Three Other Pyrus Species. Int J Mol Sci. 2018;19(10):3262. https://doi.org/10.3390/ijms19103262.
Wu L, Wu J, Liu Y, Gong X, Xu J, Lin D, et al. The Rice Pentatricopeptide repeat gene TCD10 is needed for chloroplast development under cold stress. Rice (N Y). 2016;9:67.
Mullet JE. Dynamic regulation of chloroplast transcription. Plant Physiol. 1993;103:309–13.
Hajdukiewicz PT, Allison LA, Maliga P. The two RNA polymerases encoded by the nuclear and the plastid compartments transcribe distinct groups of genes in tobacco plastids. EMBO J. 1997;16:4041–8.
De Santis-MacIossek G, Kofer W, Bock A, Schoch S, Maier RM, Wanner G, et al. Targeted disruption of the plastid RNA polymerase genes rpoA, B and C1: molecular biology, biochemistry and ultrastructure. Plant J. 1999;18:477–89.
Pfalz J, Pfannschmidt T. Essential nucleoid proteins in early chloroplast development. Trends Plant Sci. 2013;18:186–94.
Akiyama Y, Yoshihisa T, Ito K. FtsH, a membrane-bound ATPase, forms a complex in the cytoplasmic membrane of Escherichia coli. J Biol Chem. 1995;270:23485–90.
Janska H, Kwasniak M, Szczepanowska J. Protein quality control in organelles - AAA/FtsH story. Biochim Biophys Acta. 2013;1833:381–7.
Neuwald AF, Aravind L, Spouge JL, Koonin EV. AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999;9:27–43.
Granger LL, O’Hara EB, Wang RF, Meffen FV, Armstrong K, Yancey SD, et al. The Escherichia coli mrsC gene is required for cell growth and mRNA decay. J Bacteriol. 1998;180:1920–8.
Gottesman S, Wickner S, Maurizi MR. Protein quality control: triage by chaperones and proteases. Genes Dev. 1997;11:815–23.
Akiyama Y, Ogura T, Ito K. Involvement of FtsH in protein assembly into and through the membrane. I. Mutations that reduce retention efficiency of a cytoplasmic reporter. J Biol Chem. 1994;269:5218–24.
Malnoë A, Wang F, Girard-Bascou J, Wollman F-A, de Vitry C. Thylakoid FtsH protease contributes to photosystem II and cytochrome b6f remodeling in Chlamydomonas reinhardtii under stress conditions. Plant Cell. 2014;26:373–90.
Adam Z, Zaltsman A, Sinvany-Villalobo G, Sakamoto W. FtsH proteases in chloroplasts and cyanobacteria. Physiol Plant. 2005;123:386–90 https://doi.org/10.1111/j.1399-3054.2004.00436.x.
Mann NH, Novac N, Mullineaux CW, Newman J, Bailey S, Robinson C. Involvement of an FtsH homologue in the assembly of functional photosystem I in the cyanobacterium Synechocystis sp. PCC 6803. FEBS Lett. 2000;479:72–7.
Komenda J, Barker M, Kuviková S, de Vries R, Mullineaux CW, Tichy M, et al. The FtsH protease slr0228 is important for quality control of photosystem II in the thylakoid membrane of Synechocystis sp. PCC 6803. J Biol Chem. 2006;281:1145–51.
Silva P, Thompson E, Bailey S, Kruse O, Mullineaux CW, Robinson C, et al. FtsH is involved in the early stages of repair of photosystem II in Synechocystis sp PCC 6803. Plant Cell. 2003;15:2152–64.
Yin Z, Meng F, Song H, Wang X, Chao M, Zhang G, et al. GmFtsH9 expression correlates with in vivo photosystem II function: chlorophyll a fluorescence transient analysis and eQTL mapping in soybean. Planta. 2011;234:815–27.
Urantowka A, Knorpp C, Olczak T, Kolodziejczak M, Janska H. Plant mitochondria contain at least two i-AAA-like complexes. Plant Mol Biol. 2005;59:239–52.
Sakamoto W, Zaltsman A, Adam Z, Takahashi Y. Coordinated regulation and complex formation of yellow variegated1 and yellow variegated2, chloroplastic FtsH metalloproteases involved in the repair cycle of photosystem II in Arabidopsis thylakoid membranes. Plant Cell. 2003;15:2843–55.
Sinvany-Villalobo G, Davydov O, Ben-Ari G, Zaltsman A, Raskind A, Adam Z. Expression in multigene families. Analysis of chloroplast and mitochondrial proteases. Plant Physiol. 2004;135:1336–45.
Zaltsman A, Ori N, Adam Z. Two types of FtsH protease subunits are required for chloroplast biogenesis and photosystem II repair in Arabidopsis. Plant Cell. 2005;17:2782–90.
Kato Y, Miura E, Ido K, Ifuku K, Sakamoto W. The variegated mutants lacking chloroplastic FtsHs are defective in D1 degradation and accumulate reactive oxygen species. Plant Physiol. 2009;151:1790–801.
Liu X, Yu F, Rodermel S. Arabidopsis chloroplast FtsH, var2 and suppressors of var2 leaf variegation: a review. J Integr Plant Biol. 2010;52:750–61.
Lindahl M, Spetea C, Hundal T, Oppenheim AB, Adam Z, Andersson B. The thylakoid FtsH protease plays a role in the light-induced turnover of the photosystem II D1 protein. Plant Cell. 2000;12:419–31.
Bailey S, Thompson E, Nixon PJ, Horton P, Mullineaux CW, Robinson C, et al. A critical role for the Var2 FtsH homologue of Arabidopsis thaliana in the photosystem II repair cycle in vivo. J Biol Chem. 2002;277:2006–11.
Yu F, Park S, Rodermel SR. Functional redundancy of AtFtsH metalloproteases in thylakoid membrane complexes. Plant Physiol. 2005;138:1957–66.
Ostersetzer O, Adam Z. Light-stimulated degradation of an unassembled Rieske FeS protein by a thylakoid-bound protease: the possible role of the FtsH protease. Plant Cell. 1997;9:957–65.
Sakamoto W, Tamura T, Hanba-Tomita Y, Murata M. The VAR1 locus of Arabidopsis encodes a chloroplastic FtsH and is responsible for leaf variegation in the mutant alleles. Genes Cells. 2002;7:769–80.
Takechi K, Sodmergen, Murata M, Motoyoshi F, Sakamoto W. The YELLOW VARIEGATED (VAR2) locus encodes a homologue of FtsH, an ATP-dependent protease in Arabidopsis. Plant Cell Physiol. 2000;41:1334–46.
Chen M, Choi Y, Voytas DF, Rodermel S. Mutations in the Arabidopsis VAR2 locus cause leaf variegation due to the loss of a chloroplast FtsH protease. Plant J. 2000;22:303–13.
Yu F, Park S, Rodermel SR. The Arabidopsis FtsH metalloprotease gene family: interchangeability of subunits in chloroplast oligomeric complexes. Plant J. 2004;37:864–76.
Marta K, Marta G, Adam U, Hanna J. The significance of Arabidopsis AAA proteases for activity and assembly/stability of mitochondrial OXPHOS complexes. Physiol Plant. 2007;129:139-42. https://doi.org/10.1111/j.1399-3054.2006.00835.
Gibala M, Kicia M, Sakamoto W, Gola EM, Kubrakiewicz J, Smakowska E, et al. The lack of mitochondrial AtFtsH4 protease alters Arabidopsis leaf morphology at the late stage of rosette development under short-day photoperiod. Plant J. 2009;59:685–99.
Zelisko A, García-Lorenzo M, Jackowski G, Jansson S, Funk C. AtFtsH6 is involved in the degradation of the light-harvesting complex II during high-light acclimation and senescence. Proc Natl Acad Sci U S A. 2005;102:13699–704.
Sedaghatmehr M, Mueller-Roeber B, Balazadeh S. The plastid metalloprotease FtsH6 and small heat shock protein HSP21 jointly regulate thermomemory in Arabidopsis. Nat Commun. 2016;7:12439.
Adam Z, Aviv-Sharon E, Keren-Paz A, Naveh L, Rozenberg M, Savidor A, et al. The chloroplast envelope protease FTSH11 - interaction with CPN60 and identification of potential substrates. Front Plant Sci. 2019;10:428.
Chen J, Burke JJ, Xin Z. Chlorophyll fluorescence analysis revealed essential roles of FtsH11 protease in regulation of the adaptive responses of photosynthetic systems to high temperature. BMC Plant Biol. 2018;18:11.
Chen J, Burke JJ, Velten J, Xin Z. FtsH11 protease plays a critical role in Arabidopsis thermotolerance. Plant J. 2006;48:73–84.
Mielke K, Wagner R, Mishra LS, Demir F, Perrar A, Huesgen PF, et al. Abundance of metalloprotease FtsH12 modulates chloroplast development in Arabidopsis thaliana. J Exp Bot. 2021;72:3455–73.
Zhang J, Sun A. Genome-wide comparative analysis of the metalloprotease ftsH gene families between Arabidopsis thaliana and rice. Chin J Biotechnol. 2009;25:1402.
Baker NR. Chlorophyll fluorescence: a probe of photosynthesis in vivo. Annu Rev Plant Biol. 2008;59:89–113.
Wang P, Grimm B. Organization of chlorophyll biosynthesis and insertion of chlorophyll into the chlorophyll-binding proteins in chloroplasts. Photosynth Res. 2015;126:189–202.
Wang M, Zhu X, Li Y, Xia Z. Transcriptome analysis of a new maize albino mutant reveals that zeta-carotene desaturase is involved in chloroplast development and retrograde signaling. Plant Physiol Biochem PPB. 2020;156:407–19.
Nott A, Jung H-S, Koussevitzky S, Chory J. Plastid-to-nucleus retrograde signaling. Annu Rev Plant Biol. 2006;57:739–59.
Miura E, Kato Y, Sakamoto W. Comparative transcriptome analysis of green/white variegated sectors in Arabidopsis yellow variegated2: responses to oxidative and other stresses in white sectors. J Exp Bot. 2010;61:2433–45.
Järvi S, Suorsa M, Tadini L, Ivanauskaite A, Rantala S, Allahverdiyeva Y, et al. Thylakoid-bound FtsH proteins facilitate proper biosynthesis of photosystem I. Plant Physiol. 2016;171:1333–43.
Malnoë A. A genetic suppressor approach to the biogenesis, quality control and function of photosynthetic complexes in Chlamydomonas reinhardtii. 2011.
Singh R, Singh S, Parihar P, Singh VP, Prasad SM. Retrograde signaling between plastid and nucleus: a review. J Plant Physiol. 2015;181:55–66.
Chi W, Sun X, Zhang L. Intracellular signaling from plastid to nucleus. Annu Rev Plant Biol. 2013;64:559–82.
Dogra V, Duan J, Lee KP, Kim C. Impaired PSII proteostasis triggers a UPR-like response in the var2 mutant of Arabidopsis. J Exp Bot. 2019;70:3075–88.
Wang D, Li X-F, Zhou Z-J, Feng X-P, Yang W-J, Jiang D-A. Two Rubisco activase isoforms may play different roles in photosynthetic heat acclimation in the rice plant. Physiol Plant. 2010;139:55–67.
Ma X, Zhang Q, Zhu Q, Liu W, Chen Y, Qiu R, et al. A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot plants. Mol Plant. 2015;8:1274–84.
Hiei Y, Ohta S, Komari T, Kumashiro T. Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of the boundaries of the T-DNA. Plant J. 1994;6:271–82.
Wellburn AR. The spectral determination of chlorophylls a and b, as well as Total carotenoids, using various solvents with spectrophotometers of different resolution. J Plant Physiol. 1994; https://doi.org/10.1016/S0176-1617(11)81192-2.
Guo H, Hong C, Chen X, Xu Y, Liu Y, Jiang D, et al. Different growth and physiological responses to cadmium of the three Miscanthus species. PLoS One. 2016;11:e0153475.
Yu C, Wang L, Chen C, He C, Hu J, Zhu Y, et al. Protoplast: a more efficient system to study nucleo-cytoplasmic interactions. Biochem Biophys Res Commun. 2014;450:1575–80.
Dong L, Qin L, Dai X, Ding Z, Bi R, Liu P, et al. Transcriptomic analysis of leaf sheath maturation in maize. Int J Mol Sci. 2019;20:2472. https://doi.org/10.3390/ijms20102472.
Acknowledgements
We thank all the lab members for their help discussion.
Research involving plants
Experimental research and field studies on plants in this work comply with the IUCN Policy Statement on Research Involving Species at Risk of Extinction and the Convention on the Trade in Endangered Species of Wild Fauna and Flora.
Funding
This research was funded by the National Natural Science Foundation of China, grant No. 31670631, 32071509, Zhejiang Provincial Natural Science Foundation, grant No. LQ19C020003, LQ19C140002, and Science and Technology Bureau of Ningbo, grant No. 2019C10094, 2019C10008, 202002 N3083, 202002 N3028.The funders were not involved in the study design, data collection and analyses, data interpretation, or in the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
RX and WQ conceived and designed the research. WQF, HTT and YL performed the experiments. JDA, WQ and RX analyzed the data. ZYX and WQF wrote the manuscript with inputs from other authors. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Chlorophyll fluorescence analysis of osftsh2 mutants.
Additional file 2: Fig. S2.
GO annotation analysis of DEGs in osftsh2 mutants.
Additional file 3: Table S1.
All DEGs in osftsh2 mutants were listed.
Additional file 4: Table S2.
DEGs in photosynthesis-related pathways.
Additional file 5: Table S3.
DEGs in plant hormone signal transduction.
Additional file 6: Table S4.
Primers sequence in this study were listed.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
Cite this article
Wu, Q., Han, T., Yang, L. et al. The essential roles of OsFtsH2 in developing the chloroplast of rice. BMC Plant Biol 21, 445 (2021). https://doi.org/10.1186/s12870-021-03222-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-021-03222-z