
Abstract
Background
Drought is a major environmental constraint to plant growth, development and productivity. Compared with most willows that are generally susceptible to drought, the desert willow Salix psammophila has extraordinary adaptation to drought stress. However, its molecular basis of drought tolerance is still largely unknown.
Results
During polyethylene glycol 6000 (PEG 6000)-simulated drought stress, we found that the osmotic adjustment substances were accumulated and the antioxidant enzyme activities were enhanced in S. psammophila roots. A total of 8172 differentially expressed genes were identified in roots of S. psammophila through RNA-Sequencing. Based on K-means clustering, their expression patterns were classified into nine clusters, which were enriched in several stress-related processes including transcriptional regulation, response to various stresses, cell death, etc. Moreover, 672 transcription factors from 45 gene families were differentially expressed under drought stress. Furthermore, a weighted gene co-expression network was constructed, and eight genes were identified as hub genes. We demonstrated the function of two hub genes, magnesium-dependent phosphatase 1 (SpMDP1) and SpWRKY33, through overexpression in Arabidopsis thaliana. Overexpression of the two hub genes enhanced the drought tolerance in transgenic plants, suggesting that the identification of candidate drought tolerance genes in this study was highly efficient and credible.
Conclusions
Our study analyzed the physiological and molecular responses to drought stress in S. psammophila, and these results contribute to dissect the mechanism of drought tolerance of S. psammophila and facilitate identification of critical genes involved in drought tolerance for willow breeding.
Similar content being viewed by others


Background
Forest ecosystems account for ~ 30% of the land surface (covering ~ 42 million km2), providing many benefits to natural systems and humankind from ecological, economic, social, and aesthetic services [1, 2]. Drought is a major environmental constraint to plant growth, development and productivity. Notably, owing to the long-life spans of forest tree and difficulty for implementing irrigation on a large scale, drought greatly reduces tree productivity and survival across many forest ecosystems [3]. The research of current climate models has predicted increased drought episodes due to the long-term effects of global warming [4]. Therefore, expediting dissection of drought tolerance mechanism and improving drought adaptation of forest trees are urgent needed for arid land utilization, environmental sustainability and improvement of economic benefits.
To accommodate drought stress, plants have evolved sophisticated mechanisms to perceive external signal and coordinate metabolic pathways and morphological traits by modulating the expression of genes [5]. Currently, many drought-inducible genes have been identified through transcriptome analysis in Arabidopsis [6,7,8], rice [9,10,11], maize [12], and other species. These genes can be classified into two classes: functional genes and regulatory genes [5]. Overexpression of some drought-responsive genes (e.g., ethylene response factor 48, calcium-dependent protein kinase 4, NAC5, MYB5, etc.) has demonstrably enhanced drought tolerance in transgenic plants [13,14,15,16]. Nevertheless, large variations occur in transcriptional responses to drought among different plant species [17, 18]. Most researches of drought responses mechanisms have concentrated in crops or model species. Research into forest trees is still at an initial stage except several species, such as Populus, Pinus, Eucalyptus and Quercus [19, 20]. Thus, studies on the species-specific molecular mechanisms of drought response are essential for molecular breeding for drought tolerance in forest trees.
Willows (Salix; Salicaceae) are a very diverse group of dioecious catkin-bearing trees and shrubs, which were the research focus as a potential source of sustainable and renewable biomass for bioenergy, biofuels and bioproducts [21, 22]. However, willows are generally susceptible to drought stress, so they commonly grow in alluvial or riparian habitats [23]. In recent years, willow regulatory responses to drought at morphological, physiological, genomic and transcriptional levels have been examined in some studies, and some loci or candidate genes associated with drought responses were identified. For example, quantitative trait loci (QTLs) associated with drought tolerance for Salix dasyclados × S. viminalis hybrid in two water regimes (normal and drought-treated) have been identified, and each QTL explained 8 to 29% of the phenotypic variation [24]. Fifteen willow genotypes are assessed the impact on the growth and leaf traits under permanent and temporary water stresses, and great variation in drought response among genotypes has been found [25]. Two willow genotypes that originated from a cross between S. viminalis and S. viminalis × S. schwerinii and displayed different responses to drought have been performed phenotypic and transcriptional investigation under drought stress, and a set of candidate genes associated with drought response have been identified [18]. Despite these advances, the comprehensive molecular mechanisms of drought resistance in willows remain poorly elucidated, and functional validation for detected candidate genes has not been carried out.
In willows, S. psammophila is an important shrub biomass willow that distributed in arid and semi-arid desert regions. It can well adapt to multiple abiotic stresses including drought, extreme temperature, sandstorm, and so on [26]. So, it is used to prevent wind erosion and control desertification, playing vital roles in local vegetation rehabilitation [27]. Furthermore, it is identified as a promising biomass feedstock [28, 29]. Drought tolerance plants that can maintain turgor and continue metabolism in cells even at low water potential are identified as potential sources for studying the drought tolerance mechanisms and exploring key regulatory genes for breeding [30]. However, the drought response mechanism of S. psammophila is still poorly understood.
The aim of this study is to investigate the drought response mechanism and identify key regulatory genes in S. psammophila. In order to achieve this, physiological responses to drought stress were analyzed. RNA-Sequencing technology was used to identify differentially expressed genes (DEGs) of root tissue of S. psammophila at different time points (0, 6, 24 and 96 h) after drought treatment. Enrichment analysis was performed to identify pivotal functional categories involved in drought response. Based on the DEGs, gene co-expression network was constructed to identify hub genes associated with drought tolerance, and two hub genes were selected for functional verification through overexpression in A. thaliana. Our results contribute to a better understanding of drought response in S. psammophila; and suggest that co-expression network has important potential to facilitate the identification of critical genes involved in drought tolerance.
Results
Physiological responses to drought stress in S. psammophila
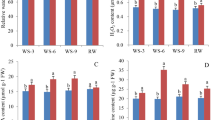
To explore the drought tolerance mechanism of S. psammophila, fine roots at four time points (0, 6, 24 and 96 h) under 22% (w/v) polyethylene glycol 6000 (PEG 6000)-simulated drought stress were used for both physiological and molecular experiments. To avoid rhythm affects and increase data comparability, the different treatments were started at corresponding time points prior to the harvest time and all the samples were collected at the same time [31]. We measured relative water content (RWC), relative electric conductivity (REC), contents of osmotic adjustment substances (proline and soluble sugars) and activities of antioxidant enzymes (superoxide dismutase, peroxidase and catalase) of roots to evaluate the physiological responses to drought stress. The RWC was detected as a measure of the ability of plants to absorb water, and it exhibited striking decrease (P < 0.01) continually from 0 h to 24 h, following slight increase at 96 h (Fig. 1). Interestingly, the REC, osmotic adjustment substances and antioxidant enzyme activities showed opposite trend of initially increase and then decrease during drought stress (Fig. 1).

Physiological responses to drought stress in the roots of S. psammophila. The experiments were performed in three biological replicates and three technical replicates. The data were analyzed with Student’s t test. * and ** represent significance of P < 0.05 and P < 0.01 of three time points (6, 24 and 96 h) compared with the control (0 h), respectively
RNA-Sequencing profiling and global transcriptome changes during drought stress in S. psammophila
To further elucidate the molecular basis for drought responses in S. psammophila, a global transcriptomic analysis was performed using RNA-Sequencing. cDNA libraries with two biological replicates at each time point were independently constructed from S. psammophila roots harvested at the four time points (0, 6, 24 and 96 h) after drought treatment (Fig. 2a), and subsequently sequenced using the Illumina HiSeq2500 platform. After quality control of raw sequencing reads, a total of 297,112,106 clean reads (37.43 Gb) with paired-ends of 125 bp were retained for further analysis. The GC content was 44.23% and the Q30 was 92.34% (Additional file 1).

Transcriptome analysis of differentially expressed genes (DEGs) during the drought stress in the roots of S. psammophila. a Harvest regime designed for a treatment-versus-control experiment with four time points (0, 6, 24 and 96 h) after 22% (w/v) polyethylene glycol 6000 (PEG 6000)-simulated drought stress. All treatments were started at different calculated times prior to the harvest day, and all root samples were designed to be harvested at the same time. b Principal component analysis (PCA) showing the uniformity between biological replicates and the relationships of samples at four stages. The PC1 and PC2 explaining 46.1 and 43.8% of the total variance, respectively. c Hierarchical cluster analysis of total DEGs. Columns and rows in the heat maps represent samples and genes, respectively. d The number of up- and down-regulated genes during drought stress with respect to the control or the preceding time point. e Venn diagram of DEGs showing common or uniquely regulated genes at different time points during drought stress
Approximately 83.50% of total reads were aligned to S. purpurea reference genome version 1.0, a well assembled Salix genome until now (Additional file 1). Principal component analysis (PCA) was carried out on gene expression dataset to confirm the uniformity between biological replicates and investigate the relationships of samples at the four stages (Fig. 2b). Tight cluster between two biological replicates indicated the reliability of our RNA-Sequencing dataset, while greatest distinctness existed between 6 h samples and control (0 h) samples, with PC1 and PC2 explaining 89.9% of the total variance. Hierarchical clustering analysis supported these results (Fig. 2c).
DEGs were identified in different comparisons, which were classified into two groups: Group I for different treatment timepoints compare to control (6 h/0 h, 24 h/0 h and 96 h/0 h) and Group II in time-course manner (6 h/0 h, 24 h/6 h and 96 h/24 h) (Fig. 2a). The largest DEG set was identified in comparison “6 h/0 h” (a total of 5308 DEGs including 2697 up- and 2611 down-regulated genes), suggesting that S. psammophila roots rapidly reprogrammed cellular response in transcriptomic level at early stage (6 h) under drought stress. In contrast, the smallest DEG set was identified in comparison “24 h/6 h” (1181 DEGs including 494 up- and 687 down-regulated genes), followed by comparison “96 h/24 h” (2010 DEGs including 1485 up- and 525 down-regulated genes) (Fig. 2d). Comparative analysis revealed that the overlapped DEGs in three comparisons of Group I (1,198 common genes, 14.7% of total DEGs) were greater than that in Group II (207 common genes, 3.2% of total DEGs) (Fig. 2a and e). The greatest subset of stage-specific DEGs was in comparison “6 h/0 h” in both two groups (2323 DEGs in Group I and 3478 DEGs in Group II). In addition, less or no overlap of up-regulated and down-regulated genes in Group II (Fig. 2e).
Functional annotation and pathway enrichment analysis of DEGs
To identify the major functional categories represented by the DEGs, gene ontology (GO) enrichment analysis was performed. These DEGs comprised three major enrichment categories: 79 biological processes (BP) (Fig. 3), 112 molecular functions (MF) (Additional file 2: Figure S1), and 28 cellular components (CC) (Additional file 2: Figure S2 and Additional file 3). Up-regulated genes were enriched in 49 BP GO terms, including several stress-responsive processes (e.g., response to stress wounding, water, stress, external or biotic stimulus) that were prominent in Group I comparisons (6 h/0 h, 24 h/0 h and 96 h/0 h), but did not enrich in 24 h/6 h and 96 h/24 h (Fig. 3). Down-regulated genes were enriched in 58 BP GO terms: the GO terms of ‘multiple carbohydrates biosynthesis and metabolic process’ and ‘cell wall organization and modification’ were mainly enriched in Group I comparisons; the GO terms of ‘chitin metabolic process’ and ‘aminoglycan metabolic process’ were prominent only in 24 h/6 h (Fig. 3). In addition, GO terms of ‘response to stimulus’, ‘response to oxidative stress’, ‘response to chemical stimulus’, ‘microtubule-based movement’ and ‘DNA replication’ were prominent in all comparisons (Fig. 3).

Biological processes in gene ontology (GO) enrichment analysis of DEGs during drought stress in S. psammophila. GO enrichment analysis was performed using Blast2GO program. Only significantly enriched terms with corrected P < 0.05 were indicated. The color and size of each point represented the -log10 (FDR) values and enrichment scores. A higher -log10 (FDR) value and enrichment score indicated a greater degree of enrichment
In MF category, the GO terms such as ‘ubiquitin-protein ligase activity’, ‘serine-type endopeptidase inhibitor activity’, ‘peptidase inhibitor activity’, ‘endopeptidase inhibitor activity’ and ‘carbohydrate binding’ were mainly enriched in Group I comparisons (6 h/0 h, 24 h/0 h and 96 h/0 h) in up-DEGs; the GO terms of ‘polysaccharide binding’, ‘pattern binding’ and ‘chitin binding’ were specifically enriched in 96 h/0 h in up-DEGs. The GO terms such as ‘glucosyltransferase activity’, ‘copper ion binding’ and ‘cellulose synthase activity’ were enriched in Group I comparisons in down-DEGs (Additional file 2: Figure S1). In CC category, up-regulated genes were significantly enriched in GO terms of ‘photosystem’, ‘thylakoid’, ‘nucleosome’, ‘microtubule’, ‘chromosome’ and ‘cell wall’ mainly in 96 h/24 h; down-regulated genes in Group I comparisons were significantly enriched in GO terms of ‘periplasmic space’, ‘microtubule’, ‘membrane’, ‘external encapsulating structure’, ‘cell wall’, ‘cell envelope’, etc. (Additional file 2: Figure S2).
To gain further insight into the molecular response to drought of S. psammophila in pathway level, we performed an enrichment analysis based on the KEGG pathways. A total of 15 pathways were significantly enriched in this process (Additional file 2: Figure S3 and Additional file 4). In particular, pathways of ‘phenypropanoid biosynthesis’, ‘metabolism of xenobiotics by cytochrome P450’, ‘phenlylalanine metabolism’ and ‘glutathione metabolism’ were significantly enriched in most comparisons, and the degree of enrichment increased with time extension under drought treatment. The pathway of ‘nitrogen metabolism’ was significantly enriched in early and medium stages (6 h/0 h and 24 h/0 h); the pathway of ‘starch and sucrose metabolism’ was significantly enriched in early and late stages (6 h/0 h and 96 h/0 h); the pathway of ‘pentose and glucuronate interconversions’ was significantly enriched in medium and late stages (24 h/0 h and 96 h/0 h). These three pathways also prominent in comparisons 24 h/6 h and 96 h/24 h. Moreover, the pathways of ‘flavonoid biosynthesis’, ‘cell cycle’, ‘gap junction’, ‘tyrosine metabolism’, ‘cyanoamino acid metabolism’ and ‘alpha-linolenic acid metabolism’ were specifically enriched in different stages in Group I comparisons (Additional file 2: Figure S3).
K-means clustering of DEGs
K-means clustering algorithm was applied to divide the 8172 DEGs into nine clusters with characteristic expression patterns and dynamics (Fig. 4a). Each cluster was distinctively composed of genes with particular biological functions, as highlighted by the cluster-specific GO enrichment (Fig. 4b and Additional file 5). In cluster 1, a total of 437 genes showed gradual up-regulation during the treatment and were enriched in ‘response to oxidative stress’ and ‘carbohydrate metabolic process’ terms. The expression of 982 genes in cluster 2 were dramatically induced at 6 h and maintained at an elevated level during drought stress, whereas 1651 genes in cluster 3 were abundantly expressed at early stage and then gradually reduced at 24 h and 96 h. Genes in both clusters 2 and 3 were enriched in transcriptional regulation-related GO terms. Notably, genes in cluster 2 were specifically enriched in ‘response to stress/external stimulus’; cluster 3 genes were enriched in the GO terms of ‘response to water’, ‘cell death’, ‘post-translational modification’, ‘protein ubiquitination’ and ‘phosphorylation’. The gene expression profiles were similar in clusters 4 (858 genes) and 6 (1078 genes), albeit with slightly different degrees of increase at 96 h. GO terms of ‘negative regulation’ were enriched in these two clusters, meaning negative regulation-related genes were inhibited during drought treatment. Compared with cluster 6, genes that encode proteins for the ‘DNA metabolic’ and ‘microtubule-based process and movement’ were highly enriched in cluster 4.

Cluster analysis of DEGs based on the K-means method and cluster-specific GO enrichment analysis. a K-means clustering analysis identified nine clusters of DEGs according to their expression patterns. Mean expression values of each cluster were highlighted. b Gene ontology (GO) terms enrichment analysis of nine DEG clusters
Transcription factors (TFs) act as a pivotal part of transcriptional regulation in plant development and stress response. We identified 672 TFs from 45 gene families were differentially expressed in the nine clusters (Additional file 2: Figure S4). The largest number of TFs were distributed in cluster 3 (164 TFs) and cluster 2 (109 TFs). TF families WRKY, NAC, MYB and ERF were abundant in these two clusters representing genes that were quickly induced under the early stage during drought stress (Fig. 4). Except for them, the members of LBD, HD-ZIP, bZIP, GRAS, G2-like, C2H2 and bHLH families were widely and unevenly distributed among the nine clusters (Additional file 2: Figure S4).
Construction of co-expression network and identification of hub genes
To investigate the interrelationships among drought-responsive genes and identify the key regulators, we constructed a co-expression network through weighted gene co-expression network analysis (WGCNA). According to the DEGs, their expression patterns were divided into six co-expression modules (Additional file 2: Figure S5a). Hierarchical clustering analysis clustered the six modules into three branches: module 1 and module 6, module 3 and module 4, module 2 and module 5 (Additional file 2: Figure S5b). Based on the co-expression relationships, eight genes with the highest connectivity values in this network were identified as hub genes (Fig. 5a), including magnesium-dependent phosphatase 1 (MDP1, SapurV1A.0463 s0100), RING domain ligase2 (RGLG2, SapurV1A.1481 s0050), modifier of rudimentary (Mod(r)) protein (SapurV1A.0226 s0250), calmodulin-like 8 (CML8, SapurV1A.0006 s1450), receptor-like cytoplasmic kinase (RLCK, SapurV1A.0062 s0090), GRAM domain-containing protein (SapurV1A.2984 s0010), transcription factor WRKY DNA-binding protein 33 (WRKY33, SapurV1A.0433 s0130) and an unknown gene (SapurV1A.1521 s0040). These genes displayed connectivity with 456 co-expressed genes, which were classified into nine categories according to their functional annotation: 113 metabolism-related genes, 43 signal-related genes, 34 transcription factors, 29 stress-related genes, 20 transport-related genes, 13 protein modification genes, 5 hormone-related genes, 5 cell death-related genes and 192 unclassified genes (Fig. 5a and Additional file 6). The functional classification of co-expressed genes including eight hub genes was performed based on the functional annotation. The expression profiles of the eight hub genes showed rapidly increase at 6 h, and gradually decline but were higher than the control at 24 h and 96 h (Fig. 5b). These eight hub genes were regarded as promising candidate genes for drought tolerance.

Identification of hub genes in co-expression network and their expression levels under drought stress. a Eight hub genes and their connectivity with co-expressed genes in the network. The co-expression network was analyzed using WGCNA software and the graphic network was created by Cytoscape software. This co-expression network was constructed from 464 genes from six co-expression modules (Additional file 2: Figure S5). b Expression confirmation of eight Hub genes using qRT-PCR. Each sample was conducted three biological replicates and four technical replicates. FPKM values of eight Hub genes from RNA-Sequencing at four time points were shown as grey columns, and relative expression of target genes by qRT-PCR were shown as red lines with standard error. Transcript level at control (0 h) was set to 1. Details of the 464 co-expressed genes was listed in Additional file 6
Overexpression of hub genes SpMDP1 or SpWRKY33 confers drought tolerance in Arabidopsis
To verify the reliability of hub genes identified through WGCNA, we selected two hub genes (SpMDP1 and SpWRKY33) that displayed high connectivity and low connectivity respectively in eight hub genes to conduct functional verification through overexpression in A. thaliana. T3 generation of three independent transgenic Arabidopsis lines (OE-5, OE-15 and OE-17) with high abundance of SpMDP1 and wild type (WT) were chosen to assess their drought tolerance (Fig. 6a-c). In petri plate, the root length and fresh weight of transgenic lines were significantly higher than that of WT under drought stress simulated by mannitol (200 mmol/L), but no significant difference was observed between WT and transgenic lines under control condition (Fig. 6d-e). In soil drought treatment, the transgenic lines also showed stronger drought tolerance than WT (Fig. 6f-g). These results of two parallel experiments both indicated that SpMDP1 improved the tolerance to drought stress in transgenic Arabidopsis.

Overexpression of SpMDP1 enhanced drought tolerance in transgenic Arabidopsis plants. a and b Phenotypic characteristics of wild type (WT) and three independent SpMDP1-overexpression lines (OE-5, OE-15 and OE-17) in control and mannitol-simulated drought treatment for 10 days, respectively. WT and three independent transgenic lines were plated in per plate, with four seedlings of each genotype. The drought treatments in petri plate were performed five biological replicates. Scale bar: 1 cm. c Semi-quantitative RT-PCR analysis of SpMDP1 expression in WT and transgenic plants. d and e Statistics of root length and fresh weight of WT and SpMDP1-overexpression plants, respectively. Values were means ± SD (n = 20). ** on the histograms indicates statistical difference between WT and transgenic plants at P < 0.01. f and g Phenotypic characteristics of WT and SpMDP1-overexpression plants under normal water condition and withholding watering for 14 days, respectively. WT and three independent transgenic lines were grown in soil block, with four seedlings of each genotype. The drought treatments in soil were performed five biological replicates
In the co-expression network, SpMDP1 had the highest connectivity with 380 genes, most of which have previously been reported to participate in drought response or improve drought tolerance, for instance, several transcription factors (ERF, bHLH, WRKY, C2H2, bZIP, MYB, NAC, etc.), late embryogenesis abundant (LEA) hydroxyproline-rich glycoprotein (LEA-HRGP), CBL-interacting protein kinase, mitogen-activated protein kinase (Additional file 7). The Arabidopsis homologous genes of SpMDP1-co-expressed genes were searched using sequences alignment (Additional file 7). To validate the relationship of SpMDP1 and its co-expressed genes, the expression patterns of 30 randomly selected Arabidopsis homologous genes in the co-expression sub-network of SpMDP1 were compared between two-week-old SpMDP1-overexpressing plants and WT plants. Among them, the expression levels of 29 genes were unambiguously detected: 13 genes were up-regulated, four genes were down-regulated, and the expression levels of other 12 genes did not change (Additional file 2: Figure S6). Of the up-regulated genes, most genes were up-regulated by > 2-fold in transgenic plants, such as WRKY75 (2.3- to 3.2-fold), LEA-HRGP (2.1- to 3.1-fold), HSP17.6 (3.2- to 4.5-fold), GSTU25 (3.6- to 4.8-fold), and NAC90 (18- to 24-fold) (Additional file 2: Figure S6).
Furthermore, we also investigated the role of SpWRKY33 in drought tolerance using the same method. As shown in Fig. 7, three independent transgenic lines (OE-2, OE-3 and OE-8) with high abundance of SpWRKY33 also exhibited increased drought tolerance. In the co-expression network, SpWRKY33 was co-expressed with 15 genes (Additional file 8), and the expression patterns of their Arabidopsis homologous genes were compared between two-week-old SpWRKY33-overexpressing plants and WT plants. Among them, the expression levels of 13 genes were unambiguously detected; six genes were up-regulated, including COR47 (2.1- to 2.7-fold), EXL2 (2.4- to 2.7-fold), HSPRO2 (1.7- to 2.3-fold), VQ (2.4- to 2.7-fold), STP1 (2.4- to 2.9-fold), and TPS9 (1.3- to 1.4-fold) (Additional file 2: Figure S7).

Overexpression of transcription factor SpWRKY33 enhanced drought tolerance in transgenic Arabidopsis plants. a and b Phenotypic characteristics of wild type (WT) and three independent overexpression lines (OE-2, OE-3 and OE-8) in control and mannitol-simulated drought treatment for 10 days, respectively. WT and three independent transgenic lines were plated in per plate, with four seedlings of each genotype. The drought treatments in petri plate were performed five biological replicates. Scale bar: 1 cm. c Semi-quantitative RT-PCR analysis of SpWRKY33 expression in WT and transgenic plants. d and e Statistics of root length and fresh weight of WT and SpWRKY33-overexpression plants, respectively. Values were means ± SD (n = 20). ** on the histograms indicates statistical difference between WT and transgenic plants at P < 0.01. f and g Phenotypic characteristics of WT and SpWRKY33-overexpression plants under normal water condition and withholding watering for 14 days, respectively. WT and three independent transgenic lines were grown in soil block, with four seedlings of each genotype. The drought treatments in soil were performed five biological replicates
Discussion
Accumulation of osmotic adjustment and enhancement of antioxidant enzyme activities are vital mechanisms of drought adaptation in plants [32, 33]. Osmotic adjustment through accumulating compatible solutes can promote water absorption from hypertonic solution or drought soils [34, 35]. Soluble sugars and proline are important compatible solutes, which regulate cell osmotic status, scavenge free radicals and protect cell membranes [33, 36]. Moreover, proline is proposed as a molecular chaperone functioning in stabilizing protein structure and as a component contributing to buffering of cytosolic pH [37, 38]. Reactive oxygen species (ROS) production is enhanced in cellular compartments under drought stress, and overproduction of ROS initiated uncontrolled oxidative cascades that damage cellular membranes and other cellular components and eventually cell death [39]. Antioxidant enzymes (e.g., POD, SOD, and CAT) have the ability to scavenge ROS and maintain redox equilibrium [40, 41]. Accumulation of osmotic adjustment and increase of scavenging enzyme activity have been detected during drought in wheat, maize, soybean, rice, potato, etc. [33, 39]. In our study, the findings of accumulation of soluble sugars and proline and enhancement of antioxidant enzyme activities suggested S. psammophila might resist drought through osmotic adjustment and ROS homeostasis to provide protection from cellular damage.
Besides of morphological and physiological adaptation, extensive transcriptomic reprogramming occurred to strive for survival under drought condition [42, 43]. Thus, understanding molecular mechanisms of drought response of S. psammophila is crucial to breed drought-tolerance willows. Based on the RNA-Sequencing profiling, we identified 8172 DEGs during drought stress in roots of S. psammophila, most of which enriched in multiple stress-related processes including transcriptional regulation, response to various stresses, cell death, protein phosphorylation and ubiquitination, ncRNA and DNA metabolic, etc. Thereinto, some processes have been confirmed as essential regulators or associated with plant tolerance to drought or other osmotic stresses. For instance, transcriptional regulation has been identified as an important regulatory mechanism of transcriptomic reprogramming in response to drought [10]. Transcriptional regulation was enriched in both clusters 2 and 3 (Fig. 4), and largest number of transcription factors existed in these two clusters (Additional file 2: Figure S4), suggesting this mechanism was dramatically evoked and executed function after short-term drought stress. Phosphoproteins have been identified under drought stress in several plants, and some proteins have been demonstrated to regulate drought tolerance via mediated phosphorylation [44, 45]. Furthermore, temporal patterns of the enriched GO terms showed that these biological processes occurred according to the time stages. These results revealed that comprehensive and well-ordered biological events motivated S. psammophila to resist and adapt to drought stress.
Based on whole-transcriptome datasets, WGCNA provides an effective approach to identify key regulators and it is widely used in various studies to explain molecular mechanisms of complex traits, such as growth, yield and stress-resistance. For instance, two potential key drivers for seed trait formation, GA20OX and NFYA, have been screened from co-expression network in soybean [46]. WRKY40 and copper transport protein have been identified to be the hub genes involved in defense against whitefly infestation in cotton [47]. Three major hubs including MYB2, phosphoglycerate kinase and heat shock protein have been identified during salinity and drought stresses in rice [48]. In our study, we identified eight hub genes (MDP-1, RGLG2, Mod(r) protein, CML8, RLCK, GRAM domain-containing protein, WRKY33, and an unknown gene) that were co-expressed with a large number of genes in S. psammophila, suggesting their key positions in drought stress response. Through functional validation, two hub genes SpMDP1 and SpWRKY33 were proved to participate in plant drought tolerance. Thus, our study indicated that identification of candidate drought tolerance genes through co-expression network was highly efficient and credible.
As a member of haloacid dehalogenase superfamily, MDP1 contains a conserved motif (Asp-X-Asp-X-Thr) [49], and its specific function in abiotic stress tolerance in plants remains unknown until now. In our study, SpMDP1 had the highest connectivity with 380 genes, and its overexpression transgenic Arabidopsis could improve the tolerance to drought. This result provides new progress and evidence for MDP1 involved in plant drought tolerance. Glycosylation is an important posttranslational modification of proteins, and it plays a crucial role in impacting protein functions and regulating biological processes [50]. For example, the N-glycosylation occurs in the proteins mainly involved in cell wall metabolism, stress response and proteolysis under drought in common bean [36]. Studies have demonstrated that MDP1 can cooperate with fructosamine-3-kinase to free proteins from the glycation products derived from Glu-6-P, indicating that MDP1 may be the key component for glycosylation-involved protein repair [51, 52]. Whether the drought tolerance improved by SpMDP1 is mediated by protein repair is needed to further research.
WRKY33 belongs to the evolutionarily conserved WRKY transcription factor. The expression of WRKY33 is induced by pathogens and environmental stimuli, and its function in plant tolerance stresses has been confirmed through gene silencing and complementation [53, 54]. In A. thaliana, atwrky33 mutants are compromised not only in plant tolerance to necrotrophic pathogens and Botrytis cinerea infection, but also in tolerance to salinity and heat stresses [53, 55, 56]. Complementation of Arabidopsis atwrky33 mutant plants by tomato (Solanum lycopersicum) SlWRKY33A and SlWRKY33B can fully restore resistance to Botrytis and heat tolerance [57]. In this study, we demonstrated that overexpression of the hub gene SpWRKY33 could enhance the tolerance to drought stress in transgenic Arabidopsis plants. Recent study has indicated that AtWRKY33 interacts with two VQ proteins, SIGMA FACTOR-INTERACTING PROTEIN1 (SIB1) and SIB2, which act as activators of AtWRKY33 in plant defence against necrotrophic pathogens [58]. Our study also identified a VQ motif-containing protein (SapurV1A.0943 s0020) was co-expressed with SpWRKY33 in the co-expression network (Additional file 8), and its expression level was up-regulated in SpWRKY33-overexpressing plants (Additional file 2: Figure S7). Moreover, the analysis of genome-wide transcriptional responses to drought in two willow genotypes revealed that WRKY33 has been identified as one of candidate genes with a putative function in drought response [18]. These results indicate that SpWRKY33 is an ideal gene used on genetic engineering for the creation of new varieties with high tolerance to stresses in plants.
Except SpMDP1 and SpWRKY33 which were performed functional validation in this study, the other six hub genes might also participate in drought tolerance in plants. For example, AtRGLG2 negatively regulates the drought stress response by mediating AtERF53 transcriptional activity in A. thaliana [59]. The expression level of AtCML8 is strongly and transiently induced by Pseudomonas syringae and overexpression of AtCML8 confers on plants a better resistance to pathogenic bacteria compared to WT, suggesting that AtCML8 plays a role in plant immunity [60]. Although there are no reports of function of CML8 in drought tolerance; other members of the CML family, such as Oryza sativa OsCML4 and Solanum habrochaites ShCML44, have been confirmed to enhance tolerance to drought stress in plants [61, 62]. Further studies are needed to investigate whether these hub genes contribute to drought tolerance in S. psammophila.
Conclusions
In this study, we analyzed DEGs in S. psammophila roots under a time-course drought stresses, performed analysis of biological processes and pathways, and identified eight hub genes based on the co-expression network. We found the accumulation of osmotic adjustment substances and enhancement of antioxidant enzyme activities under drought stress in S. psammophila. Abundant DEGs were enriched in several stress-related processes including transcriptional regulation, response to various stresses, cell death, etc. Through overexpression in A. thaliana, two hub genes, SpMDP1 and SpWRKY33, have been confirmed to enhance the drought tolerance in transgenic plants. Our results will contribute to parse the mechanism of drought tolerance of S. psammophila and facilitate identification of critical genes involved in drought tolerance for breeding willows.
Methods
Cultivation of S. psammophila and drought stress treatment
In our study, S. psammophila materials were obtained from the germplasm collection base of S. psammophila (E 110°38′59.1″, N 40°14′15.5″) in Ordos Dalad, Inner Mongolia. One-year-old twigs of S. psammophila were cut into 15 cm cuttings, water cultured in 1/2 strength Hoagland’s solution, and grown in a growth chamber under long-day conditions with 16 h light and 8 h dark at 24–25 °C. Ten-week-old seedlings were subjected to 22% (w/v) polyethylene glycol 6000 (PEG 6000)-simulated drought stress for four time points (0, 6, 24, and 96 h). At the end of each time point, fine roots were harvested, frozen immediately in liquid nitrogen, and stored in − 80 °C refrigerator for further analysis and sequencing. We performed three biological replicates.
Measurement of physiological parameters of S. psammophila under drought stress
Electric conductivity (EC) of S. psammophila roots was measured by conductivity meter (DDS-307). Relative water content (RWC) was calculated as previous study [63]. The proline content was measured with ninhydrin method [64]. The soluble sugar content was measured with anthrone method [65]. For enzyme assays, superoxide dismutase (SOD) activity was determined by measuring its ability to inhibit the photochemical reduction of nitroblue tetrazolium at 560 nm [66], peroxidase (POD) activity was measured based on the change in absorbance of 470 nm due to guaiacol oxidation [67], catalase (CAT) activity was determined by monitoring the disappearance of H2O2 [68]. All experiments were performed three technical replicates, and the data were analyzed with Student’s t test.
RNA isolation, library preparation, Illumina sequencing
Total RNA was extracted from roots using the RNeasy Plant Mini Kit (Qiagen, Germany) based on the manufacturer’s protocol. The RNA quality and quantity were detected by the NanoDrop system and Agilent2100 Bioanalyzer. The cDNA library was constructed and sequenced using Illumina HiSeq2500 platform by Biomarker Technologies Corporation (Beijing, China), with paired-end sequencing and read lengths of 125 bp. After quality-control procedure with filtering adapter sequences and low-quality reads, the clean data were obtained for subsequent analysis.
Mapping and quantification of RNA-Sequencing data
We selected S. purpurea genome version 1.0 from Phytozome (https://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Spurpurea) as reference genome, which is 392 Mb in size with 37,865 protein-coding loci and 61,520 protein coding transcripts. Clean reads were mapped to the reference genome with allowance for 5% mismatches using TopHat2 software [69]. Gene expression levels were calculated as reads per kilobase of transcript sequence per million base pairs sequenced (FPKM) using Cufflinks [70]. The differentially expressed genes (DEGs) were identified by DESeq [71]. A threshold of false discovery rate (FDR) ≤ 0.01 and an absolute value of log2 ratio > 2 were used to retrieve DEGs.
Pathway analysis and functional categorization
To study the biological function of DEGs, gene set enrichment with gene ontology (GO) terms was performed using Blast2GO [72]. Metabolic pathways were analyzed using kyoto encyclopedia of genes and genomes (KEGG) program [73]. Fischer’s exact test was used to assess the significance of GO categories and KEGG pathways.
Co-expression gene network generation
The co-expression network was analyzed using the weighted gene co-expression network analysis (WGCNA) R package [74]. The graphic network was created by Cytoscape 3.7.0 software [75].
Quantitative real-time PCR (qRT-PCR)
Primer 3 software was used to design the primers, and qRT-PCR was performed according to the previous described method [76]. SpUBC (Ubiquitin-conjugating enzyme E2) and AtActin were used as the reference genes in S. psammophila [26] and A. thaliana, respectively. Four technical replicates and three biological replicates of each sample were performed. All of the primer sequences were listed in Additional file 9. For homologous genes identification between Arabidopsis and Salix, the BLASTp best hit of Salix genes from the co-expression network in Arabidopsis genome were used for qRT-PCR analysis in Arabidopsis transgenic lines.
Plasmid construction and transformation in A. thaliana
The full length CDS sequences of SpMDP1 and SpWRKY33 were independently amplified from S. psammophila and cloned into pDONR222.1 for sequencing, and the correct coding sequences were sub-cloned into pMDC32 under driven of the cauliflower mosaic virus (CaMV) 35S promoter using Gateway system, respectively. Both above constructs were transferred into A. thaliana by floral dip method [77]. After screened on medium containing 25 mg·L− 1 hygromycin, more than 30 transgenic lines were obtained and the expression levels of these two target genes were detected using qRT-PCR.
Drought tolerance analysis of overexpression plants
WT and homozygous T3 generation transgenic Arabidopsis lines were used in drought tolerance experiments. One-week-old sterilized seedlings were transferred into 1/2 strength MS medium containing 0 mmol/L (control) and 200 mmol/L mannitol (simulated drought stress), respectively. Ten days later, phenotypic traits were compared, and the root length and fresh weight of seedlings were counted. Drought treatment was also applied to three-week-old seedlings in soil with sufficient water by withholding watering for 14 days. Five biological replicates were performed to ensure results’ reliability, and data were analyzed by t test.
Availability of data and materials
The RNA-Sequencing raw data have been deposited to the National Centre for Biotechnology Information (NCBI) BioProject database under accession number PRJNA485903. All the supporting data are included as additional files.
Abbreviations
- BP:
-
Biological processes
- CaMV:
-
Cauliflower mosaic virus
- CAT:
-
Catalase
- CC:
-
Cellular components
- CML8:
-
Calmodulin-like 8
- DEGs:
-
Differentially expressed genes
- EC:
-
Electric conductivity
- FDR:
-
False discovery rate
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- LEA-HRGP:
-
Late embryogenesis abundant hydroxyproline-rich glycoprotein
- MDP1:
-
Magnesium-dependent phosphatase 1
- MF:
-
Molecular functions
- Mod(r):
-
Modifier of rudimentary
- PEG:
-
Polyethylene glycol
- POD:
-
Peroxidase
- qRT-PCR:
-
Quantitative real-time PCR
- QTL:
-
Quantitative trait loci
- RGLG2:
-
RING domain ligase2
- RLCK:
-
Receptor-like cytoplasmic kinase
- RWC:
-
Relative water content
- SIB1:
-
SIGMA FACTOR-INTERACTING PROTEIN1
- SOD:
-
Superoxide dismutase
- TFs:
-
Transcription factors
- UBC:
-
Ubiquitin-conjugating enzyme E2
- WGCNA:
-
Weighted gene co-expression network analysis
- WT:
-
Wild type
References
Hassan R, Scholes R, Ash N. Ecosystems and human well-being: current state and trends. Washington DC: Island Press; 2005.
Bonan GB. Forests and climate change: forcings, feedbacks, and the climate benefits of forests. Science. 2008;320(5882):1444–9.
Williams AP, Allen CD, Macalady AK, Griffin D, Woodhouse CA, Meko DM, Swetnam TW, Rauscher SA, Seager R, Grissinomayer HD. Temperature as a potent driver of regional forest drought stress and tree mortality. Nat Clim Chang. 2013;3(3):292–7.
Dai A. Increasing drought under global warming in observations and models. Nat Clim Chang. 2013;3(1):52–8.
Shinozaki K, Yamaguchishinozaki K. Gene networks involved in drought stress response and tolerance. J Exp Bot. 2006;58(2):221–7.
Seki M, Narusaka M, Ishida J, Nanjo T, Fujita M, Oono Y, Kamiya A, Nakajima M, Enju A, Sakurai T. Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high–salinity stresses using a full–length cDNA microarray. Plant J. 2002;31(3):279–92.
Matsui A, Ishida J, Morosawa T, Mochizuki Y, Kaminuma E, Endo TA, Okamoto M, Nambara E, Nakajima M, Kawashima M. Arabidopsis transcriptome analysis under drought, cold, high-salinity and ABA treatment conditions using a tiling array. Plant Cell Physiol. 2008;49(8):1135–49.
Coolen S, Proietti S, Hickman R, Olivas NHD, Huang P, Van Verk MC, Van Pelt JA, Wittenberg AHJ, De Vos M, Prins M. Transcriptome dynamics of Arabidopsis during sequential biotic and abiotic stresses. Plant J. 2016;86(3):249–67.
Zhou J, Wang X, Jiao Y, Qin Y, Liu X, He K, Chen C, Ma L, Wang J, Xiong L, et al. Global genome expression analysis of rice in response to drought and high-salinity stresses in shoot, flag leaf, and panicle. Plant Mol Biol. 2007;63(5):591–608.
Lenka SK, Katiyar A, Chinnusamy V, Bansal KC. Comparative analysis of drought-responsive transcriptome in Indica rice genotypes with contrasting drought tolerance. Plant Biotechnol J. 2015;9(3):315–27.
Fei L, Zhang H, Zhao H, Gao T, Song A, Jiang J, Chen F, Chen S. Chrysanthemum CmHSFA4 gene positively regulates salt stress tolerance in transgenic chrysanthemum. Plant Biotechnol J. 2018;16(7):1311–21.
Zheng J, Fu J, Gou M, Huai J, Liu Y, Jian M, Huang Q, Guo X, Dong Z, Wang H. Genome-wide transcriptome analysis of two maize inbred lines under drought stress. Plant Mol Biol. 2010;72:407–21.
Jeong S, Trotochaud AE, Clark SE. The Arabidopsis CLAVATA2 gene encodes a receptor-like protein required for the stability of the CLAVATA1 receptor-like kinase. Plant Cell. 1999;11(10):1925–34.
Campo S, Baldrich P, Messeguer J, Lalanne E, Coca M, Segundo BS. Overexpression of a calcium-dependent protein kinase confers salt and drought tolerance in rice by preventing membrane lipid peroxidation. Plant Physiol. 2014;165(2):688–704.
Chen T, Li W, Hu X, Guo J, Liu A, Zhang B. A cotton MYB transcription factor, GbMYB5, is positively involved in plant adaptive response to drought stress. Plant Cell Physiol. 2015;56(5):917–29.
Jung H, Chung PJ, Park SH, Kim YS, Suh JW, Kim JK. Overexpression of OsERF48 causes regulation of OsCML16, a calmodulin–like protein gene that enhances root growth and drought tolerance. Plant Biotechnol J. 2017;15(10):1295–308.
Yan DH, Fenning T, Tang S, Xia X, Yin W. Genome-wide transcriptional response of Populus euphratica to long-term drought stress. Plant Sci. 2012;195(3):24–35.
Pucholt P, Sjödin P, Weih M, Berlin S. Genome-wide transcriptional and physiological responses to drought stress in leaves and roots of two willow genotypes. BMC Plant Biol. 2015;15(1):244.
Qiu Q, Ma T, Hu Q, Liu B, Wu Y, Zhou H, Wang Q, Wang J, Liu J. Genome-scale transcriptome analysis of the desert poplar, Populus euphratica. Tree Physiol. 2011;31(4):452.
Hamanishi ET, Barchet GL, Dauwe R, Mansfield SD, Campbell MM. Poplar trees reconfigure the transcriptome and metabolome in response to drought in a genotype- and time-of-day-dependent manner. BMC Genomics. 2015;16(1):329.
Karp A, Hanley SJ, Trybush SO, Macalpine W, Pei M. Shield I (2011) genetic improvement of willow for bioenergy and biofuels. J Integr Plant Biol. 2011;53(2):151–65.
Hanley SJ, Karp A. Genetic strategies for dissecting complex traits in biomass willows (Salix spp.). Tree Physiol. 2014;34(11):1167–80.
Kuzovkina YA, Weih M, Romero MA, Charles J, Hust S, Mcivor I, Karp A, Trybush S, Labrecque M, Teodorescu TI. Salix: botany and global horticulture. Hortic Rev. 2008;34(1):447–89.
Weih M. QTL analyses of drought tolerance and growth for a Salix dasyclados × Salix viminalis hybrid in contrasting water regimes. Theor Appl Genet. 2005;110(3):537–49.
Bonosi L, Ghelardini L, Weih M. Growth responses of 15 Salix genotypes to temporary water stress are different from the responses to permanent water shortage. Trees. 2010;24(5):843–54.
Li J, Jia H, Han X, Zhang J, Sun P, Lu M, Hu J. Selection of reliable reference genes for gene expression analysis under abiotic stresses in the desert biomass willow, Salix psammophila. Front Plant Sci. 2016;7:1505.
Huang J, Zhou Y, Yin L, Wenninger J, Zhang J, Hou G, Zhang E, Uhlenbrook S. Climatic controls on sap flow dynamics and used water sources of Salix psammophila in a semi-arid environment in Northwest China. Environ Earth Sci. 2015;73(1):289–301.
Li CJ, Yang X, Zhang Z, Zhou D, Zhang L, Zhang SC, Chen JM. Hydrothermal liquefaction of desert shrub Salix psammophila to high value-added chemicals and hydrochar with recycled processing water. Bioresources. 2013;8(2):2981–97.
Yang X, Lyu H, Chen K, Zhu X, Zhang S, Chen J. Selective extraction of bio-oil from hydrothermal liquefaction of Salix psammophila by organic solvents with different polarities through multistep extraction separation. Bioresources. 2014;9(3):5219–33.
Zeng X, Bai L, Wei Z, Yuan H, Wang Y, Xu Q, Tang Y, Nyima T. Transcriptome analysis revealed the drought-responsive genes in Tibetan hulless barley. BMC Genomics. 2016;17(1):386.
Yang C, Wei H. Designing microarray and RNA-Seq experiments for greater systems biology discovery in modern plant genomics. Mol Plant. 2015;8(2):196–206.
Hsiao TC, Acevedo E, Fereres E, Henderson DW. Stress metabolism: water stress, growth, and osmotic adjustment. Philos Trans R Soc Lond Ser B Biol Sci. 1976;273:479–500.
Zivcak M, Brestic M, Sytar O. Osmotic adjustment and plant adaptation to drought stress. In: Hossain MA, Wani SH, Bhattacharjee S, Burritt DJ, Tran LP, editors. Drought stress tolerance in plants. Switzerland: Springer International Publishing; 2016. p. 105–143.
Chaves MM, Maroco J, Pereira JS. Understanding plant responses to drought-from genes to the whole plant. Funct Plant Biol. 2003;30(3):239–64.
Liu C, Liu Y, Guo K, Fan D, Li G, Zheng Y, Yu L, Yang R. Effect of drought on pigments, osmotic adjustment and antioxidant enzymes in six woody plant species in karst habitats of southwestern China. Environ Exp Bot. 2011;71(2):174–83.
Zadražnik T, Moen A, Eggejacobsen W, Meglic V, Sustarvozlic J. Towards a better understanding of protein changes in common bean under drought: a case study of N-glycoproteins. Plant Physiol Biochem. 2017;118:400–12.
Verbruggen N, Hermans C. Proline accumulation in plants: a review. Amino Acids. 2008;35(4):753–9.
Filippou P, Bouchagier P, Skotti E, Fotopoulos V. Proline and reactive oxygen/nitrogen species metabolism is involved in the tolerant response of the invasive plant species Ailanthus altissima to drought and salinity. Environ Exp Bot. 2014;97:1–10.
De Carvalho MHC. Drought stress and reactive oxygen species: production, scavenging and signaling. Plant Signal Behav. 2008;3(3):156–65.
Jiang MY, Zhang JH. Water stress-induced abscisic acid accumulation triggers the increased generation of reactive oxygen species and up-regulates the activities of antioxidant enzymes in maize leaves. J Exp Bot. 2002;53(379):2401–10.
Miller G, Suzuki N, Ciftci-yilmaz S, Mittler R. Reactive oxygen species homeostasis and signalling during drought and salinity stresses. Plant Cell Environ. 2010;33(4):453–67.
Bechtold U, Penfold CA, Jenkins DJ, Legaie R, Moore JD, Lawson T, Matthews JS, Vialet-Chabrand SR, Baxter L, Subramaniam S. Time-series transcriptomics reveals that AGAMOUS-LIKE22 links primary metabolism to developmental processes in drought-stressed Arabidopsis. Plant Cell. 2016;28(2):345–55.
Iqbal A, Wang T, Wu G, Tang W, Zhu C, Wang D, Li Y, Wang H. Physiological and transcriptome analysis of heteromorphic leaves and hydrophilic roots in response to soil drying in desert Populus euphratica. Sci Rep. 2017;7(1):12188.
Zhang M, Lv D, Ge P, Bian Y, Chen G, Zhu G, Li X, Yan Y. Phosphoproteome analysis reveals new drought response and defense mechanisms of seedling leaves in bread wheat (Triticum aestivum L.). J Proteome. 2014;109:290–308.
Ding S, Zhang B, Qin F. Arabidopsis RZFP34/CHYR1, a ubiquitin E3 ligase, regulates stomatal movement and drought tolerance via SnRK2.6-mediated phosphorylation. Plant Cell. 2015;27(11):3228–44.
Lu X, Li QT, Xiong Q, Li W, Bi YD, Lai YC, Liu XL, Man WQ, Zhang WK, Ma B. The transcriptomic signature of developing soybean seeds reveals the genetic basis of seed trait adaptation during domestication. Plant J. 2016;86(6):530–44.
Li J, Zhu L, Hull JJ, Liang S, Daniell H, Jin S, Zhang X. Transcriptome analysis reveals a comprehensive insect resistance response mechanism in cotton to infestation by the phloem feeding insect Bemisia tabaci (whitefly). Plant Biotechnol J. 2016;14(10):1956–75.
Mishra P, Singh N, Jain A, Jain N, Mishra V, G P, Sandhya KP, Singh NK, Rai V. Identification of cis-regulatory elements associated with salinity and drought stress tolerance in rice from co-expressed gene interaction networks. Bioinformation. 2018;14(3):123–31.
Selengut JD. MDP-1 is a new and distinct member of the haloacid dehalogenase family of aspartate-dependent phosphohydrolases. Biochemistry. 2001;40(42):12704–11.
Fitchette A, Tran O, Dinh L, Bardor M. Plant proteomics and glycosylation. Methods Mol Biol. 2007;355:317–42.
Fortpied J, Maliekal P, Vertommen D, Van Schaftingen E. Magnesium-dependent phosphatase-1 is a protein-fructosamine-6-phosphatase potentially involved in glycation repair. J Biol Chem. 2006;281(27):18378–85.
Chondrogianni N, Petropoulos I, Grimm S, Georgila K, Catalgol B, Friguet B, Grune T, Gonos ES. Protein damage, repair and proteolysis. Mol Asp Med. 2014;35:1–71.
Zheng Z, Qamar SA, Chen Z, Mengiste T. Arabidopsis WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens. Plant J. 2010;48(4):592–605.
Lippok B, Birkenbihl RP, Rivory G, Brummer J, Schmelzer E, Logemann E, Somssich IE. Expression of AtWRKY33 encoding a pathogen- or PAMP-responsive WRKY transcription factor is regulated by a composite dna motif containing W box elements. Mol Plant-Microbe Interact. 2007;20(4):420–9.
Jiang YQ, Deyholos MK. Functional characterization of Arabidopsis NaCl-inducible WRKY25 and WRKY33 transcription factors in abiotic stresses. Plant Mol Biol. 2009;69(1–2):91–105.
Birkenbihl RP, Diezel C, Somssich IE. Arabidopsis WRKY33 is a key transcriptional regulator of hormonal and metabolic responses toward Botrytis cinerea infection. Plant Physiol. 2012;159(1):266–85.
Zhou J, Wang J, Zheng Z, Fan B, Yu JQ, Chen Z. Characterization of the promoter and extended C-terminal domain of Arabidopsis WRKY33 and functional analysis of tomato WRKY33 homologues in plant stress responses. J Exp Bot. 2015;66(15):4567.
Lai Z, Li Y, Wang F, Cheng Y, Fan B, Yu JQ, Chen Z. Arabidopsis sigma factor binding proteins are activators of the WRKY33 transcription factor in plant defense. Plant Cell. 2011;23(10):3824–41.
Cheng M, Hsieh E, Chen J, Chen H, Lin T. Arabidopsis RGLG2, functioning as a RING E3 ligase, interacts with AtERF53 and negatively regulates the plant drought stress response. Plant Physiol. 2012;158(1):363–75.
Zhu X, Robe E, Jomat L, Aldon D, Mazars C, Galaud J. CML8, an Arabidopsis calmodulin-like protein, plays a role in Pseudomonas syringae plant immunity. Plant Cell Physiol. 2017;58(2):307–19.
Yin XM, Huang LF, Zhang X, Wang ML, Xu GY, Xia X. OsCML4 improves drought tolerance through scavenging of reactive oxygen species in rice. J Plant Biol. 2015;58(1):68–73.
Shoaib M, Liu H, Xing Y, Saddam H, Ouyang B, Zhang Y, Li H, Ye Z. Overexpression of calmodulin-like (ShCML44) stress-responsive gene from Solanum habrochaites enhances tolerance to multiple abiotic stresses. Sci Rep. 2016;6:31772.
Smart RE, Bingham GE. Rapid estimates of relative water content. Plant Physiol. 1974;53(2):258–60.
Bates LS, Waldren RP, Teare ID. Rapid determination of free proline for water-stress studies. Plant Soil. 1973;39(1):205–7.
Dubois M, Gilles KA, Hamilton JK, Rebers PA, Smith F. Colorimetric method for determination of sugars and related substances. Anal Chem. 1956;28(3):350–6.
Beauchamp C, Fridovich I. Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Anal Biochem. 1971;44:276–87.
Beers RF, Sizer IW. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J Biol Chem. 1952;195(1):133–40.
Kar M, Mishra D. Catalase, peroxidase, and polyphenoloxidase activities during rice leaf senescence. Plant Physiol. 1976;57(2):315–9.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):1–13.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, Van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–5.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):1–12.
Conesa A, Gotz S, Garciagomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–6.
Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999;28(1):27–30.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9(1):559.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Jia H, Li J, Zhang J, Ren Y, Hu J, Lu M. Genome-wide survey and expression analysis of the stress-associated protein gene family in desert poplar, Populus euphratica. Tree Genet Genomes. 2016;12(4):78.
Clough SJ, Bent AF. Floral dip: a simplified method for agrobacterium–mediated transformation of Arabidopsis thaliana. Plant J. 1998;16(6):735–43.
Acknowledgements
We would like to thank the reviewers and editors for their careful reading and helpful comments on this manuscript.
Funding
This work was supported by National Nonprofit Institute Research Grant of CAF (CAFYBB2017ZY008), (CAFYBB2018ZY001–9), and Natural Science Foundation of China (31800570). The founders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
HJ, JZ, JL and JH conceived and designed the research; JL, PS and YZ performed all experiments; HJ and JZ conducted data analysis; HJ, JZ and JL wrote the manuscript; XX, ML and JH contributed with valuable discussions. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Output statistics of RNA-Sequencing. (XLSX 11 kb)
Additional file 2:
Figure S1. GO terms for molecular functions of DEGs during drought stress in S. psammophila. Only significantly enriched terms with corrected P < 0.05 were indicated. The color and size of each point represented the -log10 (FDR) values and enrichment scores. Figure S2. GO terms for cellular components of DEGs during drought stress in S. psammophila. Only significantly enriched terms with corrected P < 0.05 were indicated. The color and size of each point represented the -log10 (FDR) values and enrichment scores. Figure S3. KEGG enrichment analysis of DEGs during drought stress in S. psammophila. Only significantly enriched pathways with corrected P < 0.05 were indicated. The color and size of each point represented the -log10 (FDR) values and enrichment scores. Figure S4. Identification of 672 transcription factors (TFs) representing 45 gene families in the nine clusters. The color and size of each point both represented TF number. Figure S5. Identification of co-expression modules and their relationships. a Hierarchical cluster tree showing six co-expression modules identified by weighted gene co-expression network analysis (WGCNA). Modules corresponding to branches are labelled with colours indicated by the colour bands underneath the tree. b Cluster relationships of these six modules. Figure S6. Expression patterns of co-expression genes with SpMDP1 in WT and transgenic plants. Expression patterns of up-regulated (a) and down-regulated (b) genes in SpMDP1-overexpressing plants. The expression level of each gene in the WT was set to 1. Figure S7. Expression patterns of co-expression genes with SpWRKY33 in WT and transgenic plants. The expression level of each gene in the WT was set to 1. (PDF 1002 kb)
Additional file 3:
GO enrichment analysis of all DEGs. (XLSX 82 kb)
Additional file 4:
KEGG enrichment analysis of all DEGs. (XLSX 14 kb)
Additional file 5:
GO enrichment analysis of nine clusters. (XLSX 45 kb)
Additional file 6:
The genes in co-expression network. (XLSX 30 kb)
Additional file 7:
The co-expressed genes with SpMDP1. (XLSX 31 kb)
Additional file 8:
The co-expressed genes with SpWRKY33. (XLSX 10 kb)
Additional file 9:
The primers of eight hub genes and co-expressed genes used in qRT-PCR analysis. (XLSX 12 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jia, H., Zhang, J., Li, J. et al. Genome-wide transcriptomic analysis of a desert willow, Salix psammophila, reveals the function of hub genes SpMDP1 and SpWRKY33 in drought tolerance. BMC Plant Biol 19, 356 (2019). https://doi.org/10.1186/s12870-019-1900-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-019-1900-1