
Abstract
Background
Drought is a major environmental stress that can have severe impacts on plant productivity and survival. Understanding molecular mechanisms of drought responses is crucial in order to breed for drought adapted plant cultivars. The aim of the present study was to investigate phenotypic and transcriptional drought responses in two willow genotypes (520 and 592) originating from an experimental cross between S. viminalis × (S. viminalis × S. schwerinii). Willows are woody perennials in the Salicaceae plant family that are grown as bioenergy crops worldwide.
Methods
An experiment was conducted where plants were exposed to drought and different eco-physiological parameters were assessed. RNA-seq data was furthermore generated with the Illumina technology from root tips and leaves from plants grown in drought and well-watered (WW) conditions. The RNA-seq data was assembled de novo with the Trinity assembler to create a reference gene set to which the reads were mapped in order to obtain differentially expressed genes (DEGs) between the drought and WW conditions. To investigate molecular mechanisms involved in the drought response, GO enrichment analyses were conducted. Candidate genes with a putative function in the drought response were also identified.
Results
A total of 52,599 gene models were obtained and after filtering on gene expression (FPKM ≥ 1), 35,733 gene models remained, of which 24,421 contained open reading frames. A total of 5,112 unique DEGs were identified between drought and WW conditions, of which the majority were found in the root tips. Phenotypically, genotype 592 displayed less growth reduction in response to drought compared to genotype 520. At the transcriptional level, genotype 520 displayed a greater response in the leaves as more DEGs were found in genotype 520 compared to genotype 592. In contrast, the transcriptional responses in the root tips were rather similar between the two genotypes. A core set of candidate genes encoding proteins with a putative function in drought response was identified, for example MYBs and bZIPs as well as chlorophyll a/b binding proteins.
Discussion
We found substantial differences in drought responses between the genotypes, both at the phenotypic and transcriptional levels. In addition to the genotypic variation in several traits, we also found indications for genotypic variation in trait plasticity, which could play a role in drought adaptation. Furthermore, the two genotypes displayed overall similar transcriptional responses in the root tips, but more variation in the leaves. It is thus possible that the observed phenotypic differences could be a result of transcriptional differences mostly at the leaf level.
Conclusions
This study has contributed to a better general understanding of drought responses in woody plants, specifically in willows, and has implications for breeding research towards more drought adapted plants.
Similar content being viewed by others


Background
Drought is a major environmental stress that can have severe impacts on plant productivity and survival. Upon drought, plants will perform an array of complex responses, involving molecular, biochemical, physiological and morphological changes [1, 2]. As a result of adaptation to environmental conditions, for example water availability and drought, various drought avoidance and tolerance mechanisms have evolved. These mechanisms include whole-plant changes such as shoot-root allocation, growth rate, leaf morphology, leaf abscission, stomatal conductance and photosynthetic rate, as well as molecular changes underpinned by remodelling of the transcriptome that may include upregulation of stress signalling, transcription factors and defence processes [1, 3]. Due to the availability of the Populus trichocarpa genome sequence [4], transcriptional responses to abiotic stresses in woody plants have primarily been studied in various poplar species. Comparisons of gene expression levels between samples from different tissues, time points and treatments has been performed using microarrays [5–7] or more recently massively parallel sequencing of total RNA, i.e. RNA-seq [8–10]. As a result, a wealth of information on genes and processes involved in the molecular mechanisms of drought responses in different poplar species is available. These studies have revealed large variation in transcriptional drought responses between different species. For example, in the leaves of P. trichocarpa, 5689 genes were differentially expressed between drought and control conditions [10], while in the leaves of P. balsamifera only 98 probe sets displayed increased transcript abundance under drought [11]. Other studies have revealed variation in drought responses between populations [12–14] and even between genotypes of the same species [11]. These observed differences could originate from genetic diversification as a result of adaption to different drought conditions [15]. Such diversifying selection should be visible as genetic variation in important genes involved in drought response. Thus, identifying genetic variation associated with variation in phenotypic responses, could provide valuable insight into the genetic basis of drought adaptation. The ultimate goal would be to pinpoint genetic variants that are associated with phenotypic responses to drought that could then be used in marker assisted selection to produce cultivars better adapted to drought conditions. In order to do so, there is an urgent need for information on phenotypic and transcriptional responses from species other than model-species. In this work we have examined and compared drought responses in willow genotypes (genus Salix, the sister genus to Populus) that, like poplars, are woody perennials in the Salicaceae family. Species from both genera have for a long time been grown as bioenergy crops worldwide and there are active breeding programs for developing new high performing varieties [16, 17]. Willows are particularly suitable for cultivations in regions with good water supply such as most of Northern Europe [18]. However, considering that willows appear to be prone to drought stress [19, 20], drought tolerant varieties are desired for cultivations in southern Europe [21]. For the purpose of bioenergy, Salix viminalis, S. dascyclados and S. schwerinii and their hybrids are the mostly used species in Europe as they display rapid growth and high biomass yields [16, 22]. There is relatively high genetic diversity in natural populations of willows [23], which have proven useful for the identification of quantitative trait loci (QTLs) of different traits such as frost and rust resistance and phenology [24–28], growth, water-use efficiency and drought tolerance [19, 29, 30].
The aim of the present study was to investigate phenotypic and transcriptional drought responses in two willow genotypes originating from an experimental cross between S. viminalis × (S. viminalis × S. schwerinii) [31]. In order to achieve this, we performed a controlled experiment in a phytotrone where a number of phenotypic measurements were assessed. Since different organs show very different responses to drought [32–34] we estimated the effects of drought separately in mature leaves and root tips. We performed massive parallel sequencing of RNA (RNA-seq) on the Illumina platform, assembled the sequencing reads de novo into gene models and then mapped the reads back to the gene models in order to quantify the level of gene expression per gene model among the different samples. We then performed GO enrichment analyses on the set of differentially expressed genes to identify important functional categories involved in the response to drought in the two tissues and genotypes.
Results
Phenotypic responses to drought
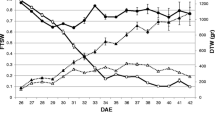
Plants grown in WW conditions were both taller (mean (± standard deviation (SD)) 135.5 ± 6.4 cm), had more sylleptic shoots and greater total root, shoot and leaf biomasses compared to the drought stressed plants (height mean 84.3 ± 10.5 cm; Fig. 1a, d, e, i, m; Table 1, main effect treatment). These differences were established already shortly after the onset of the experimental treatments, and the same overall pattern remained across the whole experimental period (Fig. 2a for plant height). Leaf chlorophyll concentration, here assessed in terms of SPAD i.e. leaf nitrogen (N) per leaf area [35], was significantly higher in the drought treatment compared to the WW condition (Table 1), and also that pattern was similar across the entire experimental period (Fig. 2b). In contrast to the leaf chlorophyll (or leaf N) concentration, the total accumulation of N was reduced by drought. Thus, at the end of the experiment, total number of leaves and total N accumulation in leaves, which is a main driver for growth, were greatly reduced by drought (Fig. 1b; Table 1, main effect treatment). The drought-exposed plants also had greater relative biomass allocation to roots compared to the WW plants (Fig. 1n; Table 1, main effect treatment). In contrast to roots, relative shoot and leaf allocation (SW/W, LW/W) was not significantly affected by drought stress (Fig. 1f, k; Table 1).

Means (± SD) for various eco-physiological traits in genotype 592 and and 520. The genotypes were grown in a growth chamber (three blocks) and exposed to two experimental conditions (well-watered (white) and drought (black)). DW = dry weight (biomass), RGR = relative growth rate, LAR = leaf area ratio, LAP = leaf area productivity, SLA = specific leaf area, RWC = relative water content, SPAD values indicate leaf chlorophyll content

Means for shoot height (a) and SPAD value (b) in genotype 592 and 520 assessed non-destructively during the experimental treatment period in April. The genotypes were grown in a growth chamber (three blocks) and exposed to two experimental conditions, i.e. well-watered (open symbols) and drought (closed symbols). Mean standard deviation (SD) across all treatments and genotypes is indicated in the lower left corner
Many traits differed between the two genotypes (Table 1, main effect genotype). At the end of the experiment, mean height growth was significantly higher for genotype 592 compared to 520, both in the WW and in the drought conditions (Fig. 2a; ANOVA: main effect genotype, p-value (p) = 0.007, not shown). Genotype 592 also produced many more sylleptic shoots than genotype 520, but had fewer leaves and lower total leaf N pool (Fig. 1b, d, o; Table 1, main effect genotype). Mean total biomass at the start of the experimental treatments was similar in the two treatments (ANOVA: main effect genotype p = 0.231). The biomass allocation to roots (RW/W) was similar between the genotypes, whilst leaf and shoot allocation varied significantly between the genotypes (Fig. 1f, j, n; Table 1 main effects genotype). There were also several significant interaction effects between genotype and treatment, indicating differential drought responses for the two genotypes. For example, genotype 592 displayed greater drought-induced increase in root biomass allocation (RW/W) than 520; leaf biomass allocation decreased (i.e. 520) and increased (i.e. 592) in response to drought; and shoot biomass allocation increased in response to drought only in genotype 520 (Fig. 1f, j, n; Table 1 Genotype x Treatment interaction (G x T) effects). In contrast, genotype 520 was characterised by stronger drought-induced reduction of leaf number and total leaf N pool, whilst 592 showed stronger drought-induced reduction in the number of sylleptic shoots (Fig. 1b, d, o; Table 1, G x T effects).
Relative growth rate (RGR) was greatly reduced by drought condition (Table 1, main effect treatment), but similar between the genotypes (main effect genotype). The drought-induced reduction in RGR was accomplished by corresponding reductions in leaf area ratio (LAR), specific leaf area (SLA) and biomass productivity per unit leaf area (LAP) (Eqn. 1 & 2) (Table 1, main effect treatment). As indicated by significant genotype by treatment interactions, the reduction of RGR, total plant DW and total leaf N pool by drought was more pronounced in 520 than 592 (Fig. 1a, b, c; Table 1, G x T effects). That genotype (i.e. 520) was characterized by generally higher leaf area ratio (LAR) and lower leaf area productivity (LAP) compared to 592 (eqn 1; Fig. 1g, k; Table 1, main effect genotype). Following eqn. 2, the greater LAR of 520 was mostly a consequence of greater leaf biomass allocation (LW/W; Table 1, main effect genotype; Fig. 1f). In contrast, the higher LAP of 592 was a consequence of greater leaf chlorophyll content (SPAD index; Fig. 1l; Table 1, main effect genotype) in that genotype. Ultimately, the RGR of the high-SPAD genotype (i.e. 592), was less affected by drought compared to the high-LAR genotype (i.e. 520).
Leaf temperature and relative water content (RWC) varied between growth conditions, but were similar between genotypes (Fig. 1p, q; Table 1, main effect treatment and genotype). In drought condition, RWC decreased, leaf temperature increased, stomata were more closed (higher leaf Δ18O indicates lower stomatal conductance, g s ), intrinsic water use efficiency was higher (lower leaf Δ13C indicates higher intrinsic water use efficiency), and also carboxylation efficiency was higher (lower ratio of Δ13C and Δ18O indicates higher carboxylation efficiency) (Fig. 1p-t, Table 1, main effect treatment). Genotype affected isotope ratios, and the genotype 592 had lower intrinsic water use efficiency (i.e. higher leaf Δ13C) and lower carboxylation efficiency (i.e. higher ratio of Δ13C and Δ18O) than genotype 520 (Table 1, main effect genotype). In general, leaf gas exchange as reflected by the isotope data indicates small genotype differences under WW conditions along with great genotype differences in drought conditions. This pattern was statistically supported by highly significant interaction effects in all isotope measures (Fig. 1r-t, Table 1). More specifically, the drought-induced decrease in stomatal conductance (i.e. increase in leaf Δ18O) was less pronounced in 592 compared to 520. Also drought-induced increase in intrinsic water use efficiency (i.e. decrease in leaf Δ13C) was less pronounced in 592 than in 520 (Table 1, G x T effects).
De novo assembly and identification of DEGs
Illumina sequencing of 36 libraries prepared from RNA extracted from leaf and root tip tissues from two willow genotypes generated 7.91 × 108 paired-end sequencing read pairs that corresponds to 158 × 109 base pairs (bp) (Table 2). Sequencing read pair numbers per library varied between 18.1 and 33.3 million and did not differ significantly between tissues (ANOVA: p = 0.60), genotypes (ANOVA: p = 0.55) or treatment (ANOVA: p = 0.39). When all sequencing reads were combined and assembled de novo using the Trinity assembler, 91,701 contigs were obtained, which represented 52,599 gene models. After filtering on gene expression(fragments per kilobase of transcript per million mapped fragments (FPKM) ≥ 1), 35,733 gene models remained of which 24,421 contained open reading frames (Additional file 1). The gene models in this high confidence gene set ranged in size from 301 to 16,559 bp with a mean length of 1485 bp and a median length of 1273 bp (Additional file 2). 40.6 % of all reads mapped to the high confidence gene set with the parameters used for expression analysis. The majority of them (99.7 %) mapped to one unique position. More detailed mapping statistics can be found in Table 2. The program edgeR was used with the “calcNormFactors()” function to identify DEGs between drought and WW conditions in: 1) leaves of genotype 592 (five replicates), 2) leaves of genotype 520 (five replicates), 3) root tips of genotype 592 (four replicates) and 4) root tips of genotype 520 (four replicates). Normalized read counts are listed in Additional file 3 and normalization factors used in the edgeR analyses are listed in Additional file 4. Genes were defined as upregulated DEGs if false discovery rate (FDR) was ≤ 0.05 and log2 fold change (FC) ≥ 1 and as downregulated DEGs if FDR was ≤ 0.05 and log2 FC ≤ −1. A total of 6935 DEGs were identified across genotypes and tissues, representing 5112 unique gene models. 3082 DEGs were upregulated (of which 2175 were unique) and 3853 were downregulated (of which 2974 were unique) (Table 3, Additional file 5). Much fewer DEGs were found in the leaves than in the root tips (Table 3, Additional file 5), a pattern that was consistent across both genotypes. This result manifests the small effects of drought on the overall transcription in the young mature leaves in these two genotypes and shows that root tips instead have a much stronger transcriptional response to drought than leaves do. When comparing the two genotypes, more DEGs were found in the leaves of genotype 520 compared to genotype 592, while the number of DEGs in the root tips was more similar between the genotypes (this was particularly evident for the upregulated DEGs) (Table 3). Twenty-eight genes (Additional file 5) displayed significant G x T effects when using a generalized linear model and the gene ontology (GO) term ADP-binding was significantly enriched among these genes. When comparing the drought responses in the leaves and the root tips of the two genotypes for these genes, 25 displayed greater drought responses in leaves than in the root tips. The hypothesis that there was an equal chance that the effect is stronger in either tissue could thus be rejected (p = 2.74 × 10−5, binomial test see Additional file 6). This result further strengthens the finding of greater genotypic differences in the drought responses in the leaves than in the roots.
Functional annotation and GO enrichment analysis
The Blast2GO software tool was used to functionally annotate the de novo assembled gene products and for GO enrichment analyses in order to identify functions and genes involved in drought stress responses in the leaves and root tips. Of the 24,421 genes in the high confidence gene set, 15,980 were annotated with a GO term.
The GO enrichment analysis was done for up and downregulated annotated DEGs in leaves and root tips for each genotype separately using the annotated high confidence gene set as reference. All enriched GO terms in every comparison are presented in Additional file 7. Overall, few GO terms were enriched for the DEGs in the leaves, in fact none was significantly enriched for the DEGs in genotype 592, suggesting that drought had little impact on the function of the leaves in this genotype. However, genotype 520 showed a greater functional drought response as several GO terms were significantly enriched, particularly for the upregulated DEGs. For example at the molecular function (F) ontology level, “peptidyl-prolyl cis-trans isomerase activity” was one of the most significantly overrepresented terms and at the biological process (P) ontology level, different response and regulation terms were significantly overrepresented (Additional file 7). At the cellular component (C) ontology level, several terms related to the thylakoid and the cell wall were overrepresented (Additional file 7). For the downregulated DEGs at the F ontology level, “oxygen binding” was the most significantly overrepresented term (Additional file 7). At the P ontology level, “arachidonic acid metabolic process” and “epoxygenase P450 pathway” were overrepresented. No term was significantly overrepresented at the C ontology level.
Many more GO terms were enriched for the DEGs in the root tips compared to the leaves, which demonstrates the greater drought responses in this tissue compared to the leaves. In Fig. 3 we present overrepresented GO terms (FDR < 0.0001) in the P ontology level associated with upregulated (Fig. 3a) and downregulated (Fig. 3b) DEGs for both genotypes. Many GO terms were significantly or nearly significantly enriched in both genotypes, which means that the genotypes displayed overall similar responses to drought in the root tips. For the upregulated DEGs, the most affected functions were associated with biosynthetic and metabolic processes (Fig. 3a).

Over-represented GO terms at the biological process ontology level for up- (a) and downregulated (b) DEGs in the root tips of genotype 592 and genotype 520. The red bars show the number of DEGs annotated with the GO terms in the root tips of genotype 592 and the blue bars show the number of DEGs annotated with the GO terms in the root tips of genotype 520. GO terms are presented if they were enriched (FDR < 1 × 10−4) in at least one of the two genotypes and if at least ten genes were enriched for that term. Solid bars represent a significant over-representation of the GO term in this genotype while fainted bars are given as reference if the significance level was not reached
A core set of candidate genes involved in the drought response
To identify candidate genes involved in drought stress responses in the two willow genotypes, we extracted the upregulated DEGs in the root tips of both genotypes annotated with any of the 15 “response” GO terms listed in Table 4. This resulted in a list with 115 genes for genotype 592 and 141 genes for genotype 520 (Additional file 8). Based on annotations and reported functions in stress responses, a core set of 28 candidate genes were identified (Table 5) that could function as targets for detailed functional studies of drought responses at the molecular level in willows and related species. Several of the genes were homologous to genes encoding transcription factors, e.g. MYBs, WRKYs, bZIPs and heat stress transcription factors with known functions in stress responses. Other genes were homologous to genes encoding dehydrin and chlorophyll a/b binding proteins.
Discussion
Genotype specific physiological responses to drought
This study accommodated a growth experiment in which cuttings of two genotypes were grown in an irrigation contrast for sufficiently long time to develop considerable variation both at the transcriptional and phenotypic levels. We demonstrated that the two genotypes displayed contrasting phenotypic responses to drought. Genotype 592 was overall less affected by drought as it displayed a weaker growth reduction. The weaker growth reduction of 592 was associated with greater increase in root biomass allocation (RW/W) in response to drought, enhancing the capacity to explore available water and nutrient resources. Interestingly, genotype 592 also displayed generally higher foliar SPAD values, which is in agreement with the hypothesis that higher area-based leaf N content is an acclimation to drought [36]. In contrast to 592, genotype 520 displayed generally higher evapotranspiration area (e.g. LAR) and responded to drought with strong leaf area reduction, greatly decreased stomatal conductance and increased intrinsic water use efficiency. Thus, in addition to the observed genotypic variation in mean traits (e.g. LAR, LAP, SPAD), we also found indications for genotype differences in trait variability in a drought contrast, especially for the stomatal physiology traits (leaf Δ13C and Δ18O). Genotypic differences in trait variability, observed in a drought contrast, may be indicative of genotypic variation in trait plasticity in relation to drought. Large genotypic variation in trait plasticity of leaf traits similar to the ones in our study was found among Eucalyptus provenances originating from a rainfall gradient in Australia, and interpreted as important features in the climate adaptation of these trees [37].
Generation of the high confidence gene set
De novo assembly of all sequencing reads generated 52,599 gene models and after filtering out lowly expressed gene models, 35,733 remained. In a next filtering step, gene models that did not contain an open reading frame were filtered out, leaving 24,421 gene models in the high confidence gene set. Out of these, 18,128 were functionally annotated with a GO term in Blast2Go. Although it is problematic to compare these numbers with those from other studies as different tissues and also filtering criteria are often used, de novo assembly of RNA-seq data in P. pruinosa resulted in 48,653 gene models [38] and in S. matsudana 48,817 unigenes were retrieved [39], showing that the figures were overall rather similar across the different species. The number of annotated genes in this high confidence set was considerably higher than the number of gene models annotated with a GO term in P. pruinosa and P. euphratica where 11,587 [40] and 9296 in [9] were reported respectively, possibly reflecting improved annotations tools.
Variation in the number of DEGs and enriched GO terms between the two genotypes and tissues
When comparing the responses to drought between the two tissues we found that drought affected gene expression of many more genes in the root tips compared to the leaves, a result that was consistent across both genotypes. A similar pattern was previously observed in hybrid poplars (P. deltoides × P. nigra) [7] where about twice as many probe sets displayed a significant change in response to drought in the root tips compared to the mature leaves. The authors proposed that this difference could in part be explained by the higher sensitivity to water deprivation in an actively growing tissue, i.e. the root tips. Judging from the number of DEGs in the root tips, the two genotypes displayed rather similar responses. This similarity was also evident in the enriched GO terms associated with these DEGs that were present in both genotypes. A somehow more unexpected result was the numerous enriched terms in the cellular component level that was related to the chloroplast. The overall lack of response of drought on gene expression in the mature leaves was also unexpected as several previous studies have reported a greater response in leaves. For instance, in P. trichocarpa 5689 DEGs were identified in leaves [10], in P. euphratica, where 5083 genes were differentially expressed in leaves [8] and in hybrid poplar (P. deltoides × P. nigra) 2120 were affected in leaves [7]. Results from the present study instead resemble the results in P. balsamifera where 98 probe sets displayed increased transcript abundance to drought [11]. Less responses were also seen in shoots of P. balsamifera [6]. In contrast to the root tips, the genotypes displayed a greater difference in the responses in the leaves with much fewer DEGs and enriched GO terms in genotype 592 than in genotype 520. Strong genotypic effects were previously found in leaves of hybrid poplars [7] and P. balsamifera (six genotypes) [11]. This was also seen in shoots of hybrid poplar clones (P. deltoides × P. nigra and P. nigra × P. maximowiczii) [6]. Interestingly, genotype 592 also displayed less physiological responses at the leaf level compared to genotype 520 supporting a correlation between transcriptional and physiological responses. For example, the drought-induced shifts in stomatal physiology (as reflected by Δ13C and Δ18O) were more pronounced in the genotype 520, which coincides with the overrepresented GO terms “thylakoid” and cell wall in the cellular component ontology level. Among the 28 genes that displayed significant G x T effects, several encoded disease resistant proteins and leucine-rich repeat proteins. An exciting hypothesis is that the two genotypes carry different alleles at these genes rendering different drought responses.
A core set of candidate genes involved in drought stress responses
We have produced a core set of candidate genes that we find particularly interesting and that should be further examined for their role in the molecular stress responses and we discuss some of them here. Among our candidate genes were several transcription factors i.e. MYBs, bZIPs and WRKYs, with known functions in ABA-dependent stress responses [41]. They among other things regulate stress responses via modulation of transcription of downstream genes. Furthermore, several candidate genes encoded chlorophyll a/b binding proteins (i.e. CABs). Typical for this protein family is the CAB domain, containing the amino acid residues involved in pigment binding. There are some indications that they function in high-light protection in a broad sense and that they encode homologs to light-harvesting-like proteins (LILs). All organisms performing oxygenic photosynthesis also contain LILs [42], which are highly homologous to the higher plant light-harvesting antenna and contain the CAB domain. The LILs are regulated opposite to the light-harvesting complex proteins (LHCs); under high light condition - when the expression of LHCs is repressed - the LILs are upregulated. While in cyanobacteria LILs have been found to respond to different stresses, their function in plant was thought to be restricted to light stress [43]. It is thus possible that these genes and gene products play an important role in the stress response – even in non-photosynthetic tissue. Also in Arabidopsis [44] and rice [45] enhanced expression of such genes was observed in roots exposed to drought stress, indicating a general importance of these genes independent of the species.
Conclusions
In this study we report the first large transcriptome study in willows where we have compared physiological and transcriptional responses to drought between two genotypes and two tissues. A de novo assembly and subsequent filtering of root tip and leaf transcriptomes across the two genotypes and two conditions, generated a set with 24,421 genes. A total of 5112 unique DEGs were identified between the drought and WW conditions, of which the majority were found in the root tips. We also found substantial differences in drought responses between the genotypes, both at the phenotypic and trancriptional level. Phenotypically, genotype 592 displayed less growth reduction in response to drought compared to genotype 520, which suggests that this genotype is more drought adapted than genotype 520. In addition to the genotypic variation in several mean traits, we also found considerable genotypic variation in trait plasticity (especially leaf traits), which may play a role in drought adaptation. At the transcriptional level, genotype 520 displayed a greater response in the leaves as many more DEGs were found compared to genotype 592. As the two genotypes displayed overall similar transcriptional responses in the root tips, but more variation in the leaves, it is possible that the observed phenotypic differences between the genotypes are due to transcriptional differences in their leaves. We also identified a core set of candidate genes encoding proteins with a putative function in drought response, for example MYBs, bZIPs and WRKYs and chlorophyll a/b binding proteins. Knowledge from this study increases our understanding of the physiological and molecular basis of drought responses in willows, however these results are applicable to all woody plants.
Methods
Plant material
Two Salix genotypes (520 and 592) from the S1 pedigree (N = 463) [31] were used in this study. S1 is a cross between the female clone 78183, a diploid S. viminalis and Björn, a diploid Salix viminalis L × S. schwerinii E Wolf hybrid male. Results from previous greenhouse and phytotron experiments showed that both genotype 520 and 592 were high producing clones, both in WW and in drought conditions with repeated periods of drought [30]. Interestingly, they displayed different responses to drought, as an example, genotype 592 lost many more leaves than 520 [30].
The S1 population is planted and conserved in a plant archive owned by the Swedish University of Agricultural Sciences, near Uppsala (59°49’ N 17°40’ E, central Sweden) where dormant shoots from the two genotypes were collected the 2nd of February 2011 and stored in −4 °C. On the 17th of February, fifty, six cm long cuttings were prepared for each genotype and planted in 3-L pots filled with commercial soil (“Weibull’s Krukväxtjord Lera & Kisel”, organic matter 95 %; pH 5.5-6.5; 182 g/m3 N, 91 g/m3 P, 195 g/m3 K, maximum 50 g/m3 micro nutrients). The relatively small pot size and the resulting high biomass to substrate volume ratio at final harvest has probably decreased growth in all genotypes and treatments compared to field conditions [46], but visible observation revealed that roots never occupied more than approximately half of the substrate volume per pot at final harvest. The experiment was conducted in one walk-in growth chamber where the plants were organized in three groups. Each group consisted of equal number of plants per genotype, thus each group represented one block or replicate. The plants were grown for 38 days under 20 °C constant temperature, 70 % relative humidity and 16 h photoperiod (300 μmol PAR m−2 s−1) with regular watering. The positions of the plants within the blocks as well as the positions of the blocks in the chamber were changed every day and the plants were pruned so that only the main shoots were preserved. For the drought treatments starting the 28th of March, 30 plants at the same growth stage of each genotype were selected, of which 15 were assigned as controls with regular watering (well-watered or WW condition) and 15 subjected to drought stress (drought condition) for 31 days. The aim was to keep the drought stressed plants just above their wilting point throughout the treatment period. The wilting point or the minimal amount of soil moisture the plants required not to wilt was therefore studied in six plants for each genotype prior to the drought stress treatment was initiated. The plants were weighed daily until they started to wilt (no water was supplied). To apply drought stress, the onset of visible wilting signs (“wilting point”) of individual plants was used as a physiological drought stress indicator. Thus, in the drought treatment, any water supply to individual pots was initially withheld until the plants had reached their individual wilting points, at which pot weights were determined. At a daily basis throughout the experimental period in April, all pots were weighed and water was added in the necessary amounts to keep the substrate at a more or less constant minimum water supply level. Applying this procedure, all plants in the drought stress condition were kept close to their wilting points throughout the whole treatment period. The WW plants were regularly watered to field capacity of the soil throughout the treatment period and weighed every day. To account for increased consumption as the plants grew, watering quantities were slightly increased throughout the treatment period. When the drought stressed plants were kept near the wilting point, they received approximately 50 % of the water supplied to the WW plants.
Eco-physiological measurements
Two harvests were carried out during the experiment, one initial harvest just before the start of the treatment period (28th of March) and a final harvest at the end of the treatment period (27th - 28th of April). The harvested plants (five plants per block, treatment and genotype) were separated into leaves, stems, the original cutting, roots, sylleptic shoots and sylleptic leaves and all parts were oven dried for 48 h at 70 °C. Excised leaves were not included. Leaf areas were determined on fresh leaves with an area meter (LI-3100, LiCor Inc., Lincoln, NE, USA). Both for the initial and the final harvest, the dry weights of each plant compartment as well as leaf areas of all plants were assessed. The total dry weight (DW) per plant was estimated as the sum of all plant compartments excluding the cutting. Height was assessed non-destructively throughout the treatment period. Height was defined as length from the base of the shoot to the thickest part of the apical bud. Leaf chlorophyll content (SPAD index) of one leaf (per plant) in the upper middle canopy was measured non-destructively at the same occasions as plant height by using a portable chlorophyll meter (SPAD-502, Konica Minolta Sensing Inc. Japan). In Salix spp. the SPAD index has previously been shown to be closely related to leaf nitrogen (N) content [35]. Also leaf temperature was measured on the same leaf using a portable infrared thermometer (IR400, Extech Instruments). An average of five measurements per leaf was used for both assessments. In general, relative water content (RWC) is a measure of drought resistance [47] and was measured on leaves sampled from all plants at the day of the final harvest. For determination of RWC, two fully developed leaves per plant were sampled. A simplified measure of RWC was determined as described by Weih and Nordh [48] and indicates here the rate of water loss from the leaf tissue. Leaves were weighted directly after sampling (wt0) and placed on sheets of filter paper in room temperature. Leaves were weighted again after 4 h (wt4) and after they had oven dried for 24 h at 70 °C (wt24). RWC was calculated according to the following formula: RWC (%) = (wt4 – wt24) / (wt0 – wt24) × 100.
The sampled leaves were used for total nitrogen (N); carbon and oxygen isotope analysis performed by Iso-Analytical Limited, Crewe, UK. All analyses were conducted by Elemental Analyser Isotope Ratio Mass Spectrometry (EA-IRMS, Europa Scientific 20-20). Leaf N concentrations were multiplied with total leaf DW of all leaves per plant to obtain total leaf N pool. Leaf carbon and oxygen isotope measurements reflect important characteristics of gas exchange at leaf level and integrate over the lifetime of the leaf. Thus, leaf Δ13C is correlated with intercellular CO2 concentration C i , and higher C i indicates lower intrinsic water use efficiency [49]. Leaf Δ18O is a measure of stomatal conductance (g s ), and increased leaf Δ18O is correlated with decreased g s [49, 50]. Finally, the ratio of Δ13C and Δ18O (i.e. C i /g s ) represents an estimate of carboxylation efficiency, and higher C i /g s is correlated with lower carboxylation efficiency [51]. The reference material used for δ13C analysis of the samples was IA-R001 (wheat flour, δ13CV-PDB = −26.43 ‰). Carbon isotope ratio of leaf samples (δ13Csample) is expressed as: δ13Csample (‰) = (Rsample / RPDB – 1) × 1000, where Rsample and RPDB are the 13C/12C molar abundance ratio of the leaf material and PeeDee Belemnite standard. Carbon isotope discrimination (Δ13C, ‰) was calculated assuming the isotope composition of atmospheric air (δ13Cair) to be −8 ‰ [51] and computed as: Δ13C = (δ13Cair – δ13Csample) / (1 + δ13Csample/1000). The reference material used for δ18O analysis was IAEA-CH-6 (sucrose, δ18OV-SMOW = 36.4 ‰). The δ18O of the irrigation water (CO2:H2O equilibrium technique) was −13.8 ‰. The 18O enrichment over the irrigation water (Δ18Osample) was computed as: Δ18Osample (‰) = δ18Osample – δ18Oirrigation water. The isotope ratio was calculated as Δ13C / Δ18Osample [51].
Changes in plant growth over time were analyzed using the methods of classical growth analysis [52] and based on biomass and leaf area changes between the two consecutive harvests. Thus possible differences in relative growth rate (RGR, final plant biomass per initial plant biomass and week, g g−1 wk−1) between genotypes were related to differences in leaf area ratio (LAR, leaf area per total plant biomass, m2 g−1); leaf area productivity (LAP, total biomass growth per leaf area and week, g m−2 wk−1); specific leaf area (SLA, leaf area per leaf biomass, m2 kg−1); and leaf biomass fraction (LW/W, leaf biomass per total plant biomass, %). The following relationships among growth traits were utilized to analyze differences among the treatments and clones [53]:
RNA extractions and sequencing
The day before the final harvest, two fully developed young leaves and about one centimetre of several root tips were collected from each plant that were immediately after harvesting snap frozen in liquid nitrogen and stored in −80 °C awaiting RNA extractions. Total RNA was extracted from 40 plants representing five biological replicates from leaves and root tips from the two genotypes in drought and WW conditions. Approximately 100 mg of leaves and 30 mg of root tips were taken for RNA extractions using Spectrum Plant Total RNA Kit (Sigma-Aldrich) with the On-Column DNase I digestion set (Sigma-Aldrich). After quality checking on a 2100 Bioanalyzer (Agilent), four root tip samples were discarded, leaving a total of 36 RNA samples for sequencing. One sequencing library was prepared for each of the 36 samples. The RNA samples were first treated with DNase, then one library per sample was prepared using Illumina’s TruSeq RNA Sample Prep Kit v1, where polyA-fragments were selected, followed by cDNA synthesis and ligation of amplification and sequencing adapters. Sequencing libraries were individually barcoded and then pooled with nine libraries per lane on an Illumina HiSeq 2000 instrument. All samples were sequenced as paired-end, and for 27 samples the read lengths were 100 bp and for nine samples the read lengths were 107 bp for read 1 and 144 bp for read 2. Library preparations and sequencing was performed by the SNP&SEQ Technology Platform in Uppsala.
De novo assembly and filtering
Sequencing reads from all 36 libraries were combined and assembled de novo using the Trinity software (version 20140717) [54] with the read trimming and digital normalization options. Default parameters were implemented except for maximum coverage of 50 in the normalization step and a minimum k-mer coverage of 20 as well as a minimum contig length of 300 in the assembly step. The contigs generated in Trinity are by default clustered in “trinity components” or gene models, where each gene model represents a putative gene. The assembly was filtered based on gene expression levels where gene models that had no contig with FPKM (fragments (read pairs) per kilobase of transcript per million mapped fragments) above or equal to 1 were filtered out. For every gene model, the contig with the highest FPKM value was retained. FPKM values were obtained by mapping the sequencing reads (from all libraries) to the contigs with the program RNA-Seq by Expectation-Maximization (RSEM) v. 1.2.12 [55]. In a second filtering step, only gene models that contained and open reading frame was retained. TransDecoder version 20140704 [56] was used with default options to predict open reading frames in the gene models from the filtered de novo assembly. This assembly is named the high confidence gene set, which was the dataset used for gene expression analyses.
Differential gene expression analysis
In order to identify genes with a putative function in the drought response, differentially expressed genes between drought and WW conditions were identified. Gene specific read counts for the genes in the high confidence gene set were obtained using the RSEM v. 1.2.12 [55], that internally uses bowtie v. 1.0.0 [57]. All sequencing libraries were mapped separately to this assembly. The applied mapping parameters allowed for up to two mismatches to reflect the allelic variation. Using the read counts obtained by RSEM, the R/Bioconductor package edgeR [58] was used to detect differentially expressed genes (DEGs) among the genes in the high confidence gene set between the drought and WW condition in leaves of genotype 592, leaves of genotype 520, root tips of genotype 592 and root tips of genotype 520. The four pairwise comparisons were made using a classic Fisher exact test. For all the analyses in edgeR, the read counts were normalized for RNA composition using the “calcNormFactors()” function. Genes that showed significant G x T effects were identified using a generalized linear model (GLM) in edgeR with an interaction model (Treatment + Tissue + Genotype + (Treatment × Tissue) + (Genotype × Treatment) + (Tissue × Genotype) + (Treatment × Tissue × Genotype)). Genotype × Treatment interaction terms were retained. In order to quantify whether the genotypes differed more in their drought responses (drought vs. WW conditions) in the leaves compared to the root tips, a statistic test was constructed and implemented to the genes with significant G x T effects (the test is described in Additional file 6). For all tests, genes were defined as differentially expressed if false discovery rate (FDR) was ≤ 0.05 and log2 fold change (FC) ≥ 1.
Functional annotation and GO enrichment analysis
Genes in the high confidence gene set were functionally annotated with GO terms in Blast2GO (version 3.1) [59]. The blast step was performed locally against the Viridiplantae subset of the NCBI nr database downloaded 2015-08-06. All up- and downregulated DEGs for leaves, root tips in the two genotypes were tested for GO enrichment using the Fisher's exact test, which uses the Gossip software integrated in the Blast2GO software. The rationale was to test for over- or underrepresentation of GO terms among the DEGs compared to all annotated genes in the high confidence gene set, using a cut-off threshold of FDR ≤ 0.05. The analyses were performed for the three ontology levels, biological processes (P), molecular function (F) and cellular component (C).
Availability of supporting data
The raw sequencing reads are available in the European Nucleotide Archive (ENA) with the reference number PRJEB10883 (http://www.ebi.ac.uk/ena/data/view/PRJEB10883).
Abbreviations
- ANOVA:
-
Analysis of variance
- bp:
-
base pair
- C ontology:
-
Cellular component ontology
- cm:
-
Centimetre
- DEG:
-
Differentially expressed gene
- df:
-
Degrees of freedom
- DW:
-
Dry weight
- F ontology:
-
Molecular function ontology
- FC:
-
Fold change
- FDR:
-
False discovery rate
- FPKM:
-
fragments (read pairs) per kilobase of transcript per million mapped fragments
- G x T:
-
Genotype x Treatment interaction
- GO:
-
Gene ontology
- h:
-
hours
- LAP:
-
Leaf area productivity
- LAR:
-
Leaf area ratio
- LW/W:
-
Leaf biomass per total plant biomass
- N:
-
Nitrogen
- p :
-
p-value
- P ontology:
-
Biological process ontology
- QTL:
-
Quantitative trait locus
- RGR:
-
Relative growth rate
- RNA-seq:
-
RNA sequencing
- RW/W:
-
Root biomass per total plant biomass
- RWC:
-
Relative water content
- SD:
-
Standard deviation
- SLA:
-
Specific leaf area
- SPAD:
-
Leaf chlorophyll content
- SW/W:
-
Shoot biomass per total plant biomass
- WW:
-
Well-watered
- Δ13C:
-
Carbon isotope ratio 13C/12C
- Δ18O:
-
Oxygen isotope ratio 18O/16O
References
Chaves MM, Maroco JP, Pereira JS. Understanding plant responses to drought - from genes to the whole plant. Funct Plant Biol. 2003;30:239–64.
Henckel PA. Physiology of plants under drought. Annu Rev Plant Physiol. 1964;15:363–86.
Hamanishi ET, Campbell MM. Genome-wide responses to drought in forest trees. Forestry. 2011;84:273–83.
Tuskan GA, Difazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science. 2006;313:1596–604.
Street NR, Skogström O, Sjödin A, Tucker J, Rodriguez-Acosta M, Nilsson P, et al. The genetics and genomics of the drought response in Populus. Plant J. 2006;48:321–41.
Wilkins O, Waldron L, Nahal H, Provart NJ, Campbell MM. Genotype and time of day shape the Populus drought response. Plant J. 2009;60:703–15.
Cohen D, Bogeat-Triboulot MB, Tisserant E, Balzergue S, Martin-Magniette ML, Lelandais G, et al. Comparative transcriptomics of drought responses in Populus: a meta-analysis of genome-wide expression profiling in mature leaves and root apices across two genotypes. BMC Genomics. 2010;11:630.
Tang S, Liang H, Yan D, Zhao Y, Han X, Carlson JE, et al. Populus euphratica: the transcriptomic response to drought stress. Plant Mol Biol. 2013;83:539–57.
Qiu Q, Ma T, Hu QJ, Liu BB, Wu YX, Zhou HH, et al. Genome-scale transcriptome analysis of the desert poplar, Populus euphratica. Tree Physiol. 2011;31:452–61.
Tang S, Dong Y, Liang D, Zhang Z, Ye C-Y, Shuai P, et al. Analysis of the drought stress-responsive trancriptome of Black Cottonwood (Populus trichocarpa) using deep RNA sequencing. Plant Mol Biol Rep. 2014.
Hamanishi ET, Raj S, Wilkins O, Thomas BR, Mansfield SD, Plant AL, et al. Intraspecific variation in the Populus balsamifera drought transcriptome. Plant Cell Environ. 2010;33:1742–55.
Zhang X, Zang R, Li C. Population differences in physiological and morphological adaptations of Populus davidiana seedlings in response to progressive drought stress. Plant Sci. 2004;66:791–7.
Xiao XW, Xu X, Yang F. Adaptive responses to progressive drought stress in two Populus cathayana populations. Silva Fenn. 2008;42:705–19.
Lei YB, Yin CY, Li CY. Differences in some morphological, physiological, and biochemical responses to drought stress in two contrasting populations of Populus przewalskii. Physiol Plantarum. 2006;127:182–91.
Kozlowski TT, Pallardy SG. Acclimation and adaptive responses of woody plants to environmental stresses. Bot Rev. 2002;68:270–334.
Karp A, Hanley SJ, Trybush SO, Macalpine W, Pei M, Shield I. Genetic improvement of willow for bioenergy and biofuels. J Integr Plant Biol. 2011;53:151–65.
Stanton BJ, Neale DB, Shanwen L: Populus Breeding: From the Classical to the Genomic Approach. In: Genetics and Genomics of Populus. Edited by Jansson S, Bhalerao RP, Groover AT. New York, US: Springer; 2010: 309–48.
Kuzovkina Y, Weih M, Abalos Romero M, Belyaeva I, Charles J, Hurst S, et al. Salix: Botany and global horticulture. Hortic Revs. 2008;34:447–89.
Rönnberg-Wästljung AC, Glynn C, Weih M. QTL analyses of drought tolerance and growth for a Salix dasyclados x Salix viminalis hybrid in contrasting water regimes. Theor Appl Genet. 2005;110:537–49.
Turtola S, Rousi M, Pusenius J, Yamaji K, Heiska S, Tirkkonen V, et al. Genotypic variation in drought response of willows grown under ambient and enhanced UV-B radiation. Environ Exp Bot. 2006;56:80–6.
Bonosi L, Ghelardini L, Weih M. Towards making willows potential bio-resources in the South: Northern Salix hybrids can cope with warm and dry climate when irrigated. Biomass Bioenerg. 2013;51:136–44.
Karp A, Shield I. Bioenergy from plants and the sustainable yield challenge. New Phytol. 2008;179:15–32.
Berlin S, Fogelqvist J, Lascoux M, Lagercrantz U, Rönnberg-Wästljung AC. Polymorphism and divergence in two willow species, Salix viminalis L. and Salix schwerinii E. Wolf. G3: Genes Genom Genet. 2011;1:387–400.
Tsarouhas V, Gullberg U, Lagercrantz U. Mapping of quantitative trait loci controlling timing of bud flush in Salix. Hereditas. 2003;138:172–8.
Tsarouhas V, Gullberg U, Lagercrantz U. Mapping of quantitative trait loci (QTLs) affecting autumn freezing resistance and phenology in Salix. Theor Appl Genet. 2004;108:1335–42.
Samils B, Rönnberg-Wästljung AC, Stenlid J. QTL mapping of resistance to leaf rust in Salix. Tree Genet Genomes. 2011;7:1219–35.
Rönnberg-Wästljung AC, Samils B, Tsarouhas V, Gullberg U. Resistance to Melampsora larici-epitea leaf rust in Salix: analyses of quantitative trait loci. J Appl Genet. 2008;49:321–31.
Ghelardini L, Berlin S, Weih M, Lagercrantz U, Gyllenstrand N, Rönnberg-Wästljung AC. Genetic architecture of spring and autumn phenology in Salix. BMC Plant Biol. 2014;14:31.
Weih M, Rönnberg-Wästljung AC, Glynn C. Genetic basis of phenotypic correlations among growth traits in hybrid willow (Salix dasyclados x S. viminalis) grown under two water regimes. New Phytol. 2006;170:467–77.
Berlin S, Ghelardini L, Bonosi L, Weih M, Rönnberg-Wästljung AC. QTL mapping of biomass and nitrogen economy traits in willows (Salix spp.) grown under contrasting water and nutrient conditions. Mol Breeding. 2014;34:1987–2003.
Berlin S, Lagercrantz U, von Arnold S, Öst T, Rönnberg-Wästljung AC. High-density linkage mapping and evolution of paralogs and orthologs in Salix and Populus. BMC Genomics. 2010;11:129.
Kreps JA, Wu Y, Chang HS, Zhu T, Wang X, Harper JF. Transcriptome changes for Arabidopsis in response to salt, osmotic, and cold stress. Plant Physiol. 2002;130:2129–41.
Zhou JL, Wang XF, Jiao YL, Qin YH, Liu XG, He K, et al. Global genome expression analysis of rice in response to drought and high-salinity stresses in shoot, flag leaf, and panicle. Plant Mol Biol. 2007;63:591–608.
Bogeat-Triboulot MB, Brosche M, Renaut J, Jouve L, Le Thiec D, Fayyaz P, et al. Gradual soil water depletion results in reversible changes of gene expression, protein profiles, ecophysiology, and growth performance in Populus euphratica, a poplar growing in arid regions. Plant Physiol. 2007;143:876–92.
Weih M, Rönnberg-Wästljung AC. Shoot biomass growth is related to the vertical leaf nitrogen gradient in Salix canopies. Tree Physiol. 2007;27:1551–9.
Weih M, Bonosi L, Ghelardini L, Rönnberg-Wästljung AC. Optimizing nitrogen economy under drought: increased leaf nitrogen is an acclimation to water stress in willow (Salix spp.). Ann Bot 2011;108:1347–53.
McLean EH, Prober SM, Stock WD, Steane DA, Potts BM, Vaillancourt RE, et al. Plasticity of functional traits varies clinally along a rainfall gradient in Eucalyptus tricarpa. Plant Cell Environ. 2014;37:1440–51.
Zhang J, Jiang DC, Liu BB, Luo WC, Lu J, Ma T, et al. Transcriptome dynamics of a desert poplar (Populus pruinosa) in response to continuous salinity stress. Plant Cell Rep. 2014;33:1565–79.
Rao GD, Sui JK, Zeng YF, He CY, Duan AG, Zhang JG. De novo transcriptome and small RNA analysis of two Chinese willow cultivars reveals stress response genes in Salix matsudana. PLoS One. 2014;9.
Zhang J, Xie P, Lascoux M, Meagher TR, Liu J. Rapidly evolving genes and stress adaptation of two desert poplars. Populus euphratica and P pruinosa PLoS One. 2013;8:e66370.
Abe H, Urao T, Ito T, Seki M, Shinozaki K, Yamaguchi-Shinozaki K. Arabidopsis AtMYC2 (bHLH) and AtMYB2 (MYB) function as transcriptional activators in abscisic acid signaling. Plant Cell. 2003;15:63–78.
Neilson JA, Durnford DG. Evolutionary distribution of light-harvesting complex-like proteins in photosynthetic eukaryotes. Genome. 2010;53:68–78.
Jansson S. A protein family saga: from photoprotection to light-harvesting (and back?). In: Demmig-Adams B, Adams III WW, Mattoo AK, editors. Photoprotection, Photoinhibition, Gene Regulation, and Environment Advances in Photosynthesis and Respiration. 22nd ed. Dordrecht: Springer; 2006. p. 145–53.
Xu YH, Liu R, Yan L, Liu ZQ, Jiang SC, Shen YY, et al. Light-harvesting chlorophyll a/b-binding proteins are required for stomatal response to abscisic acid in Arabidopsis. J Exp Bot. 2012;63:1095–106.
Minh-Thu PT, Hwang DJ, Jeon JS, Nahm BH, Kim YK. Transcriptome analysis of leaf and root of rice seedling to acute dehydration. Rice. 2013;6:38.
Poorter H, Buhler J, van Dusschoten D, Climent J, Postma JA. Pot size matters: a meta-analysis of the effects of rooting volume on plant growth. Funct Plant Biol. 2012;39:839–50.
Ritchie SW, Nguyen HT, Holaday AS. Leaf water content and gas-exchange parameters of two wheat genotypes differing in drought resistance. Crop Sci. 1990;30:105–11.
Weih M, Nordh NE. Characterising willows for biomass and phytoremediation: growth, nitrogen and water use of 14 willow clones under different irrigation and fertilisation regimes. Biomass Bioenerg. 2002;23:397–413.
Sullivan PF, Welker JM. Variation in leaf physiology of Salix arctica within and across ecosystems in the High Arctic: test of a dual isotope (Δ13C and Δ18O) conceptual model. Oecologia. 2007;151:372–86.
Farquhar GD, Ehleringer JR, Hubick KT. Carbon Isotope Discrimination and Photosynthesis. Annu Rev Plant Phys. 1989;40:503–37.
Bindumadhava H, Sheshshayee MS, Shashidhar G, Prasad TG, Udayakumar M. Ratio of stable carbon and oxygen isotope discrimination (Δ13C / Δ18O) reflects variability in leaf intrinsic carboxylation efficiency in plants. Curr Sci India. 2005;89:1256–8.
Hunt R. Plant Growth Curves: The Functional Approach to Plant Growth Analysis. London: Edward Arnold; 1982.
Lambers H, Cambridge ML, Konings H, Pons TL. Causes and Consequences of Variation in Growth Rate and Productivity of Higher Plants. The Hague: SPB Academic Publishing; 1990.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–U130.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323.
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494–512.
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40.
Gotz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36:3420–35.
Acknowledgements
Financial support for this work was provided by the Swedish Energy Agency (Grant number P30599-2). We thank Miguel Nemesio Gorrez and Marie Melander for assistance during the experiment and Luisa Ghelardini for advise on the experimental design and for selecting the genotypes for the study. Sequencing was performed by the SNP&SEQ Technology Platform in Uppsala. The facility is part of the National Genomics Infrastructure (NGI) Sweden and Science for Life Laboratory. The SNP&SEQ Platform is also supported by the Swedish Research Council and the Knut and Alice Wallenberg Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
PP and PS carried out the bioinformatic analyses and drafted parts of the manuscript. MW analysed physiological data and drafted parts of the manuscript. ACRW carried out experiments, acquired funding and drafted parts of the manuscript. SB conceived and designed the study, performed the experiments, acquired funding and had the main responsibility for writing the manuscript. All authors read and approved the final manuscript.
Authors’ information
Not applicable.
Availability of data and materials
Not applicable.
An erratum to this article is available at http://dx.doi.org/10.1186/s12870-015-0665-4.
Additional files
Additional file 1:
Sequences of all gene models in the high confidence gene set. (TXT 40356 kb)
Additional file 2:
Length distribution of transcripts in filtered and high confidence gene set. (EPS 33 kb)
Additional file 3:
Normalized read counts used in egdeR. (XLSX 12012 kb)
Additional file 4:
Normalization factors used in egdeR. (XLSX 58 kb)
Additional file 5:
Differentially expressed gene models (DEGs). (XLSX 1313 kb)
Additional file 6:
Description of statistical test. (DOCX 89 kb)
Additional file 7:
Enriched GO terms. (XLSX 203 kb)
Additional file 8:
Genes associated with response GO terms for upregulated DEGs in 592-roots and 520-roots. (XLSX 72 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Pucholt, P., Sjödin, P., Weih, M. et al. Genome-wide transcriptional and physiological responses to drought stress in leaves and roots of two willow genotypes. BMC Plant Biol 15, 244 (2015). https://doi.org/10.1186/s12870-015-0630-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-015-0630-2