
Abstract
Background
Fusarium crown rot (FCR) is a severe and chronic disease in common wheat and is able to cause serious yield loss and health problems to human and livestock.
Results
Here, 234 Chinese wheat cultivars were evaluated in four greenhouse experiments for FCR resistance and genome-wide association studies (GWAS) were performed using the wheat 660 K genotyping assay. The results indicated that most cultivars evaluated showed FCR disease index (DI) of 40–60, while some cultivars showed stably good FCR resistance (DI < 30). GWAS identified 286 SNPs to be significantly associated with FCR resistance, of which 266, 6 and 8 were distributed on chromosomes 6A, 6B and 6D, respectively. The significant SNPs on 6A were located in a 7.0-Mb region containing 51 annotated genes. On the other hand, QTL mapping using a bi-parental population derived from UC1110 and PI610750 detected three QTLs on chromosomes 6A (explaining 7.77–10.17% of phenotypic variation), 2D (7.15–9.29%) and 2A (5.24–6.92%). The 6A QTL in the UC1110/PI610750 population falls into the same chromosomal region as those detected from GWAS, demonstrating its importance in Chinese materials for FCR resistance.
Conclusion
This study could provide useful information for utilization of FCR-resistant wheat germplasm and further understanding of molecular and genetics basis of FCR resistance in common wheat.
Similar content being viewed by others

Background
Fusarium crown rot (FCR), also known as foot rot or root rot, is one of the most seriously insidious disease of wheat and barley [1, 2]. This disease is mainly caused by the fungal pathogen F. Pseudograminearum, which often co-exists with other FCR-causing Fusarium species, such as F. graminearum, F. culmorum and F. avenaceum [3]. Infected seedlings are usually characterized by browning in coleoptile, leaf sheath and stem base, which can become evident after planting and throughout plant development [4].
FCR is widespread in many parts of the arid and semi-arid regions of the world and is of economic concern in Australia, Canada, the Pacific Northwest of the USA, North Africa, South Africa, China and the Middle East [4]. In Australia, FCR caused an estimated annual yield loss of $97 million Australian dollars in wheat and barley [5, 6], while in the Pacific Northwest of the USA, it reduced yields of winter wheat by up to 35% and on barely an average of 13% in commercial fields [7]. In addition, wheat grains infected by Fusarium species were usually associated with the accumulation of mycotoxins like deoxynivalenol (DON) [8] and nivalenol (NIV) [9], which are harmful to human and livestock [10].
In China, wheat is one of the main food crops accounting for a considerable proportion of planting area and crop production. The Yellow and Huai wheat region, with 60–70% of both total harvested area and production, is the largest and most important wheat production zone in China [11]. In recent years, wheat FCR has rapidly increased in this region, including main production regions of Henan and Shandong, south-central Hebei, northern Anhui and Jiangsu, southern Shanxi and east-central Shaanxi [12]. According to Li et al. [13], more than 10% planting areas in Henan province were affected with FCR and more than 30% of yield loss was reported in Xuchang, Jiaozuo and other epidemic places. FCR has occurred seriously in Xinxiang with an incidence area of 3334 ha and yield loss of 10–20%, even exceeding 50% in some sites [14]. Meanwhile, previous studies showed that wheat germplasm with high degree of FCR resistance are rare [4], and almost all current popular varieties in the Yellow and Huai wheat region are susceptible or highly susceptible to FCR [15]. Therefore, developing FCR-resistant cultivars has been vital to prevent the yield damage in cereals [16, 17].
Significant progress in identifying FCR-resistance QTL has been made in wheat and barley. There are three large effect QTLs on the long arms of chromosomes 1H, 3H and 4H in barley [18, 19]. According to the research from CIMMYT and other research institutes, FCR QTLs conferring partial resistance have been identified in different wheat varieties worldwide [20]. These QTLs are distributed on 13 of the 21 wheat chromosomes [2]. One of the most important FCR-resistant QTL is located on 3BL in a RIL population of ‘CSCR6/Lang’ and explained the phenotypic variation up to 49% [21], and limited data further shows that it confers field resistance to FCR and reduces whitehead incidence. The QTL on 4B identified from a cross of ‘Kukri/Janz’ was near the dwarfing gene Rht1 [22] and this QTL showed limited effect of FCR resistance. Additional studies have identified significant QTLs on various chromosomes, e.g. 2B, 3B, 4B, 4D and 7A in RIL populations ‘Sunco/Macon’ and ‘Sunco/Otis’ [23], and QTLs on 2D and 5D in a RIL population ‘Wylie/Sumai 3’ [24]. Genomic analysis of FCR resistance in bread wheat revealed 6, 25 and 11 FCR-resistant QTLs in the A, B and D sub-genome, respectively [2].
As an effective method to study the associations between nucleotide polymorphisms and phenotypic variation: GWAS which has been widely used for analyzing the inheritance of agronomic traits and disease resistance [25, 26]. The newly developed wheat 660 K SNP genotyping assay is generally more abundant and effective than the 90 K SNP [27]. The advances in high-throughput sequencing technologies have enabled rapid and accurate sequencing of a large number of genomes and facilitated direct searches for the causal variation underlying phenotypic diversity [28, 29]. To date, GWAS has been conducted in many plant species, including Arabidopsis, maize, rice, barley, sorghum and wheat [30,31,32]. In wheat, GWAS has been used to study agronomic traits, quality traits and disease resistance [33]. In a previous study, Sun et al. [34] used GWAS to investigate the distribution of superior alleles of 13 agronomic traits in bread wheat from the Yellow and Huai wheat region of China. Marco et al. [33] used GWAS to map strip rust resistance QTL in a worldwide collection of hexaploid spring wheat. In the research by Gurung et al. [35], novel QTL associated with resistance to multiple leaf spot diseases were revealed by GWAS analysis. GWAS for wheat FCR has rarely been reported, and the molecular mechanisms for FCR remain poorly understood.
The Yellow and Huai wheat region is the largest and most important wheat production zone of China [11]. Varieties in this region play a major role in national wheat production. FCR is becoming more prevalent in this region due to a lack of varietal resistance. In this study, we evaluated FCR resistance in bread wheat accessions from the Yellow and Huai wheat region, and then performed GWAS and bi-parental QTL analysis. Our aims were: 1) to identify FCR-resistant wheat germplasms that could be used as resistance donors in breeding, 2) to uncover novel FCR-resistant loci that could be used in marker-assisted selection, and 3) to provide useful information for understanding of molecular and genetic basis of FCR resistance in common wheat.
Results
Resistance investigation of Fusarium crown rot
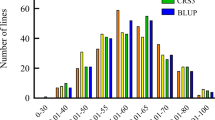
The schematic diagram of FCR inoculation and resistance investigation was shown in Fig. 1. The assessment of FCR resistance in Chinese common wheat showed a very broad range of DI from 20.6 to 84.4 with a normal distribution in the four environments (Additional file 1: Table S1, Fig. 2). Correlation coefficients among the four environments ranged from 0.71 to 0.95 (Additional file 2: Table S2). ANOVA showed that environment had no significant effect on the seedling FCR resistance in the panel (Additional file 3: Table S3). Further analysis indicated that only 7 accessions (Yanke 316, Xunmai 118, Kaimai 26, Jiyanmai 7, Zhonglemai 9, Shenzhou 209 and Jinmai 1) showed an averaged DI of less than 30, exhibiting their potentiality in wheat FCR resistance breeding. There were 31, 87, 70, 27 and 11 accessions showing averaged DI ranges of 30.01–40, 40.01–50, 50.01–60, 60.01–70 and 70.01–80, respectively, and only one cultivar (Zhengmai 082) showed an averaged DI of more than 80. Results indicated that majority of the varieties fell into the range of 40.01–60 (67.09%), indicating an urgent need of improving FCR resistance in the Yellow and Huai wheat region. Representative materials with different grades of resistance to FCR were showed in Table 1.

Schematic diagram for FCR inoculation and screening. (a) Isolate preservation, experimental design and inoculation; (b) typical FCR symptoms on seedlings in greenhouse experiments, scaling from 0 to 9

Number of cultivars at different levels of resistance to FCR in the four environments. ZZ1, ZZ2, ZMD1, ZMD2 represent 2015–2016 Zhengzhou, 2016–2017 Zhengzhou, 2015–2016 Zhumadian and 2016–2017 Zhumadian, respectively
Genome-wide association study
The structure results were integrated by uploading to Structure Harvester http://taylor0.biology.ucla.edu/structureHarvester/. Figure 3 (a, b) shows that peak of the broken line graph was observed at k = 10, indicating ten subpopulations were observed. After filtering, 395,783 SNPs were used for GWAS analysis, and a total of 286 significant SNPs was identified from seven chromosomes, of which 266, 6 and 8 were distributed on 6A, 6B and 6D, respectively (Table 2; Figs. 4 and 5). Phenotypic variation explained by these SNPs ranged from 9.89 to 15.16% for SNPs on 6A, from 10.17 to 12.61% for SNPs on 6B and from 9.90 to 12.39% for SNPs on 6D. There were 24 SNPs being significant in all of the 4 environments (Fig. 6), and 22 of them were detected on 6A. Haplotype analysis showed that these SNPs were clustered into two blocks, where block 1 comprised 16 SNPs forming 3 haplotypes and block 2 comprised 4 SNPs forming 4 haplotypes (Fig. 7a, b). In block 1, the three haplotypes showed significantly different DI, where Hapl_1A showed the lowest averaged DI (46.57), followed by Hapl_1B (51.53) and Hap_1C (59.35) (Fig. 7b, c). In block 2, the four haplotypes also showed significantly different DI, where Hapl_2A and Hapl_2B showed low DI (46.8 and 46.98, respectively), followed by Hapl_2C (51.41) and Hapl_2D (58.91) (Fig. 7b, c). BLAST of the significant SNPs on 6A in the genome database of Chinese Spring showed that these SNPs ranged from 490,486,046 to 497,462,135 (≈ 7.0 Mb) containing 51 annotation genes.

Population structure and principal component analysis (PCA) of the 234 wheat cultivars. (a) plot of cross. validation. error; (b) stacked bar plot of ancestry relationship of the 234 cultivars

Manhattan and Q-Q plots for FCR identified by genome-wide association study (GWAS) in four environments, respectively

Distribution of significant SNPs revealed by GWAS on various chromosomes

The significant SNP intersections among the four environments

Haplotype analysis of the repeatable SNPs on chromosome 6A. (a) local Manhattan plot (top) and LD heatmap (bottom) surrounding the peak on chromosome 6A; (b) haplotypes with different alleles in the two blocks; (c) phenotypic effects of different haplotypes
The repeatable SNPs which were detected in at least two environments were verified based on their polymorphism in the natural population, and the differences of phenotypic values of these SNPs reached highly significant levels in each environment (p < 0.01) (Table 3). Of all these SNPs, AX-111106634, AX-94534539, AX-111704011, AX-109474774, AX-112290591, AX-111013769 and AX-110077933 showed a mean phenotypic variation explained of 14.79, 13.01, 13.19, 13.28, 14.12, 13.62 and 12.31%, respectively (Additional file 4: Table S4). Cultivars with allele AA at AX-111106634 locus, allele GG at AX-112290591, allele TT at AX-111013769, allele AA at AX-94534539, allele AA at AX-111704011, allele AA at AX-109474774 and allele TT at AX-110077933 all showed relatively higher FCR resistance, respectively, and they are regarded as superior alleles (Table 3). At these seven loci, cultivars with all superior alleles showed the averaged DI of 48.58 (ranging from 48.15 to 48.70), while cultivars with all inferior alleles showed the averaged DI of 59.02 (ranging from 57.72 to 60.68) (Fig. 8). Of all varieties surveyed, six (Yanke 316, Jiyanmai 7, Jinmai 1, Zhonglemai 9, Shenzhou 209 and Fannong 1) with all these 7 superior alleles showed DI lower than 30, whereas six (Yunong 805, Zhengmai 082, Saidemai 7, Dongfanghongmai 6, Jinmai 109 and Wenmai 29) with all these 7 inferior alleles showed DI higher than 70.

The phenotype values of cultivars with superior alleles and inferior alleles in significant loci for FCR
Bi-parental QTL mapping for FCR
Three QTLs for FCR resistance were detected in the population derived from UC1110 (DI: 55.16%) and PI610750 (DI: 54.15%), which were significant in at least three of the four environments. The resistance QTLs on chromosomes 2D and 6A were derived from UC1110, whereas the one on chromosome 2A was from PI610750. The most stable QTL, designated as QFCR.heau-6A, was located between wmc754-6A and barc1055 on 6A and was significant in all four environments (ZZ1, ZZ2, ZMD1, ZMD2), explaining 7.77–10.17% of the phenotypic variation (Table 4; Fig. 9a). Another QTL designated as QFCR.heau-2D and flanked by markers cfd53 and cfd43, was significant in 3 environments (ZZ1, ZZ2, ZMD1) with phenotypic variation explained of 7.15–9.29% (Table 4, Fig. 9b). The QTL named as QFCR.heau-2A was between gwm301 and wPt-4861 and was significant in 3 environments (ZZ1, ZZ2, ZMD1), explaining 5.24–6.92% of phenotypic variation (Table 4; Fig. 9c). Further analysis of averaged DI of FCR for the 3 QTLs showed that wheat lines with QFCR.heau-6A, QFCR.heau-2D and QFCR.heau-2A displayed DI of 37.11, 36.51 and 37.67, respectively, whereas wheat lines without QFCR.heau-6A, QFCR.heau-2D and QFCR.heau-2A displayed DI of 41.71, 41.66 and 42.08, respectively (Fig. 10, Table 5). Furthermore, wheat lines with the three FCR-resistance QTLs showed the averaged disease index of 34.67, whereas those with none of the FCR-resistance QTLs showed an averaged DI of 47.44 (Table 5), and their differences reached extremely significant levels in each environment.

LOD contours for FCR QTL on chromosomes 6A, 2D and 2A identified in the UC1110 × PI610750 population. The significant LOD thresholds were calculated from 1000 permutations and finally set at 2.5

The averaged disease index of FCR resistance for the 3 QTLs present or absent
Discussion
FCR is a serious soil-borne disease in wheat and barley that not only causes yield loss but also brings health problem [4]. The pathogens of FCR (Fusarium. species) are tenacious fungus that can survive in stubble for more than 3 years which results in continued disease incidence [12]. Therefore, the genetic improvement of growing resistant and tolerant varieties is a key component in effectively managing the disease of FCR [36].
Previously, only limited efforts have been spent in screening sources of resistance and no cultivar showing fully resistance or immunity to FCR has been identified so far [2, 12]. Wildermuth and Puss [37] screened 400 wheat varieties for the resistance to FCR and found that only four showed moderate resistance. In the study of Zhang [38], there were only 13 varieties had moderate resistance to FCR, and most of the 82 tested varieties were susceptible. In this study, there was no immunity nor high resistance identified, and approximately 80% of the screened materials showed highly susceptible reactions to FCR. This was consistent with the result of Yang et al. [15] who performed a screening study on 88 wheat cultivars from the Yellow and Huai wheat region in 2015. Though the resistance resources to FCR are rare, it is helpful to divided the cultivars or advanced lines into different groups in terms of FCR resistance. In this regard, varieties such as Xunmai 118, Kaimai 26, Yanke 316, Xuke 732, Zhonglemai 9, Jinmai 1, Shenzhou 209, Fannong 1 and Jiyanmai 7 that showed stably moderate resistance (DI < 30) to FCR in multiple environments could be important resistant resources and should be considered for application in wheat FCR resistance breeding. Cultivars like Yanfeng 712, Yunong 805, Zhengmai 082, Jinmai 109 and Saidemai 7 that showed relative high susceptibility (DI > 70) to FCR in all environments should be avoid in FCR epidemic regions. In addition, cultivars like Fannong 3, Pumai 27, Nongda 399, Yunong 99 and Xuyou 46 that showed intermediate reactions (40 < DI < 60) should be paid sufficient attention to prevent FCR. Our study expanded the screened resources of FCR and highlighted a few resistant varieties for FCR resistance.
The accuracy of the phenotypic characterization for the disease over multi-environments are essential for assessing stability of FCR resistance and identification of stably expressed genomic regions [39]. So far, there is no uniform standard for the inoculation method and classification of resistance for wheat FCR [2, 4]. In this study, we used fungal colonized millet grains as inoculum to inoculate wheat seedlings, which has been considered to be the most reliable method to achieve an adequate level of FCR for obtaining consistent cultivar ranking [2, 40]. In addition, a key point in inoculation was to ensure that at least one millet grain was inoculated to a seedling, leading to the uniformity in infection and sufficient disease stress obtained in this study. This was reflected in the better correlation coefficient among experiments obtained in the current study (0.71 to 0.95) than in Erginbas-Orakci et al. [40] (r2 = 0.53) and Huo et al. [41] (r2 = 0.659). As seedling resistance is an important component of field performance [2], our seedling FCR experiments should be able to predicted the field results to a certain extent.
Up to date, FCR-resistant QTLs have been reported to distribute on 13 of the 21 chromosomes in wheat [2], but only QTLs on 2DL, 3BL and 5DS could be consistently detected in different genetic backgrounds [24]. Erginbas-Orakci et al. [20] identified that the QTLs related to crown rot resistance in CIMMYT spring wheat were located on chromosomes 3B and 2D with phenotypic effects of 11.4 and 11.6%, respectively. In this study, FCR-resistant QTLs were identified on 6A, 2D and 2A, and QFCR.heau-6A with a PVE of 7.77–10.17% between wmc754-6A and barc1005 was a novel QTL because it was not reported previously. QFCR.hau-2D with PVE of 7.15–9.29% and QFCR.hau-2A with PVE of 5.24–6.92% could be detected in 3 environments, but they could be the same QTLs as reported in Zheng [24], Bovill et al. [42] and Martin [43]. These three QTLs showed significant additive effects (Fig. 10), and lines with all three QTLs exhibited good FCR resistance, implying the effectiveness of pyramiding all three QTLs to improve FCR resistance in wheat breeding programs.
In this study, significant SNPs closely associated with FCR were mainly distributed on chromosome 6A, e.g. AX-111106634, AX-94534539, AX-11170401, AX-109474774, AX-112290591, AX-111013769 and AX-110077933. The DI difference between cultivars with superior and inferior alleles at these SNP loci all reached extremely significant levels (p < 0.01). A total of 17 cultivars (i.e. Yanke 316, Jiyanmai 7, Jinmai 1, Zhonglemai 9, Shenzhou 209, Fannong 1, Hongmai 618, Zimai 627, Gengmai 237, Ximai 505, Zhoukang 918, Jinmai 108, Xuke 732, L 668, Xianmai 522, Yufeng 1 and Xinyanmai 98) with all superior alleles at these 7 SNP loci showed an average DI of less than 35, whereas 6 cultivars (i.e. Yunong 805, Zhengmai 082, Saidemai 7, Dongfanghongmai 6, Jinmai 109 and Wenmai 29) with all 7 inferior alleles at these loci showed an averaged DI of more than 70. Compared the DI of cultivars with only the 7 alleles (mentioned above) to the cultivars with 16 alleles from block1, the results of haplotypes Hapl-1A (a total of 16 superior alleles, including the 7 superior alleles) and Hapl-1C (a total of 16 inferior alleles, include the 7 inferior alleles) with the 7 alleles showed the same resistance to FCR with the results of 16 alleles. It suggested that these 7 alleles were with great effect to FCR and these loci usually closely linked and provided a guidance for pyramid breeding. It was consistent with the results of Sun [34] that revealed the superior alleles for pyramid breeding in 14 environments of 13 important agronomic traits in the Yellow and Huai valley of China.
Planting resistant cultivars is a valuable method to prevent FCR damage in wheat breeding program, and investigation of wheat FCR resistance requires effective and repeatable screening methods [44]. This study improved the inoculation method, screened some FCR-resistant wheat germplasm and identified some important FCR-resistant genetic loci that could be utilized in current elite cultivars. For example, we have identified several stable FCR-resistant genetic loci (i.e. AX-111106634, AX-94534539, AX-11170401, AX-109474774, AX-112290591, AX-111013769 and AX-110077933) and they could be utilized in marker-assisted selection in FCR disease resistance. Among these genetic loci, AX-111106634 on 6A was the most significant and stable locus.
QTL mapping indicated that the QFCR.hau-6A was located on 6A based on the genetic map of UC1110/PI610750. The physical position of the markers (≈ 495,246,396 bp) near the QFCR.hau-6A on 6A fell into the same chromosomal region as those detected from GWAS (490486046–497,462,135 bp) in the genome of Chinese Spring. Combination of GWAS and QTL mapping showed that the QFCR.hau-6A was the most significant QTL to modulate FCR resistance in this study and should be paid more attention to use for improvement of FCR resistance. All in all, both of the QTL mapping and GWAS in this study showed that there is an important genetic loci or QTL on the chromosome 6A with a relative largest effect of resistance to FCR, and thus this QTL could be considered as a priority for marker-assisted selection to improve FCR resistance in wheat cultivars from the Yellow and Huai wheat region. In addition, pyramiding several QTLs or genetic loci conferring partial FCR resistance in multiply environments by GWAS and QTL mapping will be able to improve FCR resistance for wheat pyramiding breeding.
Conclusions
In this study, we investigated the FCR resistance of wheat cultivars or advanced lines from the Yellow and Huai wheat region and discovered some valuable FCR-resistant wheat germplasms. GWAS analysis showed that a total of 286 SNPs was significantly associated with FCR resistance in wheat, and 266 of them was on chromosome 6A. Physical mapping in a RIL population showed that there were 3 FCR-resistant QTLs, i.e. QFCR.heau-6A, QFCR.heau-2D and QFCR.heau-2A. The QFCR.heau-6A is a novel FCR-resistant QTL with the largest effect among them. Combination of GWAS and QTL mapping indicated that the FCR-resistant gene ranged from 490,486,046 to 497,462,135 (≈ 7.0 Mb) in Chinese Spring database.
Methods
Plant materials
A total of 234 common wheat cultivars or advanced lines were selected as an association panel for investigation of FCR resistance and GWAS analysis. They were released or developed after 2014, representing current breeding situation in the Yellow and Huai wheat region for the last 5 years. Seeds were harvested in Zhengzhou Scientific Research and Education Center of Henan Agricultural University (N 34.87°, E 113.60°) and Zhumadian Academy of Agricultural Science (N 33.01°, E 114.05°) during the cropping seasons 2015–2016 and 2016–2017.
A recombinant inbred lines (RIL) population with 187 F10 lines derived from UC1110 and PI610750 (UP) was used to map FCR resistance QTLs. Greenhouse experiments for this RIL population were the same as the GWAS panel.
Phenotyping of Fusarium crown rot resistance
Fungal isolate
All plant materials were planted in greenhouse and inoculated with a prevalent Chinese F. pseudograminearum isolate namely WZ-8A, kindly provided by Prof. Honglian Li from the College of Plant Protection of Henan Agricultural University. WZ-8A is a highly aggressive strain isolated from infected crowns of wheat in northwest of the Yellow and Huai wheat region. The strain (culture medium with mycelium of 4 mm diameter) was put on a potato dextrose agar (PDA, 200 g of peeled potato, 15 g of agar and 20 g of dextrose in 1000 mL distilled water) plate at 25 °C under fluorescent lights with a 12 h/12 h day/night photoperiod in an incubator. After 6 days, when the white mycelium grew over the plates and showed peach-red to dark red pigmentation, a piece of 15 mm × 3 mm culture medium with mycelium was taken out from the margin of the plate, which was then transferred into a 2 mL centrifuge tube full of 30% glycerin and stored under − 80 °C for long-term storage. Another piece of medium with mycelium (of 4 mm diameter) was transferred into a 1.5 mL centrifuge tube with PDA medium and placed into the incubator to grow under the same condition mentioned above. After mycelia harvest, they were stored in 4 °C refrigerator for further application. When needed, mycelia from the 1.5 mL centrifuge tubes were transferred to fresh PDA plates for multiplication.
Spawn inoculum preparation
Millet grains of uniform size were boiled for 2 min and then rinsed 3–4 times with cold distilled water. The grains were put on clean gauze in fume hood until dry up and were then transferred into flasks for sterilization at 121 °C, 0.1 Mpa for 30 min. Shake the flasks before cooling down to prevent agglomeration. After cooling, a piece of medium with F. pseudograminearum mycelium was added to the flask, mixed thoroughly, and then placed in the incubator to grow at 25 °C under fluorescent lights with a 12/12 h day/night photoperiod. The flasks were shaken twice a day to promote uniform colonization. After 7 days, the colonized grains became ready for inoculation on seedlings.
Experimental design and inoculation
The experiments were carried out in plastic containers of 7 × 7 × 7 cm. Seeds were surface disinfested with 95% ethanol for 3 min and then rinsed under running distilled water for 3 min. Soil was sterilized at 121 °C for 1 h at 0.1 Mpa. When sowing, 150 g sterilized soil was put into a plastic container, followed by placing 12 sterilized seeds that were then covered with 20 g sterilized soil. Three replicates were performed for each cultivar. All plastic containers were placed on trays and watered from the bottom of the trays. The condition of the greenhouse was controlled at 25/20 °C day/night temperatures, 16 h/8 h light/dark photoperiod, and 60–80% relative humidity [45]. Three days after germination, weak seedlings were pulled out. When seedlings grew to 3 cm long, 8–10 seedlings per cultivar that grew uniformly were inoculated, then 0.4 g colonized millet grains (about 70–80 millet grains) were scattered to each plastic container. It is important to ensure at least one millet grain was distributed to the stem of a seedling. Then 30 g sterilized soil was scattered into each container and water was applied from the bottom of the trays. Subsequently, the containers were watered every two days.
Phenotyping
Severity of FCR was evaluated 4 weeks after inoculation, with a scale from 0 (no obvious symptoms) to 9 (completely necrotic damage) according to Li et al. [46] with some modification. Disease index (DI) was calculated for each replicate according to the methods of Li et al. [46] and Zheng et al. [24].
Where, S is the scale value of each plant; ns is the number of plants in the scale S; N is the total number of plants assessed in one replicate.
Genotyping and quality control
All 234 accessions surveyed were genotyped using the wheat 660 K genotyping assay by Beijing CapitalBio Technology Company (http://cn.capitalbio.com/). The quality pretreatment of genotyping data was carried out for SNP call rate and MAF (minor allele frequency) with the PLINK software with threshold of --maf 0.02 and --geno 0.1. (http://zzz.bwh.harvard.edu/plink/tutorial.shtml) [47].
Population structure analysis
Population structure was assessed by STRUCTURE software v2.3.4 with unlinked markers (r2 = 0) [48]. The number of subpopulations (k) was implemented by a burn-in of 1000 iterations followed by 1000 Monte Carlo Markov Chain (MCMC) replicates in a putative range of 1–10.
Genome-wide association study
GWAS was implemented using the mixed linear model (PCA + K) by GAPIT packages in R software [49, 50] and the variance–covariance kinship matrix (K) was calculated using the VanRaden method [51]. Threshold for P value was calculated using a modified Bonferroni correction (Genetic type 1 Error Calculator, version 0.2) with a suggestive threshold of P value = 1.0e-4 (P = 1/n, n = effective SNP number) [52].
Bi-parental QTL mapping
In total, 1494 polymorphic markers (SSRs, ESTs and DArTs) from 558 unique loci in the RIL population UC1110 × PI610750, kindly provided by Jorge Dubcovsky [53], were used to construct genetic linkage map by software IciMapping 4.0 (http://www.isbreeding.net). QTLs were calculated with the Inclusive composite interval mapping (ICIM) algorithm in four environments. The threshold of LOD scores was set at 2.5.
Abbreviations
- Add:
-
Additive effects of the QTL
- Chr.:
-
Chromosome
- cm:
-
centimeter
- DArT:
-
Diversity arrays technology
- DI:
-
Disease index
- DON:
-
Deoxynivalenol
- EST:
-
Expressed sequence tags
- FCR:
-
Fusarium crown rot
- GWAS:
-
Genome-wide association studies
- hapl :
-
Haplotype
- ICIM:
-
Inclusive composite interval mapping
- MAF:
-
Minor allele frequency
- MCMC:
-
Monte Carlo Markov Chain
- mm:
-
millimeter
- MSS:
-
Most significant SNP
- NIV:
-
Nivalenol
- PCA:
-
Principal component analysis
- PDA:
-
Potato dextrose agar
- PVE:
-
Phenotypic variation explained
- QTL:
-
Quantitative trait loci
- RIL:
-
Recombinant inbred lines
- SNP:
-
Single nucleotide polymorphism
- SSR:
-
Simple sequence repeat
- UP:
-
UC1110 and PI610750
- ZMD1:
-
2015–2016 Zhumadian
- ZMD2:
-
2016–2017 Zhumadian
- ZZ1:
-
2015–2016 Zhengzhou
- ZZ2:
-
2016–2017 Zhengzhou
References
Smiley RW, Gourlie JA, Easley SA, Patterson LM, Whittaker RG. Crop damage estimates for crown rot of wheat and barley in the pacific northwest. Plant Dis. 2005;89:595–604.
Liu CJ, Ogbonnaya FC. Resistance to Fusarium crown rot in wheat and barley: a review. Plant Breed. 2015;134:365–72.
Zhang XX, Sun HY, Shen CM, Li W, Yu HS, Chen HG. Survey of Fusarium spp. causing wheat crown rot in major winter wheat growing regions of China. Plant Dis. 2015;99:1610–5.
Kazan K, Gardiner DM. Fusarium crown rot caused by Fusarium pseudograminearum in cereal crops: recent progress and future prospects. Mol Plant Pathol. 2018;19:1547–62.
Murray GM, Brennan JP. Estimating disease losses to the Australian wheat industry. Australas Plant Pathol. 2009;38:558–70.
Murray GM, Brennan JP. Estimating disease losses to the Australian barley industry. Australas Plant Pathol. 2010;39:85–96.
Smiley RW, Gourlie JA, Easley SA, Patterson LM. Pathogenicity of fungi associated with the wheat crown rot complex in Oregon and Washington. Plant Dis. 2005;89:949–57.
Obanor F, Chakraborty S. Aetiology and toxigenicity of Fusarium graminearum and F. pseudograminearum causing crown rot and head blight in Australia under natural and artificial infection. Plant Pathol. 2014;63:1218–29.
Monds RD, Cromey MG, Lauren DR, Menna M, Marshall J. Fusarium graminearum, F. cortaderiae and F. pseudograminearum in New Zealand: molecular phylogenetic analysis, mycotoxin chemotypes and co-existence of species. Mycol. Res. 2005;109:410–20.
Mudge AM, Dill MR, Dong Y, Gardiner DM, White RG, Manners JM. A role for the mycotoxin deoxynivalenol in stem colonisation during crown rot disease of wheat caused by Fusarium graminearum and Fusarium pseudograminearum. Physiol Mol Plant Pathol. 2006;69:73–85.
Chen F, Gao M, Zhang J, Zuo A, Shang X, Cui D. Molecular characterization of vernalization and response genes in bread wheat from the yellow and Huai Valley of China. BMC Plant Biol. 2013;13:1–12.
Zhou HF, Yang Y, Niu YJ, Yang HX, Li HL. Occurrence and control methods of crown rot of wheat. J Henan Agric Sci. 2014;43:114–7.
Li HL, Yuan HX, Fu B, Xing XP, Sun BJ, Tang WH. First report of Fusarium pseudograminearum causing crown rot of wheat in Henan. China Plant Dis. 2012;96:1065.
Zhu SM, Liu QR. The cause reason and comprehensive control of wheat crown rot in Xinxiang. China Plant Protection. 2016;36:40–2.
Yang Y, He XL, Hu YF, Hou Y, Niu YJ, Dai JL, Yuan HX, Li HL. Resistance of wheat cultivars in Huang-Huai region of China to crown rot caused by Fusarium pseudograminearum. J Triticeae Crops. 2015;35:339–45.
Ren Y, Hou WX, Lan CX, Basnet BR, Singh RP, Zhu W, Cheng XY, Cui DQ, Chen F. QTL analysis and nested association mapping for adult plant resistance to powdery mildew in two bread wheat populations. Front Plant Sci. 2017;8:1212.
Lan CX, Basnet BR, Herrera-Foessel SA, Huerta-Espino J, Ren Y, Calvo-Salazar V, Singh RP. Genetic analysis and mapping of adult plant resistance loci to leaf rust in durum wheat cultivar Bairds. Theor Appl Genet. 2017;130:609–19.
Chen G, Liu Y, Ma J, Zheng Z, Wei Y, McIntyre CL, Zheng YL, Liu C. A novel and major quantitative trait locus for Fusarium crown rot resistance in a genotype of wild barley (Hordeum spontaneum L.). PLOS One. 2013;8:e58040.
Chen GD, Liu YX, Wei YM, McIntyre CL, Zhou MX, Zheng YL, Liu CJ. Major QTL for Fusarium crown rot resistance in a barley landrace. Theor Appl Genet. 2013;126:2511–20.
Erginbas-Orakci G, Sehgal D, Sohail Q, Ogbonnaya F, Dreisigacker S, Pariyar SR, Dababat AA. Identification of novel quantitative trait loci linked to crown rot resistance in spring wheat. Int J Mol Sci. 2018;19:2666.
Ma J, Li HB, Zhang CY, Yang XM, Liu YX, Yan GJ, Liu CJ. Identification and validation of a major QTL conferring crown rot resistance in hexaploid wheat. Theor Appl Genet. 2010;120:1119–28.
Wallwork H, Butt M, Cheong JPE, Williams KJ. Resistance to crown rot in wheat identified through an improved method for screening adult plants. Plant Pathol. 2004;33:1–7.
Poole GJ, Smiley RW, Paulitz TC, Walker CA, Carter AH. Identification of quantitative trait loci (QTL) for resistance to Fusarium crown rot (Fusarium pseudograminearum) in multiple assay environments in the Pacific northwestern US. Theor Appl Genet. 2012;125:91–107.
Zheng Z, Kilian A, Yan G, Liu C. QTL conferring Fusarium crown rot resistance in the elite bread wheat variety EGA Wylie. PLoS One. 2014;9:e96011.
Nordborg M, Weigel D. Next-generation genetics in plants. Nature. 2008;456:720–3.
Kenji Y, Eiji Y, Koichiro A, Hideyuki T, Pei-ching L, Masanori Y, Shiya Y, Hidemi K, Ko H, Makoto M. Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat Genet. 2016;48:927–36.
Rasheed A, Hao Y, Xia X, Khan A, Xu Y, Varshney RK, He Z. Crop breeding chips and genotyping platforms: progress, challenges, and perspectives. Mol Plant. 2017;10:1047–64.
Huang X, Kurata N, Wei X, Wang ZX, Wang A, Zhao Q, Zhao Y, Liu K, Lu H, Li W, Guo Y, Lu Y, Zhou C, Fan D, Weng Q, Zhu C, Huang T, Zhang L, Wang Y, Feng L, Furuumi H, Kubo T, Miyabayashi T, Yuan X, Xu Q, Dong G, Zhan Q, Li C, Fujiyama A, Toyoda A, Lu T, Feng Q, Qian Q, Li J, Han B. A map of rice genome variation reveals the origin of cultivated rice. Nature. 2012;490:497–501.
Koboldt DC, Steinberg KM, Larson DE, Wilson RK, Mardis ER. The next-generation sequencing revolution and its impact on genomics. Cell. 2013;155:27–38.
Jia GQ, Huang XH, Zhi H, Zhao Y, Zhao Q. A haplotype map of genomic variations and genome-wide association studies of agronomic traits in foxtail millet (Setaria italica). Nat Genet. 2013;45:957–61.
Zhang YL, Liu P, Zhang XX, Zheng Q, Chen M, Ge F, Li ZL, Sun WT, Guan ZR, Liang TH, Zheng Y, Tan XL, Zou CY, Peng HW, Pan GT, Shen Y. Multi-locus genome-wide association study reveals the genetic architecture of stalk lodging resistance-related traits in maize. Front Plant Sci. 2018;9:611.
Morris GP, Ramu P, Deshpande SP, Hash CT, Shah T, Upadhyaya HD. Population genomic and genome-wide association studies of agroclimatic traits in sorghum. Proc Natl Acad Sci U S A. 2013;110:453–8.
Marco M, Zhang JL, Peter B, Zewdie A, Chao S, Dario C, Eligio B, Chen XM, Michael P, Jorge D. A genome-wide association study of resistance to stripe rust (Puccinia striiformis f.sp.tritici) in a worldwide collection of hexaploid spring wheat (Triticum aestivum L.). G3. 2015;5:449–65.
Sun CW, Zhang FY, Yan XF, Zhang XF, Dong ZD, Cui DQ, Chen F. Genome -wide association study for 13 agronomic traits reveals distribution of superior alleles in bread wheat from the yellow and Huai Valley of China. Plant Biotechnol J. 2017;15:953–69.
Gurung S, Mamidi S, Bonman JM, Xiong M, Brown-Guedira G, Adhikari TB. Genome-wide association study reveals novel quantitative trait loci associated with resistance to multiple leaf spot diseases of spring wheat. PLoS One. 2014;9:e108179.
Li HB, Xie GQ, Ma J, Liu GR, Wen SM, Ban T, Chakraborty S, Liu CJ. Genetic relationships between resistances to Fusarium head blight and crown rot in bread wheat (Triticum aestivum L.). Theor Appl Genet. 2010;121:941–50.
Wildermuth GB, Purss GS. Further sources of field resistance to crown rot (Gibberella zea) of cereals in Queensland. Australian J Exp Agric Animal Husbandry. 1971;11:455–9.
Zhang P, Huo Y, Zhou MP, Yao BQ, Ma HX. Identification and evaluation of wheat germplasm resistance to crown rot caused by Fusarium graminearum. J Plant Genet Resour. 2009;10:431–5.
Ogbonnaya FC, Rasheed A, Okechukwu EC, Jighly A, Makdis F, Wuletaw T. Genome-wide association study for agronomic and physiological traits in spring wheat evaluated in a range of heat prone environments. Theor Appl Genet. 2017;130:1819–35.
Erginbas-Orakci G, Poole G, Nicol JM, Paulitz T, Dababat AA, Campbell K. Assessment of inoculation methods to identify resistance to Fusarium crown rot in wheat. Plant Dis. 2016;123:19–27.
Huo Y. Evaluation and QTL analysis for crown rot resistance in wheat [D]. Nanjing: Nanjing agricultural university. 2010:22–5.
Bovill WD, Horne M, Herde D, Wildermuth GB, Sutherland MW. Pyramiding QTL increases seeding resistance to crown rot (Fusarium pseudograminearum) of wheat (Triticum aestivum). Theor Appl Genet. 2010;121:127–36.
Martin A, Bovill WD, Percy CD, Herde D, Fletcher S, Kelly A, Neate SM, Sutherland MW. Markers for seedling and adult plant crown rot resistance in four partially resistant bread wheat sources. Theor Appl Genet. 2015;128:377–85.
Wu B, Guo X, Zhang M, Jiang SS, Xin ZM, Wang SJ, Xin XQ. Identification and pathogenicity of pathogens associated with the crown rot in the southwest of Shandong Province. J Triticeae Crop. 2018;38:358–65.
Mitter V, Zhang MC, Liu CJ, Ghosh R, Ghosh M, Chakraborty S. A high-throughput glasshouse bioassay to detect crown rot resistance in wheat germplasm. Plant Pathol. 2006;55:433–41.
Li X, Liu C, Chakraborty S, Manners J, Kazan K. A simple method for the assessment of crown rot disease severity in wheat seedlings inoculated with Fusarium pseudograminearum. J Phytopathol. 2008;156:751–4.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. Plink: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59.
Zhang Z, Ersoz E, Lai CQ, Todhunter RJ, Tiwari HK, Gore MA, Bradbury PJ, Yu J, Arnett DK, Ordovas JM, Buckler ES. Mixed linear model approach adapted for genome-wide association studies. Nat Genet. 2010;42:355–60.
Lipka AE, Tian F, Wang Q, Peiffer J, Li M, Bradbury PJ, Gore MA, Buckler ES, Zhang Z. GAPIT: genome association and prediction integrated tool. Bioinformatics. 2012;28:2397–9.
Vanraden PM. Efficient methods to compute genomic predictions. J Dairy Sci. 2008;91:4414–23.
Li H, Peng Z, Yang X, Wang W, Fu J, Wang J, Han Y, Chai Y, Guo T, Yang N, Liu J, Warburton ML, Cheng Y, Hao X, Zhang P, Zhao J, Liu Y, Wang G, Li J, Yan J. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat Genet. 2013;45:43–50.
Lowe I, Jankuloski L, Chao S, Chen X, See D, Dubcovsky J. Mapping and validation of QTL which confer partial resistance to broadly virulent post-2000 north American races of stripe rust in hexaploid wheat. Theor Appl Genet. 2011;123:143–57.
Acknowledgements
The authors thank Prof. Honglian Li from Henan Agricultural University for the help of improving inoculation methods of Fusarium crown rot.
Funding
This work was supported by the National Key Research and Development Program (2016YFD0101802), National Natural Science Foundation (3181101544) and Henan Major Science and Technology Projects (181100110200) of China. The supporters did not play any role in the design, analysis, or interpretation of this study and the relevant data.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on request.
Author information
Authors and Affiliations
Contributions
FC designed the research. XY, YP, YR, LZ and NZ performed genotyping and phenotyping. XY, YP, PS and XH performed data analysis. XY and FC wrote the manuscript and all authors reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Resistance to FCR of wheat cultivars inoculated by F.pg in four environments. (XLSX 25 kb)
Additional file 2:
Table S2. Information for FCR disease index and correlation coefficients among environments. (XLSX 9 kb)
Additional file 3:
Table S3. ANOVA for FCR resistance of the GWAS panel. (XLSX 8 kb)
Additional file 4:
Table S4. The most significant SNPs on chromosome 6A that highly correlation with FCR resistance in different environments. (XLSX 9 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yang, X., Pan, Y., Singh, P.K. et al. Investigation and genome-wide association study for Fusarium crown rot resistance in Chinese common wheat. BMC Plant Biol 19, 153 (2019). https://doi.org/10.1186/s12870-019-1758-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-019-1758-2