
Abstract
Background
Notopterygium incisum is an important Chinese medicinal plant. Its mature seeds have underdeveloped embryos and are physiological dormant. We found the seeds with full developed embryos can germinate after treated by fluridone (FL), an inhibitor of abscisic acid (ABA). In order to understand the molecular mechanisms underlying seed dormancy release by FL, we compared the transcriptomic changes in dormancy release induced by two different methods, FL and cold stratification (CS) in N. incisum. We further analyzed the gene expression patterns involved in seed germination and dormancy using quantitative reverse-transcription PCR.
Results
RNA-sequence analysis revealed more dramatic changes in the transcriptomes of FL than those in CS, particularly for genes involved in the biosynthesis and regulation of gibberellins (GAs) and ABA. The down-regulation of ABA biosynthesis genes and the dramatic up-regulation of NiCYP707As, an ABA catabolic gene, contributed to the reduced ABA levels in FL. The increased GA3 levels in CS-treated seeds were due to the up-regulation of NiGA3OX. Both NiABI5 (a positive ABA regulator) and NiGAI (a negative regulator of GA) were down-regulated in FL and CS. The upregulation of strigolactones (SLs; the metabolites with the same precursor as ABA) biosynthesis and regulatory genes in both FL- and CS-treated seeds indicates that SLs contribute positively to seed dormancy release in N. incisum.
Conclusions
Our results indicated that FL- and CS-seed dormancy release possibly depends on two totally different mechanisms: alleviation of the effects of ABA and potentiation of the effects of GA, respectively. However, NiABI5 and NiGAI probably function as common factors integrating the effects of ABA and GA on seed dormancy release.
Similar content being viewed by others


Background
The underground parts of Notopterygium incisum C. C. Ting ex H. T. Chang (Apiaceae) are used to produce the popular traditional Chinese medicine Qianghuo to treat headaches, the common cold, and rheumatism [1]. Seeds of N. incisum are morpho-physiological dormant (MPD), meaning that underdeveloped embryos must grow to a species-specific critical size and that physiological dormancy must be alleviated before seed germination [2]. However, little is known about the molecular mechanisms underlying seed dormancy release induced by FL and CS in N. incisum.
Abscisic acid (ABA) and gibberellins (GAs) are two main plant hormones involved in seed germination and dormancy via their ability to inhibit and promote germination [3]. Carotenoids are precursors of the ABA biosynthesis pathway [4]. Fluridone (FL) acts as an inhibitor of phytoene desaturase (PDS) activity, which converts phytoene to phytofluene, zeta-carotene, or neurosporene in the carotenoid-biosynthesis pathway [5,6,7]. It has been widely used as an inhibitor to prevent ABA biosynthesis [5], to relieve seed dormancy, and induce seed germination [8,9,10,11]. We found that FL treatment significantly improved seed germination in fully developed N. incisum seeds. Compared to treatment with CS for 3 months, a generally used method to release seed dormancy in N. incisum [12], the use of FL greatly reduced the seed germination period.
In order to investigate whether there is a common molecular mechanism between seed dormancy release induced by FL and CS in N. incisum, we used de novo transcriptomic analysis to find genes involved in the biosynthesis and regulation of ABA and GAs and their expression patterns.
Methods
Seed treatments and germination
Notopterygium incisum seeds were provided by Sichuan Notopterygium Organic Herbs Co., Ltd. (Aba prefecture, Sichuan province, China). Mature dry seeds were pretreated using the warm-cold moist treatment method [2]. The pretreated seeds were soaked in 100 mg/L FL solution for 24 h, with pure water used as the control (Con). CS treatment was carried out by storing the pretreated seeds in a sealed plastic box containing moist sand at 5 °C for 3 months. The seeds were removed from FL or sand (CS) and washed. The embryos were sampled from the washed seeds, frozen in liquid nitrogen immediately, and stored at − 80 °C for further analysis. To test the seed dormancy release, germination tests of CS, FL, and Con were carried out. Samples of 100 fresh seeds were sown on 1% water agar in 90 mm-diameter Petri dishes and incubated at 15 °C (the optimal germination temperature among the five temperatures tested; data not shown). The incubators were set to a photoperiod of 12 h light/ 12 h dark and 1000 lux white fluorescent light.
RNA extraction, library preparation, and de novo sequencing
Total RNA was isolated from approximately 20 N. incisum embryos per sample using an RNeasy Plant kit (BioTeke Corporation, China). A total of 1.5 μg RNA per sample, with six samples from three treatments (Con, FL, and CS), was used for RNA sample preparation. The quality, purity and integrity of RNA were examined according to Ma et al. [13]. RNA library preparation was performed according to the protocol of the NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, USA). Paired-end sequencing was carried out on an Illumina HiSeq 2500 platform.
Gene functional annotation and differential expression analysis
BLAST analysis [14] was used for sequence annotation against seven databases, namely NCBI-Nr, NCBI-Nt, Pfam [15], KOG/COG, Swiss-Prot, KO, and GO, with expected E-values (E-value ≤1.0E-5 for Nr, Nt, and Swiss-Prot; E-value ≤0.01 for Pfam; E-value ≤1.0E-3 for KOG/COG; E-value ≤1.0E-10 for KEGG; E-value ≤1.0E-6 for GO). The expression levels of genes in each sample were assessed using RSEM [16]. The DESeq R package (1.10.1) was used for differential expression analysis [17]. To control the false discovery rate, the Benjamini and Hochberg’s approach was used to adjust the resulting P values. An adjusted P-value of < 0.05 was considered significant for differential gene expression.
GO enrichment analysis and KEGG pathway enrichment analysis of the DEGs
Gene function classification of differentially expressed genes (DEGs) was performed using GO analysis with three categories including molecular function, cellular component, and biological process. The GOseq R package was used for GO enrichment analysis of the DEGs based on Wallenius’ noncentral hypergeometric distribution [18]. KOBAS software was used to test the statistical enrichment of DEGs in the KEGG pathway maps [19].
Q-RT-PCR analysis
Total RNA was isolated from embryos using a Universal Plant RNeasy kit (BioTeke, China). Contaminating genomic DNA in the prepared RNA was removed using gDNA Eraser (Takara, Dalian, China) and the RNA was reverse transcribed using 1.0 μg of total RNA per reaction (20 μl) and the Prime-Script™ RT reagent Kit (Takara, China). Quantitative reverse-transcription PCR (q-RT-PCR) was carried out in an ABI 7500 system (Applied Biosystems, USA). The ABI Prism 7500 Sequence Detection System was used for data analysis according to Applied Biosystems User Bulletin. The expression levels of the genes in this study were normalized to that of endogenous control gene NiGAPDH2.
ABA and GA3 measurements
ABA and GA3 levels were measured using ultra-performance liquid chromatography coupled with a quadrupole trap mass spectrometer equipped with electrospray ionization (UHPLC-ESI-MS/MS). ABA and GA3 were extracted using a revised method describe in [20]. Approximately 50 mg embryos were extracted from N. incisum seeds. UPLC separation was performed on a Waters Acquity UPLC-I-Class system (Waters Corporation, Milford, MA) with an Acquity BEH C18 column for chromatographic separation. The precursor-to-product ion transition was m/z: 263/153 for ABA and m/z: 345.1/143.1 for GA3. The LC-MS/MS system was controlled by Analyst 1.5.2, and the data analysis was performed using Multi Quantity software.
Data analysis
All analyses were conducted with Microsoft Excel 2010 and Origin 7.0 software. The quantitative data are shown as means ± standard deviations. The data were subjected to one-way analysis of variance (ANOVA) with SPSS 16.0. Statistical significance was determined using Fisher’s least significant difference (LSD) test.
Results
Seed germination after FL and CS treatment and quantitative analysis of plant hormones in N. incisum
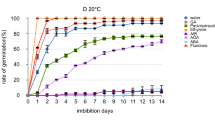
Mature seeds of N. incisum have small, underdeveloped embryos, which must grow to ~ 2/3 of seed length during the warm, moist pretreatment period. Approximately 12.8% germination was obtained for untreated seeds (Con) with developed embryos (Fig. 1a, b). In contrast, about 73.5% germination percentage was achieved upon CS for 90 days, whereas 81.3% germination was obtained after the seeds were treated by FL (Fig. 1a, b). FL is an effective treatment for improving the germination of N. incisum seeds and can be used instead of cold stratification for 90 days.

Germination and plant hormone levels in N. incisum seeds under different treatments. a Seed germination; b germination percentage; c plant hormone levels. Con, control; FL: imbibition with fluridone liquid for 24 h; CS: cold stratification for 3 months; the same in the following figures and tables. Data represent the mean ± SE (n = 3); “*” indicates a significant difference in FL or CS compared to Con at P < 0.05
We examined ABA and GA3 contents following three treatments with Con, FL, and CS via UHPLC-MS/MS. Both ABA and GA3 levels increased significantly in CS compared to Con (Fig. 1c). The accumulation of GA3 during CS is thought to be the key cause of seed dormancy release [21, 22]. FL reduces ABA levels to improve seed germination in several species [9, 10, 23]. ABA levels decreased from 118.5 ng / g to 98.7 ng / g after FL treatment in present work (Fig. 1c). GA3 level did not significantly change after FL treatment. These results suggest that sensitivity to ABA, not the ABA level, correlates directly with the dormancy status of N. incisum seeds, which is in agreement with other reports [23].
Analysis of RNA-Seq datasets
Approximately 58, 60, 54, 55, 53, and 52 million raw reads were obtained from samples Con1, Con2, FL1, FL2, CS1, and CS2, respectively. The RNA-seq datasets were deposited in the Sequence Read Archive of NCBI (Access No. SRP107325). After filtering low-quality reads and adaptor sequences, 56.22, 59.02, 52.66, 53.94, 49.82, and 49.78 million clean reads and 8.43, 8.85, 7.9, 8.09, 7.47, and 7.47 G bases for samples Con1, Con2, FL1, FL2, CS1, and CS2 were obtained, respectively (Q30 of all samples ~ 90% and GC of all samples ~ 43%). In total, 18, 1376 unigenes with an average length of 650 bases and N50 of 939 bases were obtained using Trinity assembly software [24]. Sequencing and assembly information is provided in Table 1.
BLASTX revealed that 68,236 of the unigenes (37.62%) significantly matched sequences in GO, KEGG, KOG/COG, NR, Nt, Pfam, and Swiss-Prot. The functional annotation for all genes in more detail is shown in Additional file 1: Table S1. Among the annotated unigenes, 51,594 (75.61% annotated in Nr) are predicted to be proteins. The gene sequences in N. incisum were most similar to those of Vitis vinifera (14.7%), followed by Nicotiana tomentosiformis (7.4%) and Coffea canephora (7.0%) (Additional file 2: Figure S1). The predicted unigenes were functionally categorized by GO annotation, with 39,353 unigenes assigned to GO classes (Additional file 3: Figure S2). The 17,942 unigenes were clustered into 26 functional groups by KOG analysis (Additional file 4: Figure S3). To help reconstruct the metabolic pathways in N. incisum, we performed KEGG pathway mapping for all of the unigenes. A total of 18,363 unigenes were successfully annotated and 281 KEGG pathways were mapped. Detailed pathway information is provided in Additional file 5: Table S2.
Differential gene expression in FL and CS compared to con
Based on the density plot of FPKM [25] values, the gene expression patterns of CS and FL were much more similar than those of Con and CS and Con and FL (Fig. 2a). These results are in agreement with the seed germination results, i.e., the seed germination percentage of CS and FL was higher than that of Con (Fig. 1). In total, 484 unigenes were differentially expressed between CS and Con, whereas 9156 DEGs were detected between FL and Con (Fig. 2b). There were 250 up-regulated and 234 down-regulated DEGs in CS, whereas there were 4142 up-regulated and 5014 down-regulated DEGs in FL. The top 20 enriched functional processes based on KEGG analysis between FL and CS compared to Con are shown in Additional file 6: Figure S4a, b. Among these processes, phenylalanine and phenylpropanoid metabolism were both highly enriched in CS and FL, as well as carbohydrate metabolism, amino acid and lipid metabolism, and genetic information processing. We also identified different enriched groups between FL and CS; for example, flavonoid metabolism (including flavonoid biosynthesis and flavone and flavonol biosynthesis) and photosynthesis processes are highly enriched in CS but not in FL (Additional file 6: Figure S4a). By contrast, the categories metabolism of xenobiotics by cytochrome P450, plant hormone signal transduction, and plant-pathogen interaction, and carotenoid biosynthesis were much more highly enriched in FL than in CS (Additional file 6: Figure S4b).

Differentially expressed N. incisum genes between Con, FL and CS. a FPKM density distribution; b Venn diagram
To elucidate the molecular changes that occur during seed dormancy alleviation in N. incisum, we clustered the DEGs expressed following CS and FL treatments by H-cluster and K-cluster analysis. H-cluster analysis showed that seeds of CS were much more similar to those of Con than to those of FL (Additional file 7: Figure S5a). Perhaps imbibition in FL solution is a much more crucial treatment than incubation in cold, moist conditions, as was performed during pretreatment. However, there were still some DEGs with similar expression patterns between FL and CS (Additional file 7: Figure S5a), which might be the key genes involved in seed germination and dormancy loss. K-clustering analysis identified six groups, including one group with 356 DEGs that were simultaneously down-regulated and one group of 114 ones that were simultaneously up-regulated in CS and FL (Additional file 7: Figure S5b). GO and KEGG enrichment analysis of the 468 co-expressed DEGs suggested that these DEGs function in numerous processes including mass metabolism (metabolic, proteolysis, carbohydrate metabolic, lipid metabolic, cellular metabolic, and cellular amino acid metabolic), energy metabolism (oxidation-reduction, NAD binding, and Rab GTPase binding), and genetic materials metabolism (nucleus, chromatin, chromatin binding, and nucleotidyl transferase activity) during seed dormancy release. The category “biological process of embryo development” was also enriched during this process, implying that genes involved in seed development are actively expressed (Additional file 8: Figure S6a).
We compared the biological processes occurring in FL vs. CS and found that most functional categories were more highly enriched in FL than in CS. However, flavonoid biosynthesis was much more highly enriched in CS than in FL (Additional file 8: Figure S6b). We identified ten key genes involved in flavone or flavonoid biosynthesis in N. incisum based on the RNA-Seq data and quantified their expression by reverse-transcription PCR (RT-PCR) using the primers listed in Table 2. Six of ten genes for key flavone biosynthesis enzymes, including phenylalanine ammonia-lyase 1 (NiPAL1), 4-coumarate: CoA ligase 2 (Ni4CL2), 4-coumarate: CoA ligase-like 5 (Ni4CLL5), chalcone synthase 2 (NiCHS2), p-coumarate 3-hydroxylase (NiC3H), and flavone synthase I (NiFNSI), were significantly up-regulated in CS (Additional file 9: Figure S7). The dramatically increased expression of NiPAL1 in CS suggests that secondary metabolites are biosynthesized in N. incisum seeds upon CS. The expression of NiFNSI, a flavone synthase gene particular to Apiaceae species [26], was significantly up-regulated in CS, but unchanged in FL (Additional file 9: Figure S7). The large number of up-regulated genes involved in flavone or flavonoid biosynthesis in CS suggests that one or both of these processes is induced during or after CS in N. incisum.
We primarily focused on genes involved in the biosynthesis and signal transduction pathways of key plant hormones related to seed dormancy loss, such as GA and ABA, as well as strigolactones (SLs; metabolites derived from carotenoids) during FL and CS treatments in subsequent analyses.
RNA-Seq data validation by RT-PCR
To validate the RNA-Seq results, we quantitatively assessed the expression patterns of the mRNAs using RT-PCR. We randomly selected 18 unigenes (some up-regulated, some down-regulated, and some with no change in expression; the primers listed in Table 2) involved in the biosynthesis and regulation of ABA, GA, SLs, and flavonoids. The gene expression patterns of the 16 unigenes revealed by RT-PCR agreed with those obtained by RNA-Seq (Fig. 3). However, the expression levels of ABI5 and the metabolic enzyme gene PDS obtained by RT-PCR were different from those obtained by RNA-Seq, perhaps due to the lower level of accuracy of RNA-Seq, as observed in other studies [27]. In general, the RNA-Seq data confirm the gene expression patterns during the alleviation of seed dormancy in N. incisum.

Comparison of relative gene expression levels detected by RT-PCR vs. RNA-Seq (read count). The data are log2-transformed values of the fold changes in FL compared to Con for N. incisum seeds
Gene expression of ABA and GA in FL
In total, 12 genes involved in ABA biosynthesis and signal transduction were identified in N. incisum. The PDS gene was slightly down-regulated after FL treatment (Fig. 4b, Additional file 10: Table S3). As were the genes of carotenoid beta-ring hydroxylase (NiLUT5), zeaxanthin epoxidase (NiZEP), and violaxanthin de-epoxidase (NiVDE) (down-regulated ~ 3.7-,3.8-, and 5.9-fold, respectively) (Fig. 4a, b, Additional file 10: Table S3). The subsequent downstream reaction involves the conversion of 9′-cis-violaxanthin or 9′-cis-neoxanthin to xanthoxin by 9-cis-epoxycarotenoid dioxygenase (NCED) [28]. Ten NiNCED genes were identified in the six N. incisum samples, eight of which were expressed at very low levels. In FL, the expression of NiNCED1 and NiNCED3 were up-regulated for 7.2- and 7.5-fold (Fig. 4, Additional file 10: Table S3). NiABA2 was differentially expressed in FL (Fig. 4). The gene for aldehyde oxidase 3 (NiAAO3) catalyzed the last step of ABA biosynthesis [29] was down-regulated (~ 2.1-fold) in FL (Fig. 4, Additional file 10: Table S3). The expression of ABA3, encoding a MoCo sulfurase required by AAO3 for its activity [30], remained unchanged in FL (Additional file 10: Table S3). ABA 8′-hydroxylase (CYP707A) is involved in the degradation of ABA into phaseic acid [31]. Three genes, NiCYP707A1, NiCYP707A4, and NiCYP707A7, were significantly up-regulated in FL, especially NiCYP707A1 (10.2-fold) (Fig. 4, Additional file 10: Table S3).

Metabolism and regulation of ABA and SL biosynthesis in N. incisum seeds. Up-regulated genes are shown in red, down-regulated genes are shown in blue, genes with no significant change in expression are shown in black, and genes not identified are shown in gray. The inset graphs show relative expression levels of the major ABA genes in FL compared to Con detected by RT-PCR (a) or RNA-Seq (b), and the major SLs genes in FL and CS compared to Con detected by RT-PCR (c). Data represent means ± SD (n = 3)
NiPYL5, the only differentially expressed ABA receptor gene, was down-regulated 8.9-fold in FL (Fig. 4, Additional file 10: Table S3). The gene for ABA-induced Snf1-related kinase 2 (NiSnRK2), a positive regulator of ABA signaling processes including stress tolerance and seed dormancy [32,33,34,35], was down-regulated ~ 2.5-fold in FL (Fig. 4, Additional file 10: Table S3). ABF/AREB subfamily genes ABA-insensitive 5-like protein 6 (NiABF3), NiABF4, ABA-insensitive 5-like protein 2 (NiDPBF3), and ABA-insensitive 5 (NiABI5) were significantly differentially expressed (down-regulated ~ 2.4-, 2.8-, 2.2-, and 1.9-fold, respectively) in FL (Fig. 4b, Additional file 10: Table S3).
GA and ABA affect antagonistically seed germination and dormancy. GA improves seed germination. The formation of a C19-GA skeleton from C20-GA is catalyzed by GA 20-oxidase (GA20OX) [36]. The final step in the formation of physiologically active GAs is catalyzed by GA 3-oxidase (GA3OX) [37]. GA 2-oxidase (GA2OXs) is involved in the conversion from physiologically active GAs to inactive forms [38]. NiGA20OX was dramatically up-regulated (NiGA20OX1 and NiGA20OX5 were up-regulated 10.6- and 7.9-fold, respectively) in FL (Fig. 5, Additional file 10: Table S3). Gene NiGA3OX was down-regulated for 4.5-fold. Genes NiGA2OX1, NiGA2OX2, and NiGA2OX8 were marked up-regulated for ~ 61.7-, 10.6-, and 7.9-fold, respectively (Fig. 5, Additional file 10: Table S3). Gibberellin receptor 1 (GID1) is a receptor of GAs whose accumulation is associated with seed dormancy loss in Arabidopsis [39]. NiGID1B was up-regulated 3.2-fold in FL, whereas the expression of NiGID1C in FL was similar to that of Con (Additional file 10: Table S3). The gene of DELLA protein gibberellic acid-insensitive (GAI), which represses physiological processes induced by GA [40], was down-regulated 10.5-fold in FL (Fig. 5, Additional file 10: Table S3). This is consistent with the observation that seed germination occurred after FL treatment. NiGASA14, the only differentially expressed GASA gene, was up-regulated 3.8-fold in FL (Fig. 5, Additional file 10: Table S3).

Metabolism and regulation of GA biosynthesis in N. incisum seeds. Genes down-regulated in both FL and CS are indicated in blue. The inset graph shows the relative expression levels of GA genes in FL and CS compared to Con detected by RT-PCR. Values are means ± SD (n = 3)
Genes involved in seed dormancy release upon CS in N. incisum
Three genes NiGA20OX1, NiGA20OX5, and NiGA3OX1, encoding enzymes leading to the production of bioactive GA, were up-regulated 6.2-, 2.2-, and 3.6-fold, respectively in CS (Fig. 5, Additional file 10: Table S3). However, NiGA2OX8, which function in the inactivation of GA, were up-regulated as well (11.2-fold) (Fig. 5, Additional file 10: Table S3). NiGAI was down-regulated 1.4-fold in CS. It is positive related to seed germination. Among the GASA genes, although NiGASA6 was significantly up-regulated (1.6-fold) in CS vs Con, NiGASA9 and NiGASA14 were not differentially expressed in CS (Fig. 5, Additional file 10: Table S3). Therefore, NiGASA6 might be involved with seed dormancy loss induced by CS in N. incisum.
Although ABA levels increased significantly after CS (Fig. 1c), none of the ABA metabolism genes were differentially expressed, whereas NiABI5 was significantly down-regulated (2.1- fold) (Additional file 10: Table S3).
Gene expressions of SLs in FL and CS
SLs are produced from carotenoids in plants and play key roles in stimulating seed germination in parasitic plants [41, 42]. However, whether SLs are involved in seed germination in non-parasitic plants is currently unknown. Among the metabolites derived from phytoene during carotenoid metabolism, lycopene ε-cyclase (LYCe) and lycopene β-cyclase (LYCb) convert lycopene into α-carotene, and the latter is then converted into lutein by CYP97A/C [43]. We identified NiLYCe, NiCYP97A, and NiCYP97C in N. incisum, but they were not differentially expressed in FL or CS (Additional file 10: Table S3). The gene NiLUT5 was down-regulated (3.7-fold) in FL but expressed at control levels in CS (Fig. 4a, Additional file 10: Table S3). D27, β-carotene isomerase dwarf 27, isomerizes all-trans-beta-carotene to 9-cis-beta-carotene, the first step in SL metabolism. Carotenoid cleavage dioxygenase 7 is responsible for the subsequent reaction, giving rise to the formation of 9-cis-beta-10′-carotenal, the latter being directly catalyzed into carlactone by CCD8 [44]. CYP711A functions in the conversion of carlactone to 5-deoxystrigol [45]. Among the four known genes involved in SL biosynthesis, three (NiD27, NiCCD8, and NiCYP711A1) were identified, and all were highly up-regulated (~ 2.9-, 17-, and 5.2-fold in FL vs. Con, and ~ 1.4-, 2.1-, 1.8-fold in CS) in N. incisum (Fig. 4c, Additional file 10: Table S3). By contrast, no gene encoding CCD7 was identified in any treated sample, but there might be other genes involved in the conversion from 9-cis-beta-carotene into 9-cis-beta-10′-carotenal in N. incisum. Moreover, no gene for BKT, which is involved in astaxanthin metabolism, was identified in N. incisum seeds. DAD2, a SL receptor, interacts with MAX2A in the presence of GR24 (a synthetic analogue of SLs) [46]. One DAD2 and one MAX2 gene were identified in N. incisum seeds. NiDAD2 was highly up-regulated (5.8-fold in FL and 2.0-fold in CS), but NiMAX2 was expressed at the level of the control in both FL and CS (Fig. 4c).
Discussion
Upon FL, genes involved in ABA biosynthesis (NiLUT5, NiZEP, and NiAAO3) were down-regulated and genes involved in ABA catabolism (NiCYP707As) were dramatically up-regulated, indicating that these genes contributed to the reduction of ABA levels and degrading through hydrolyzation after FL treatment. The down-regulation of ABA receptor gene and its signaling regulator gene implied that the physiological events induced by ABA were retarded after FL treatment. Among them, genes NiABF3, NiABF4, NiDPBF3, and NiABI5, together with NiPYL5 and NiSnRK2, were negatively correlated with seed dormancy release [47, 48].
The up-regulated NiGID1B suggests that NiGID1B might be important for seed dormancy loss, which also occurs when seed dormancy is broken by after-ripening and CS in Arabidopsis [39]. The dramatic down-regulation of DELLA protein gibberellic acid-insensitive (GAI) gene, which represses physiological processes induced by GA [40], is consistent with the observation that seed germination occurred after FL treatment. It indicates that GAI likely contributes to seed germination under FL treatment. GA-Stimulated Arabidopsis (GASA) proteins also play important roles in seed germination [49] and other developmental processes. Our results suggest GASA14 might play a positive role in seed dormancy release or seed germination in FL, and by positively affecting cell elongation during seed germination [50]. Although the expression patterns of GA metabolic genes implied that the level or activity of GA3 decreased after FL treatment, the expression of GA regulatory genes NiGID1B, NiGAI, and NiGASA14 was positively involved in seed dormancy release treated by FL, and it might not strongly rely on GA3 levels or activity.
Dramatically up-regulation of three genes NiGA20OX1, NiGA20OX5, and NiGA3OX1, together with down-regulation of NiGA2OX8 must attribute to the increase of GA level (Fig. 1c). Our results suggest that ABA regulators are involved in seed dormancy release induced by CS. The negative association of NiABI5 with seed dormancy release in CS was the same as in FL. The up-regulation of NiGASA6 and the down-regulation of NiGAI during CS suggest that they may be involved in CS-dormancy release.
Gene expressions of SLs biosynthesis and regulators suggest that SLs are actively synthesized after seed dormancy alleviation. Both the inhibition of ABA biosynthesis and the induction of SLs biosynthesis occurred after treated by FL in N. incisum seeds. The gene expression of SLs regulators indicates that SLs are active after FL and CS treatment in N. incisum seeds, further demonstrating that SLs might play a positive role in seed dormancy release in N. incisum.
Taken together, we propose FL- and CS-dormancy release involve completely different mechanisms involving the alleviation of ABA effects and potentiation of GA effects, respectively. Nevertheless, GA still positively regulated seed dormancy release in FL, and ABA regulators are involved in seed dormancy release induced by GA accumulation in CS as well. Moreover, down-regulation of NiABI5 and NiGAI might occur or be necessary for seed dormancy release in N. incisum; these two genes possibly function as integrators of ABA and GA signaling associated with seed dormancy alleviation or germination.
Conclusions
This study indicates that the processes involved in FL-dormancy release are much more complex than in CS-dormancy release. Different mechanisms occurred in FL- and CS-dormancy release: alleviation of ABA effects and potentiation of GA effects, respectively. Both ABA and GA regulators contributed to seed dormancy release irrespective of the dormancy treatment. It is possible that ABA and GA regulators act to integrate the effects of ABA and GA on seed dormancy release. The upregulation of SL biosynthesis and its regulatory genes in both FL- and CS-dormancy release imply that SLs are positive to seed dormancy release in N. incisum.
Abbreviations
- 4CL2:
-
4-coumarate:CoA ligase 2
- 4CLL5:
-
4-coumarate:CoA ligase-like 5
- AAO3:
-
Aldehyde oxidase 3
- ABA:
-
Abscisic acid
- ABF3:
-
ABA-insensitive 5-like protein 6
- ABI5:
-
ABA-insensitive 5
- C3H:
-
p-coumarate 3-hydroxylase
- C4H:
-
Cinnamate 4-hydroxylase
- CCD:
-
Carotenoid cleavage dioxygenase
- CHS2:
-
Chalcone synthase 2
- CS:
-
Cold stratification
- CYP707A:
-
ABA 8′-hydroxylase
- CYP75B2:
-
Flavonoid 3′-monooxygenase
- D27:
-
β-carotene isomerase dwarf 27
- DAD2:
-
Decreased apical dominance 2
- DEGs:
-
Differentially expressed genes
- DPBF3:
-
ABA-insensitive 5-like protein 2
- FAOMT:
-
Flavonoid 3′,5′-methyltransferase
- FL:
-
Fluridone
- FNSI:
-
Flavone synthase I
- FPKM:
-
Expected number of Fragments Per Kilobase of transcript sequence per Millions base pairs sequenced
- GA:
-
Gibberrellin
- GA20OX:
-
GA 20-oxidase
- GA2OX:
-
GA 2-oxidase
- GA3OX:
-
GA 3-oxidase
- GAI:
-
Gibberellic acid-insensitive
- GASA:
-
GA-Stimulated Arabidopsis
- GID1:
-
Gibberellin receptor 1
- LUT5:
-
Carotenoid beta-ring hydroxylase
- MAX:
-
More axillary growth
- MPD:
-
Morpho-physiological dormant
- NCED:
-
9-cis-epoxycarotenoid dioxygenase
- PAL:
-
Phenylalanine ammonia-lyase
- PDS:
-
Phytoene desaturase
- PYL5:
-
Abscisic acid receptor
- SL:
-
Strigolactone
- SnRK2:
-
Snf1-related kinase 2
- UHPLC-MS/MS:
-
Ultra-performance liquid chromatography coupled with a quadrupole trap mass spectrometer equipped with electrospray ionization
- VDE:
-
Violaxanthin de-epoxidase
- ZEP:
-
Zeaxanthin epoxidase
References
The State Pharmacopoeia Commission of P.R. China. Pharmacopoeia of the People's Republic of China. Vol. 1. Beijing, China: China Medical Science; 2015. p. 182–3.
Zhang EH, Chen XL, Fang ZS, Wang D. Studies on seed dormancy mechanism and breaking technique of wild Notopterygium incisum. Acta Agrestia Sinica. 2007;15(6):509–14.
Wareing PF, Saunders PF, Machlis L, Briggs WR, Park RB. Hormones and dormancy. Annu Rev Plant Physiol. 1971;22(22):261–88.
Milborrow BV. The pathway of biosynthesis of abscisic acid in vascular plants: a review of the present state of knowledge of ABA biosynthesis. J Exp Bot. 2001;52(359):1145–64.
Bartels PG, Watson CW. Inhibition of carotenoid synthesis by fluridone and norflurazon. Weed Sci. 1978;26(2):198–203.
Breitenbach J, Sandmann G. ζ-Carotene cis isomers as products and substrates in the plant poly-cis carotenoid biosynthetic pathway to lycopene. Planta. 2005;220(5):785–93.
Harada J, Nagashima KV, Takaichi S, Misawa N, Matsuura K, Shimada K. Phytoene desaturase, CrtI, of the purple photosynthetic bacterium, Rubrivivax gelatinosus, produces both neurosporene and lycopene. Plant Cell Physiol. 2001;42(10):1112–8.
Goggin DE, Powles SB. Fluridone: a combination germination stimulant and herbicide for problem fields? Pest Manag Sci. 2014;70(9):1418–24.
Feurtado JA, Yang J, Ambrose SJ, Cutler AJ, Abrams SR, Kermode AR. Disrupting abscisic acid homeostasis in western white pine (Pinus monticola Dougl. Ex D. Don) seeds induces dormancy termination and changes in abscisic acid catabolites. J Plant Growth Regul. 2007;26(1):46–54.
Ali-Rachedi S, Bouinot D, Wagner MH, Bonnet M, Sotta B, Grappin P. Changes in endogenous abscisic acid levels during dormancy release and maintenance of mature seeds: studies with the Cape Verde Islands ecotype, the dormant model of Arabidopsis thaliana. Planta. 2004;219(3):479–88.
Yoshioka T, Endo T, Satoh S. Restoration of seed germination at supraoptimal temperatures by fluridone, an inhibitor of abscisic acid biosynthesis. Plant Cell Physiol. 1998;39(3):307–12.
Ma X, Jiang S, Shi J, Sun H, Zhou Y. Breaking sleep and budding of Notopterygium incisum seeds. China: Vol. CN10104906; 2007. p. 10.
Ma Z, Tan Y, Cui G, Feng Y, Cui Q, Song X. Transcriptome and gene expression analysis of DHA producer Aurantiochytrium under low temperature conditions. Sci Rep. 2015;5:14446–58.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402.
Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K. The Pfam protein families database. Nucleic Acids Res. 2008;36(Database issue):263–6.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12(1):323.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106.
Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11(2):R14.
Mao X, Cai T, Olyarchuk JG, Wei L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21(19):3787–93.
Wang X. Quantitative analysis of major plant hormones in crude plant extracts by high-performance liquid chromatography-mass spectrometry. Nat Protoc. 2010;5(6):986–92.
Chen QL, Wang HL, Wang ZF, Shan CG, Zhai ZX, Guo YH. Effects of cold stratification and exogenous gibberellic acid (GA3) on seed germination and contents of endogenous gibberellins (GAs) and abscisic acid (ABA) in Cistanche deserticola Y.C.Ma. Plant Physiol Commun. 2009;45(3):270–2.
Paul KB, Patel CS, Biswas PK. Changes in endogenous growth kegulators in loblolly pine seeds during the process of stratification and germination. Physiol Plant. 2006;28(3):530–4.
Gianinetti A, Vernieri P. On the role of abscisic acid in seed dormancy of red rice. J Exp Bot. 2007;58(12):3449–62.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–52.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–5.
Heller W, Forkmann G. Biosynthesis of flavonoids. In: Harborne JB, editor. The flavonoids: Advances in Research since 1986. London: Chapman and Hall; 1993. p. 499–535.
Huang W, Ye J, Zhang J, Lin Y, He M, Huang J. Transcriptome analysis of Chlorella zofingiensis to identify genes and their expressions involved in astaxanthin and triacylglycerol biosynthesis. Algal Res. 2016;17:236–43.
Iuchi S, Kobayashi M, Shinozaki K. Function of Arabidopsis NCED genes in the biosynthesis of abscisic acid under drought stress. Plant Cell Physiol. 2001;42(Supplement):s90.
Su X, Li Q, Chen S, Dong C, Hu Y, Yin L, Yang J. Analysis of the transcriptome of Isodon rubescens and key enzymes involved in terpenoid biosynthesis. Biotechnol Biotechnol Equip. 2016;30(3):592–601.
Bittner F, Oreb M, Mendel RR. ABA3 is a molybdenum cofactor sulfurase required for activation of aldehyde oxidase and xanthine dehydrogenase in Arabidopsis thaliana. J Biol Chem. 2001;276(44):40381–4.
Kushiro T, Okamoto M, Nakabayashi K, Yamagishi K, Kitamura S, Asami T, Hirai N, Koshiba T, Kamiya Y, Nambara E. The Arabidopsis cytochrome P450 CYP707A encodes ABA 8′-hydroxylases: key enzymes in ABA catabolism. EMBO J. 2004;23(7):1647–56.
Johnson RR, Wagner RL, Verhey SD, Walker-Simmons MK. The abscisic acid-responsive kinase PKABA1 interacts with a seed-specific abscisic acid response element-binding factor, TaABF, and phosphorylates TaABF peptide sequences. Plant Physiol. 2002;130(2):837–46.
Fujita Y, Nakashima K, Yoshida T, Katagiri T, Kidokoro S, Kanamori N, Umezawa T, Fujita M, Maruyama K, Ishiyama K. Three SnRK2 protein kinases are the main positive regulators of abscisic acid signaling in response to water stress in Arabidopsis. Plant Cell Physiol. 2009;50(12):2123–32.
Nakashima K, Fujita Y, Kanamori N, Katagiri T, Umezawa T, Kidokoro S, Maruyama K, Yoshida T, Ishiyama K, Kobayashi M. Three Arabidopsis SnRK2 protein kinases, SRK2D/SnRK2.2, SRK2E/SnRK2.6/OST1 and SRK2I/SnRK2.3, involved in ABA signaling are essential for the control of seed development and dormancy. Plant Cell Physiol. 2009;50(7):1345–63.
Huai J, Wang M, He J, Zheng J, Dong Z, Lv H, Zhao J, Wang G. Cloning and characterization of the SnRK2 gene family from Zea mays. Plant Cell Rep. 2008;27(12):1861–8.
Lange T, Graebe JE. The partial purification and characterization of a gibberellin C-20 hydroxylase from immature Pisum sativum L. seeds. Planta. 1989;179(2):211–21.
Macmillan J, Hedden P. Gibberellin biosynthesis from gibberellin A12-aldehyde in endosperm and embryos of Marah macrocarpus. Plant Physiol. 1997;113(4):1369–77.
Ross JJ, Reid JB, Swain SM, Hasan O, Poole AT, Hedden P, Willis CL. Genetic regulation of gibberellin deactivation in Pisum. Plant J. 1995;7(3):513–23.
Hauvermale AL, Tuttle KM, Takebayashi Y, Seo M, Steber CM. Loss of Arabidopsis thaliana seed dormancy is associated with increased accumulation of the GID1 GA hormone receptors. Plant Cell Physiol. 2015;56(9):188–90.
Peng J, Carol P, Richards DE, King KE, Cowling RJ, Murphy GP, Harberd NP. The Arabidopsis GAI gene defines a signaling pathway that negatively regulates gibberellin responses. Genes Dev. 1997;11(23):3194–205.
Matusova R, Rani K, Verstappen FWA, Franssen MCR, Beale MH, Bouwmeester HJ. The strigolactone germination stimulants of the plant-parasitic striga and orobanche spp are derived from the carotenoid pathway. Plant Physiol. 2005;139(2):920–34.
Cook CE, Whichard LP, Turner B, Wall ME, Egley GH. Germination of witchweed (Striga lutea Lour.): isolation and properties of a potent stimulant. Science. 1966;154(3753):1189–90.
Lin H, Wang R, Qian Q, Yan M, Meng X, Fu Z, Yan C, Jiang B, Su Z, Li J. DWARF27, an iron-containing protein required for the biosynthesis of strigolactones, regulates rice tiller bud outgrowth. Plant Cell. 2009;21(5):1512–25.
Alder A, Jamil M, Marzorati M, Bruno M, Vermathen M, Bigler P, Ghisla S, Bouwmeester H, Beyer P, Albabili S. The path from β-carotene to carlactone, a strigolactone-like plant hormone. Science. 2012;335(6074):1348.
Seto Y, Kameoka H, Yamaguchi S, Kyozuka J. Recent advances in strigolactone research: chemical and biological aspects. Plant Cell Physiol. 2012;53(11):1843–53.
Hamiaux C, Drummond RS, Janssen BJ, Ledger SE, Cooney JM, Newcomb RD, Snowden KC. DAD2 is an α/β hydrolase likely to be involved in the perception of the plant branching hormone, strigolactone. Curr Biol. 2012;22(21):2032–6.
Liu S, Zhu M, Liu X, Ling LI. Studies on the responses to ABA and salt stress in abf3 and abf4-related mutants of Arabidopsis. J South China Normal Univ. 2012;44(4):100–4.
Lopez-Molina L, Mongrand S, Chua NH. A postgermination developmental arrest checkpoint is mediated by abscisic acid and requires the ABI5 transcription factor in Arabidopsis. PNAS. 2001;98(8):4782–7.
Rubinovich L, Weiss D. The Arabidopsis cysteine-rich protein GASA4 promotes GA responses and exhibits redox activity in bacteria and in planta. Plant J. 2010;64(6):1018–27.
Roxrud I, Lid SE, Fletcher JC, Schmidt ED, Opsahlsorteberg HG. GASA4, one of the 14-member Arabidopsis GASA family of small polypeptides, regulates flowering and seed development. Plant Cell Physiol. 2007;48(3):471–83.
Acknowledgements
The authors would like to thank Dr. Xiuteng Zhou for helping the detection of plant hormone ABA and GA3.
Funding
This research was supported by grants from the National Natural Science Foundation of China (81325023), China Postdoctoral Science Foundation (ZZ2016012), and Key project at central government level: The ability establishment of sustainable use for valuable Chinese medicine resources.
Availability of data and materials
The transcriptomic data was deposited in SRA database of NCBI (Access No. SRP107325) (https://www.ncbi.nlm.nih.gov/sra/). All the data supporting results of this article are listed in additional files.
Author information
Authors and Affiliations
Contributions
AHL and LQH conceived and designed the experiments. AHL, SYJ, GY, and YL performed the experiments. AHL, TC, NG, and LPK analyzed the data. AHL, GN, and KLP wrote the paper. LQH critically revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Plant materials were provided by Sichuan Notopterygium Organic Herbs Co., Ltd.
Competing interests
The authors declare that they have no competing interest.
Additional files
Additional file 1:
Table S1. Summary of functional annotation of genes in N. incisum seeds. (XLSX 40668 kb)
Additional file 2:
Figure S1. Gene expression patterns in N. incisum seeds aligned to those of other species in the Nr database. (TIF 6208 kb)
Additional file 3:
Figure S2. GO classification of unigenes in N. incisum. The unigenes were assigned to the three GO categories: biological process, cellular component, and molecular function. (TIF 4228 kb)
Additional file 4:
Figure S3. KOG analysis of unigenes in N. incisum seeds. (TIF 4024 kb)
Additional file 5:
Table S2. KEGG classification of assembled unigenes in N. incisum. (XLSX 137 kb)
Additional file 6:
Figure S4. KEGG enrichment analysis of DEGs in N. incisum seeds under FL (a) and CS (b) treatment compared to Con. (TIF 3608 kb).
Additional file 7:
Figure S5. H-cluster (a) and K-cluster (b) analysis of differentially expressed genes in samples Con, FL, and CS of N. incisum seeds. (TIFF 5869 kb)
Additional file 8:
Figure S6. GO (a) and KEGG (b) enrichment analysis of co-expressed DEGs in N. incisum seeds. BP, CC, and MF indicate tshe three GO categories: biological process, cellular component, and molecular function, respectively. (TIF 9584 kb)
Additional file 9:
Figure S7. Relative expression of genes encoding enzymes involved in flavone or flavonoid metabolism in N. incisum seeds under FL and CS treatment compared to Con determined by RT-PCR. Data represent means ± SD (n = 3). (TIF 2011 kb)
Additional file 10:
Table S3. The gene expression information of genes involved in ABA, GA, and SL biosynthesis and their expression under FL and CS treatments in N. incisum seeds. (XLSX 11 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Aihua, L., Shunyuan, J., Guang, Y. et al. Molecular mechanism of seed dormancy release induced by fluridone compared with cod stratification in Notopterygium incisum. BMC Plant Biol 18, 116 (2018). https://doi.org/10.1186/s12870-018-1333-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-018-1333-2