
Abstract
Background
The Calakmul Biosphere Reserve (CBR) is known for its rich animal and plant biodiversity, yet its microbial communities remain largely unknown. The reserve does not possess permanent bodies of water; nevertheless, seasonal depressions associated with fractures create wetlands, known locally as aguadas. Given the recent construction of the Maya train that crosses the CRB, it is essential to assess the biodiversity of its microorganisms and recognize their potential as a valuable source of goods. This evaluation is pivotal in mitigating potential mismanagement of the forest ecosystem. To enhance comprehension of microbial communities, we characterized the microbiota in three different wetlands. Ag-UD1 and Ag-UD2 wetlands are located in a zone without human disturbances, while the third, Ag-SU3, is in a semi-urbanized zone. Sampling was carried out over three years (2017, 2018, and 2019), enabling the monitoring of spatiotemporal variations in bacterial community diversity. The characterization of microbiome composition was conducted using 16S rRNA metabarcoding. Concurrently, the genomic potential of select samples was examined through shotgun metagenomics.
Results
Statistical analysis of alpha and beta diversity indices showed significant differences among the bacterial communities found in undisturbed sites Ag-UD1 and Ag-UD2 compared to Ag-SU3. However, no significant differences were observed among sites belonging to the undisturbed area. Furthermore, a comparative analysis at the zone level reveals substantial divergence among the communities, indicating that the geographic location of the samples significantly influences these patterns. The bacterial communities in the CBR wetlands predominantly consist of genera from phyla Actinobacteria, Acidobacteria, and Proteobacteria.
Conclusion
This characterization has identified the composition of microbial communities and provided the initial overview of the metabolic capacities of the microbiomes inhabiting the aguadas across diverse conservation zones. The three sites exhibit distinct microbial compositions, suggesting that variables such as chemical composition, natural and anthropogenic disturbances, vegetation, and fauna may play a pivotal role in determining the microbial structure of the aguadas. This study establishes a foundational baseline for evaluating the impact of climatic factors and human interventions on critical environments such as wetlands.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Background
The Calakmul Biosphere Reserve (CBR), located in southeast Campeche, Mexico, represents a tropical humid forest ecosystem. As a UNESCO World Network of Biosphere Reserves member and a recognized Cultural Heritage of Humanity site, the CBR forms a part of the Mesoamerican Biological Corridor [1] extending through Guatemala, Belize, and Chiapas. Historically, Calakmul is recognized as a significant urban center in the Mayan civilization, with approximately 1500 years of habitation (500 BC to AD 900). Designated as a protected natural area by the Mexican government since 1989, the Calakmul Biosphere Reserve stands as the second largest biome of its kind, following the Amazon [2]. The decree delineates the reserve’s zoning into two core areas, named Core Zone I and Core Zone II, in addition to a buffer zone. In 2023, there was a change in the surface area of Calakmul [3]. This modification expanded the protected natural area from 723,185-12-50 to 728,908-57-63.13 hectares, enlarging the core zones to include areas of ecological importance.
In CBR soils, rainwater filters quickly, preventing the formation of permanent surface water bodies such as rivers or lakes [2]. However, depressions associated with faults and fractures allow water accumulation during the rainy season, creating temporary water bodies known as aguadas, which serve as the only water source during the dry season. The soils near the aguadas, undergoing hydromorphic pedogenesis, develop into gleysol-type soils, whereas the formation of vertisols requires a dry season [2]. These areas mirroring wetland characteristics are fundamental for the Reserve’s macro diversity, including plants and animals, and microbiological diversity [4]. The aguadas of Selva Maya are essential for certain endangered species, leading to collaboration in their monitoring through various international initiatives [4]. Macro species like vultures and jaguars have been extensively monitored [5,6,7,8], and the impact of aguadas drought on fauna has been documented [8]. Nevertheless, the exploration of microbial diversity in these areas remains unexplored.
Soil microbial communities in freshwater and saline wetlands hold global significance due to their roles in primary production, organic decomposition, carbon cycling, and nutrient mineralization [9,10,11]. The diversity of prokaryotic microorganisms in these habits is estimated to vary, with ranges between 2,000 and 18,000 genomes per gram of soil [12,13,14]. Soil is recognized as the richest natural source of antibiotic-resistance genes. This richness is attributable to the vast biodiversity of microorganisms in both pristine ecosystems and human-modified environments, including cultivated and polluted soils [15]. Furthermore, soil bacterial communities are a treasure trove of genes and metabolites crucial in pharmaceutical and industrial applications [16].
Microorganisms in water-soil environments are widely acknowledged for producing specialized compounds, namely secondary metabolites. These microorganisms act as carbon sink and sources, contributing to climate change dynamics [17, 18]. Additionally, the decline in endemic microbial biodiversity in the soil is primarily attributed to human activities, particularly in agroecosystems, and is associated with industrialized agricultural practices and deforestation [13,14,15,16, 19]. Measuring baselined diversity is crucial for understanding how human disturbances alter microbial diversity in aguadas ecosystems. In the Yucatan Peninsula, most microbial diversity studies have concentrated on coastal areas or cenotes [20, 21]. However, despite the ecological significance of Calakmul, research focusing on the content and distribution of microorganisms in its soils or sediments remains limited or almost non-existent [22].
Chemical and physical soil properties can significantly influence microorganisms’ diversity, composition, and activity. Soils in the CBR are characterized as slightly alkaline and shallow [23], which contrasts with the typically acidic soils (average pH 5.2) of other tropical forests. The alkaline nature of these soils originates from limestone rock, which is soft in hardness and predominantly consists of calcium carbonate (CaCO3), accounting for over 60% of its mineral composition. These soils are notably deficient in iron (Fe), silica (SiO2), and aluminum (Al). The weathering of limestone through dissolution does not produce new clays, resulting in predominantly shallow soil formation. High organic matter levels characterized these soils, with fertility largely dependent on organic content rather than clay. The soil is deficient in micronutrients such as phosphorus, zinc, iron, and copper [24]. This particular soil composition, the limited human presence near the biosphere, and the minor anthropological disturbances in the reserve create a unique space for microbiological diversity research.
Integrating complementary methodologies, including 16 S rRNA amplicon sequencing and shotgun metagenomics, facilitates a more comprehensive investigation of microbial ecology [25, 26]. In a pioneering effort to investigate the diversity within the aguadas, our study focused on analyzing bacterial communities in the wetland ecosystems of CBR using next-generation sequencing (NGS) technology for 16S rRNA sequencing. Over three years, sampling was conducted at three distinct sites within the reserve, predominantly in September, coinciding with the rainy season’s peak, when the sites were flooded. Additionally, one site was also sampled in June during the dry season. Under government protection, two sites restrict human access to maintain the undisturbed state (Ag-UD1 and Ag-UD2). The third site, situated in the semi-urbanized zone, permits public transit from Calakmul (Ag-SU3).
Shotgun metagenomics was applied across all three sites in the 2019 samples to evaluate the metabolic capabilities of the community. Previous research has documented a decline in microbiome diversity transitioning from undisturbed to semi-urbanized areas. Moreover, in the Yucatan Peninsula, the Bacalar Lagoon is a notable example of reduced microbial diversity in locations impacted by tourism [27]. Although existing research addresses the impact of urbanization on a range of ecosystems, a notable gap persists in comprehending the influence of semi-urbanization — mainly when linked to large-scale infrastructure projects — on the bacterial communities of Calakmul’s wetlands. This substantial infrastructural venture, which will traverse the Reserve, could alter microbial populations as observed in a previous study in Salt Lake, Utah’s microbialites [28]. In that study, the construction of a railroad led to altered microbial community compositions on each side of the barrier, resulting in the diminished carbonate precipitation capabilities of one such population [28].
Microdiversity within Calakmul aguadas is likely to exhibit spatial and temporal variations, with the aguadas in undisturbed areas expected to be more diverse than those in semi-urbanized zones. This study aims to characterize the baseline diversity of bacterial communities in wetlands of both the undisturbed and semi-urbanized zones within the CBR. It aims to initially assess temporal shifts in bacterial community structures, discerning annual trends and variations due to environmental dynamics. Also, our results are intended to serve as a benchmark for future investigations into the impacts of the Mayan Train.
Results
Physicochemical parameters of the sampled sites
In this work, we sampled three waterholes (aguadas). Samples were collected from two sites, Ag-UD1 and Ag-UD2, within the undisturbed zone of the CBR. A third site, Ag-SU3, was located in the semi-urbanized zone (Fig. 1). Sampling occurred in September during the rainy season, which had already flooded the sites across three different years: 2017, 2018, and 2019. Notably, only one of these sites, Ag-SU3, was sampled both in September and in June, the latter being a period preceding the rainy season when the site remains dry. In September 2019, the sediment samples collected for this study were analyzed to determine physicochemical factors, including pH, total nitrogen, phosphorus, and organic matter. This analysis aimed to obtain metadata to complement the metagenomic studies of bacterial communities.
The pH values in the three waterholes ranged from 6.1 to 6.7: Ag-UD1 recorded a pH of 6.2, Ag-UD2 was 6.7, and Ag-SU3 showed 6.1. Given the calcareous nature of the Peninsula’s soils, characterized by a high CaCO3 content, the pH is slightly higher than that typically found in other tropical forest soils [29]. Total nitrogen levels were determined to be 0.36%, 0.22%, and 0.29% for Ag-UD1, Ag-UD2, and Ag-SU3, respectively. Phosphorus in sediment samples was evaluated using the Olsen method; the Ag-UD1 site exhibited 29.6 mg Kg− 1, which is approximately twice the level in Ag-UD2 wetland sediments (13.5 mg Kg− 1) and 7.4-fold times more than in the Ag-SU3 site (4.0 mg Kg− 1). The percentage of organic matter, evaluated through organic carbon content using the Walkley-Black method, exhibited varying behavior among sites. Ag-SU3 had the highest rate, at 24.20%, while Ag-UD2 recorded the lowest, at 7.8%. The Ag-UD1 site presented an intermediate percentage of 12%.

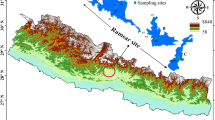
Sampling location information. A Map of the Mexican Republic, highlighting the Calakmul Biosphere Reserve (CBR). B Detailed map of CBR, delineating core and buffer zones and indicating the locations of the three sample sites, named Ag-UD1 (blue), Ag-UD2 (cyan), and Ag-SU3 (red), as well as part of section seven of the Maya Train route (blue line) C-E Photographs of the three sites, Ag-UD1, Ag-UD2 and Ag-SU3, respectively, sampled during a flood event
Alpha and beta diversity
Bacterial communities are primarily composed of populations that can be grouped genetically or ecologically, suggesting that they exhibit characteristics typical of species. Microorganisms remain a significant source of diversity and new natural products [32]. We initially explored bacterial diversity through metabarcoding (sequencing a region of the 16S rRNA gene) and shotgun metagenomics. We assess diversity in these wetland sediments within (alpha diversity) and between sites (beta diversity) by calculating alpha and beta indices.
To understand the structure of bacterial communities we computed richness, Shannon, and Simpson indices. The richness index measures the number of species present in a sample, providing insight into species abundance. The Shannon index considers species richness and evenness, offering a more comprehensive understanding of community diversity. Meanwhile, the Simpson index focuses on species dominance, reflecting the probability that two individuals randomly selected from the sample belong to the same species. The three diversity indices indicate higher values at the AG-SU3 site compared to both the Ag-UD1 and Ag-UD2 sites (Fig. 2A). This suggests more variability in the samples collected from the AG-SU3 site compared to those obtained from the Ag-UD1 and Ag-UD2 areas. This outcome contrasts with findings from other studies, which have shown that sites impacted by human activity typically exhibit lower species diversity and richness compared to pristine locations [13, 14, 19, 27, 28]. The abundance tables used to calculate these indices were glommed to the genus level. Acknowledging that the operational unit impacts the perspective of diversity, the same three indices were calculated at default OTU level and by Phylum, and in both cases, higher records were registered at Ag-SU3 (Fig S1).

Alpha and Beta diversity in aguadas of Calakmul at three sampling locations: Ag-UD1, Ag-UD2, and Ag-SU3. Phyloseq object was processed by rarefaction and agglomeration to Genus level. A) Alpha diversity indices. B) Beta diversity employing the PCoA method with Weighted UniFrac distance. The sample marked with a red box and asterisk indicates data from June when the Ag-SU3 site was dry. C) Statistical analysis of diversity, including Alpha diversity (ANOVA with Tukey’s post-hoc test) and Beta diversity (PERMANOVA with multilevel pairwise comparison)
In beta diversity analysis, the samples front the three sites clustered according to their geographic locations (Fig. 2B). The arrangement of the data suggests that all three sites cluster independently, each displaying a markedly different microorganism community. A PERMANOVA statistical analysis with multilevel pairwise comparison found significant differences between Ag-UD1 vs. Ag-SU3 and Ag-UD2 vs. Ag-SU3 (p-value of 0.038 and 0.042, respectively). This result aligns with the expectation that undisturbed sites’ diversities differed from the diversity measure of the semi-urbanized zone. No significant differences between undisturbed sites were found. Similar to the approach for alpha diversity, the analyses were performed with data agglomerated at the genus level. Results from not agglomerated data and at the phylum level show the same tendency, Fig. S1B. Beta diversity analyses with shotgun sequencing data are separated by site, Fig. S2.
Lower diversity was found statistically significant in Shannon and Simpson indices in the samples collected from the Ag-UD1 and Ag-UD2 sites compared to the measures from the Ag-SU3 area. The ANOVA and subsequent Tukey’s post hoc testing, with a significance threshold set at p < 0.05, delineated these differences. Specifically, for Shannon diversity, comparisons yielded p-values of 0.006 for Ag-SU3/Ag-UD1 and 0.039 for Ag-SU3/Ag-UD2. Similarly, for Simpson diversity, significant differences were observed with p-values of 0.002 for Ag-SU3/Ag-UD1 and 0.015 for Ag-SU3/Ag-UD2 (Fig. 2C). This indicates that the Tukey test effectively identified specific pairwise differences among the sites following the initial ANOVA analysis (Fig. 2C). Statistical tests show no significant differences between the mean alpha indices of Ag-UD1 and Ag-UD2.
Physicochemical analyses carried out in 2019 at the three sites showed that the organic matter content in Ag-SU3 is more than double that in the other sites. In contrast, the phosphorus concentration in Ag-SU3 is 7.4 and 3.4 times lower than in Ag-UD1 and Ag-UD2, respectively. These observations suggest two possibilities: firstly, the increase in diversity at Ag-SU3 might be attributed to microorganisms associated with anthropogenic activity rather than being endemic to the area. Secondly, the higher diversity observed at Ag-SU3 could be due to its higher organic matter concentration and lower phosphorus levels. Previous studies have reported that soils rich in organic matter support a greater proliferation of microorganisms. High phosphorus levels can adversely affect soil diversity due to its potential toxicity to microorganisms present in the soil [30,31,32].
Taxonomic distribution of bacterial communities in the Calakmul wetlands from 16S rRNA and shotgun sequencing
To analyze the taxonomic distribution of the samples by site and year, we determined the abundance and prevalence of microorganism groups present in the samples. The massive sequencing results of the 16S rRNA gene, employing data with genus-level taxonomic assignment (205 OTUs), reveal changes in the relative abundance patterns of phyla according to years and sites (Fig. 3A). The classification of microbial taxa at the phylum level identified 15 bacterial phyla with a relative abundance exceeding 1%. Actinobacteria, Acidobacteria, and Proteobacteria emerged as the most dominant and widely distributed phyla across the samples and years. It is also essential to notice that certain phyla are characteristic of a specific site and year; for example, the phylum Gemmatimonadota was exclusively present in the Ag-UD2 site in 2017, and Bacteroidetes were only present in 2019 at the Ag-UD2 site, while Halobacteriota was observed at the Ag-UD1 site in 2018 and Ag-SU3 in 2019 (Fig. 3A).
Four phyla exhibited more dynamic patterns of prevalence and absence across the sampled sites and years (Fig. 3B). Chloroflexi was exclusively present in undisturbed sites in 2018 and not found at any site in 2019. This phylum is commonly associated with anaerobic environments and contaminated locations [33]. It plays a crucial role in forming a specific type of biofilm called “flakes’’ and degrading carbon compounds. Another distinct phylum, Firmicutes, was consistently observed at the Ag-UD1 site over three years. It was completely absent at the Ag-UD2 site but presented at the semi-urbanized Ag-SU3 site in 2017 and 2019. Firmicutes are ubiquitous in soils and various environments, such as the intestinal microbiota [34]. Euryarchaeota, part of the Archaea domain, was absent in 2019 but observed at the Ag-UD2 and Ag-SU3 sites in 2017 and the Ag-UD1 site in 2018. Conversely, the Myxococcota phylum is distributed across all three sites and throughout the three years, with a notable abundance at the Ag-UD2 site compared to the others. These observations consider a grouping criterion of a relative abundance of 1% or more.
The predominant phyla and genera (in parentheses) present in all samples of 16s rRNA sequences were Actinobacteria (Solirubrobacter, Nocardioides, Gaiella, Streptomyces, Conexibacter), Proteobacteria (Rhodoplanes, Acidibacter, Sphingomonas, Reyranella, Pedomicrobium, Defluviicoccus, Rhodomicrobium), Myxococcota (Haliangium), Firmicutes (Bacillus), and Acidobacteriota (RB41). On the other hand, within the domain Archaea, the phylum Euryarchaeota was the most represented (Fig. 3B and C).
Furthermore, we observed by genus those that exhibited greater and lesser relative abundance at each of the Ag-UD1, Ag-UD2, and Ag-SU3 sites (Fig. 3B). Overrepresented in Ag-SU3 were: Proteobacteria (Acidibacter, Reyranella, Pedomicrobium, Rhodomicrobium), Actinobacteria (Streptomyces,Conexibacter, and Mycobacterium). In contrast, underrepresented compared to Ag-UD1 and Ag-UD2 were Proteobacteria (Sphingomonas, Defluvicoccus) and Actinobacteria (Nocardioides). In Ag-UD1, this analysis revealed that the genera with the highest relative abundance compared to Ag-UD2 and Ag-SU3 were Proteobacteria (Sphingomonas, Defluvicoccus), Actinobacteria (Solirubrobacter, Nocardioides, Gaiella), Acidobacteria: (RB41). Proteobacteria (Rhodoplanes, Acidibacter, Reyranella, Rhodomicrobium) were the least abundant. In Ag-UD2, the genera most abundant in comparison to the other two sites were Proteobacteria) Rhodoplanes, Acidibacter), Myxococcota (Haliangium), with the least abundant being Proteobacteria (Pedomicrobium), Actinobacteria (Solirubrobacter, and Mycobacterium).
A relative abundance plot (Fig. S3) was generated using rarefaction data from 35 Operational Taxonomical Units (OTU), obtained after Phylum-level agglomeration (using phyloseq::tax_glom) of 3753 OTU, from which previously empty data had been removed. Despite not being assigned at the genus level, unique phyla specific to each site were identified. For Ag-UD1, Entothonellaeota was present in 2018 and 2019, and Halobacteriota in 2018, while NB1-j was exclusive to Ag-UD2 in 2017. These three phyla are unique to the aguadas in the reserve’s undisturbed zone. The phylum Verrucomicrobiota was absent from all sites in 2017 but appeared in 2018 and 2019; it was only found in Ag-UD1.
The taxonomic distribution was also assessed based on shotgun metagenomic sequencing, focusing on reads assigned at the genus and phylum levels (Fig. 4A, S4 & S5). Kraken2 [35] and Pavian [36] analyses revealed that the most abundant and prevalent group across all three wetlands was the copiotrophic phylum Actinobacteria. This phylum includes the Streptomyces genus, the most prolific producer of secondary metabolites such as antibiotics and other natural products vital to humans. Other abundant genera within Actinobacteria were Micromonospora and the mycolic acid-containing Mycobacterium and Mycolicibacterium. Proteobacteria emerged as the second most prevalent phylum in the three sites, with the most abundant genera being the N2-fixing microsymbionts Bradyrhizobium and the cosmopolitan and diverse Pseudomonas, previously reported as dominant in North American forest soils [37]. Interestingly, the genomes of Pseudolabrys, another soil proteobacteria [38], were found to be the closest matches in the Genome Taxonomy Database (GTDB) [39] for bins Ag-UD1.001 and Ag-SU3.001, which recovered from Ag-UD1 and Ag-SU3 sites respectively. From the reconstructions using MaxBin2 [40] (as detailed in Table S1), it was observed that only two bins from each site had genome completeness exceeding 50%: Ag-UD1.001 at 55.1% and Ag-SU3.001 at 78.5%. Additionally, bin quality was assessed using CheckM [41], revealing completeness scores of 70.5% and 91.07% for Ag-UD1.001 and Ag-SU3.001, respectively (as shown in Table S2). However, both of these scores fall below the recommended threshold of 95% for considering genomes assembled from metagenomes. Both 16 S rRNA and shotgun sequencing corroborate previous findings that while most soil bacterial phylotypes are rare, relatively few are abundant, and many are found across a spectrum of soils [42].
The genus-level comparison of relative abundances across all three sites, using all reads assigned up to the genus level from shotgun metabarcoding sequencing (Fig. S4), indicates distinct patterns. In Ag-UD1, the most abundant genus compared to Ag-UD2 and Ag-SU3 were Proteobacteria (Pseudomonas), and the least abundant Proteobacteria (Bradyrhyzobium and Rhodoplanes), Actinobacteria (Streptomyces, Mycobacterium, Mycolibacterium, and Nocardioides). For Ag-UD2, the most abundant genera compared to the other two sites were Proteobacteria (Bradyryzobium, Rhodoplanes, and Rhizobium), Actinobacteria (Streptomyces, Conezybacter, Nocardioides, Rhosococcus, and Micromonospora) while the least abundant Proteobacteria (Pseudomonas, Burkholderia, and Rhodopseudomonas), Actinobacteria (Micromonospora). In Ag-SU3, the most abundant genera compared to Ag-UD1 and Ag-UD2 were Proteobacteria (Rhodopseudomonas), Actinobacteria (Micromonospora and Mycobacterium), with the least abundant being Actinobacteria (Amycolatopsis).

Composition of bacterial communities in aguadas of Calakmul at three sampling locations: Ag-UD1, Ag-UD2, and Ag-SU3. Phyloseq object was processed by rarefaction and agglomeration to Genus level. A) Relative abundance of phyla with greater than 1% presence. B) Relative abundance of the nine principal phylum-level taxa at each site for the year sampled. C) Heat map illustrating the percentages of the top 10 taxa at both phylum and genus levels
To identify each site’s characteristic bacterial genera, we created heatmaps using both amplicon and shotgun sequence data. According to 16S rRNA sequencing, the distinctive Ag-UD1 genera consistent across all sampling years are Solirubrobacter, Gaiella, Sphingomonas, Defluvicoccus, and RB41. In contrast, Ag-UD2 exhibits fewer unique genera, with Haliangium being the most significant. For Ag-SU3, the genera Streptomyces and Acidibacter differentiate it from the other sites (Fig. 5). Regarding shotgun metagenome sequencing, it is not feasible to identify a specific genus that uniquely characterizes any site. However, it is essential to note that Streptomyces and Bradyrhizobium are significantly more represented across all three sites than other genera (Fig. S4C).

Phylum-Genus-Level heatmap of communities from aguadas in Calakmul at three sampling locations: Ag-UD1, Ag-UD2, and Ag-SU3. Phyloseq object was processed by rarefaction and agglomeration to Genus level. This heat map depicts the relative abundance at the phylum and genus taxonomic levels. This analysis includes clusterization, considering the various sites and years sampled
Functional potential in the Calakmul wetlands
In an initial effort to understand the functional capabilities of these microbes, we performed a functional analysis on the environmental DNA sequenced via shotgun metagenomics. It was conducted utilizing four tools: MEBS [43], DiTinG [44], MG-RAST [45], and SUPER-FOCUS [46].
The analysis with DiTinG revealed similarities between the sites in terms of carbohydrate metabolism (Fig. S6B), although some differences were observed in the nitrogen and sulfur cycles (Fig. 5C). In the nitrogen cycle, a higher relative abundance of genes related to denitrification was observed at the Ag-SU3 site. In the sulfur cycle, more metabolic pathways were found at the Ag-SU3 site, including sulfite oxidation, thiosulfate oxidation, and sulfite reduction. The sulfate reduction and sulfite oxidation pathways were mainly present at the Ag-UD2 site, while the sulfuric acid oxidation pathway was only observed at the Ag-UD1 site.
The MG-RAST analysis indicated that both the relative abundance of taxa and functional potential is stable across the three aguadas (Fig. S6A), with the Carbohydrates subsystem primarily prevailing, a result similar to that shown by DiTinG. SUPER-FOCUS allowed us to observe functions at different subsystem levels, but similar to what was observed with previous tools, the behavior between the sites was similar, with a tendency to present greater abundance at the Ag-SU3 site (Fig. S6C). This observation aligns with previous studies, which have shown that functional redundancy is more prevalent than taxonomic redundancy across climate zones [47]. It is also known that various climate and geographical gradients can induce changes at the physicochemical and taxonomical level in the soil, yet microbial functional potentials tend to remain relatively unaffected [47, 48].
The Multigenomic Entropy Based Score (MEBS) analysis revealed numerous pathways involved in nitrogen (N) and sulfur (S) cycles, as well as some related to carbon (C) and iron (Fe). Regarding nitrogen (N) pathways (Fig. 5B), nitrate reduction and ammonia oxidation emerged as the most prevalent across all three sites. Notably, a significant abundance of Alphaproteobacteria, particularly Rizhobiales, known for their symbiotic nitrogen fixation, suggests their potential role in these functions. Additionally, dissimilatory nitrate reduction (nitrite-ammonia) and denitrification (nitrate-nitrite) pathways, predominantly expressed in Proteobacteria, including Alpha, Beta, and Gamma proteobacteria classes, along with Bacilli class, contribute to the nitrogen cycle, with no significant taxonomic differences observed between sites. The ecosystem’s capability to complete the nitrogen cycle highlights the crucial role of wetlands, which can remove up to 75% of nitrogen (N), as indicated in other studies.
Furthermore, sulfur (S) cycle pathways (Fig. 5B) are well represented, particularly those related to sulfite oxidation and dimethylsulfoniopropionate (DMSP) oxidation. Bacteria from the Delta and Gamma class of proteobacteria, along with some groups of Cyanobacteriota phylum present in all samples, are primarily responsible for these functions, especially DMSP oxidation. Notably, the presence of DMSP in Ag-UD1 suggests its importance as a carbon and energy source, with certain Cyanobacteriota groups potentially carrying out this function, as observed in other sites.
In contrast, the representation of carbon (C) cycle pathways varies significantly between sites (Fig. 5B), indicating potential bias in bacterial composition influenced by alternative energy sources. This underscores the complexity of wetland ecosystems and their susceptibility to environmental factors impacting microbial communities and biogeochemical cycling.

Shotgun Sequencing Analysis of libraries from the three sites sampled in 2019. A Sankey diagram of the Ag-SU3 site, derived from the metagenomic classification analysis using Pavian. B Heatmap from MEBS analysis showing the main pathways of S, N, Fe, and C biogeochemical cycles. C Sketch maps regarding nitrogen and sulfur cycles. The pie chart indicates the relative abundance of each pathway in each metagenomic sample. The size of the pie chart represents the total relative abundance of each pathway
Discussion
Wetlands are critical reservoirs of biodiversity, with a single gram of soil potentially harboring up to 1010 unique bacterial ‘‘species’’ [14], most of which remain uncharacterized. This study aimed to explore the microbial communities of the Calakmul wetlands, laying the foundation for potential monitoring of microdiversity adaptation mechanisms in wetlands both before and after significant changes, such as the mega infrastructure projects planned for the area.
Our results indicate that Actinobacteriota (Ag-UD1 32.7%, Ag-UD2 30%, Ag-SU3 30.2%) Proteobacteria (Ag-UD1 20.7%, Ag-UD2 23.7%, Ag-SU3 21.8%), Acidobacteriota (Ag-UD1 11%, Ag-UD2 10.3%, Ag-SU3 9.5%) and Myxococcota (Ag-UD1 5.7%, Ag-UD2 9.7%, Ag-SU3 6.2%) are the most abundant phyla across the three Calakmul sites studied. These phyla have previously been reported in higher abundance in studies focused on carbon degradation, specifically involving significant activity of glycoside hydrolases [49]. The metagenomic analysis, grouping by phylum, allowed us to compare our results with previous studies conducted in various ecosystems, ranging from different mangroves to arid and Antarctic soils, as well as a lagoon in a location geographically close to the CBR (see Table S3). Previous studies have reported that Actinobacteria and Acidobacteria are prevalent in nutrient-rich mangrove sediments [50, 51], which could be similar to the conditions in the Calakmul aguadas. When compared with similar studies in different ecosystems, our results show slight deviations from previous reports in the mangrove forests. In those environments, the microbiota composition is predominantly characterized by Proteobacteria, Bacteroidetes, Chloroflexi, and Cyanobacteria [11]. In contrast, in soils from drastically different climates, such as arid environments, Actinobacteria, Acidobacteria, Proteobacteria, and Bacteroidetes are the most predominant phyla [52], and in Antarctic soils, the most abundant phyla reported are Actinobacteriota, followed by Acidobacteriota, Chloroflexi, and Proteobacteria [53].
We also hypothesize that microbial communities are predominantly influenced by abiotic factors, such as pH, as previously described [54], in addition to the inter-kingdom biotic factors. Prior studies at the continental scale have explored how changes in soil microbial diversity are primarily driven by pH [55, 56]. In this study, we measured the pH for the three sampled sites in 2019, finding values of 6.2 in Ag-UD1, 6.7 in Ag-UD2, and 6.1 in Ag-SU3. While these pH levels do not appear to differ dramatically from one another in a way that significantly impacts community structure, specific phyla, such as Acidobacteria, were more abundant in Ag-UD1, which has a lower pH compared to Ag-SU3. This observation aligns with previous studies reporting the dominance of Acidobacteria and Alphaproteobacteria in acidic soil environments [57, 58].
Initially, we hypothesized that the structure of bacterial communities would differ between water areas in the undisturbed zone and the semi-urbanized zone over time. To investigate the differences in disturbance among the sampled zones as well as temporal variation, we conducted sampling over three different years, specifically in September 2017, 2018, and 2019. Additionally, we collected a sample for one of the sites in June 2019. Variation across different years and sites was observed in several phyla, including Chloroflexi, Bacteroidetes, Firmicutes, and Euryarcheaota.
Chloroflexi, a group of photosynthetic bacteria, is not well-characterized but is environmentally widespread in various types of subseafloor sediments. This phylum may represent one of Earth’s most abundant microbial components, potentially playing a crucial role in global sediment carbon cycling [33]. In environments more similar to those studied here, such as mangrove wetlands, Chloroflexi has been associated with sites disturbed by anthropogenic activities or contaminated ones [51]. Our study detected Chloroflexi in all three sampled sites, with a notably higher presence in one of the conserved sites, Ag-UD2, located in the undisturbed zone where anthropogenic activity and contamination are presumed to be minimal. In addition to its ecological significance, Chloroflexi has been used in water treatment plants for nitrogen and phosphorus reduction [33]. Consequently, it could be related to the cycle of these elements within the Calakmul aguadas.
Bacteroidetes are commonly associated with substrates rich in organic carbon in aquatic environments. Active members of this phylum often act as initial metabolizers of labile carbon inputs [59, 60]. Previous studies also indicate their increased abundance in soils with higher pH. Other studies have found that Chloroflexi and Bacteroidetes are more abundant in unprotected mangrove sediments [51]. As previously mentioned, both the Ag-UD1 and Ag-UD2 sites are located in the most protected area of the reserve. Nevertheless, detecting these two phyla in 2018 and 2019 at the Ag-UD2 site may suggest some disturbance.
Firmicutes, particularly the class Bacilli, is one of the most extensively studied phyla due to its potential in promoting sustainable agriculture. This is attributed to its application as a biofertilizer and biological control [34]. This phylum exhibited almost constant abundance across the sites and through the years. It is well-known for its ability to form specialized dormant cellular structures known as endospores. This ability to sporulate enables them to withstand environmental conditions in sediments and soils. However, some studies suggest that the absence or low detection of Firmicutes in amplicon sequencing approaches may be attributed to the low rate of lysis of endospores during DNA extraction procedures. This implies that vegetative Firmicutes populations may only constitute a small proportion of the total community of vegetative microbial cells [61, 62]. Conversely, the phylum Euryarchaeota was notably overrepresented in the Ag-SU3 site during 2017 but not in the others. This phylum encompasses five groups: extreme halophiles, methanogens, hyperthermophiles, extreme acidophiles, and planktonic archaea. Considering that the samples were taken from sediments, it is highly likely that the detected Euryarchaeota are methanogenic. Methanogens, a part of the Euryarchaeota group, produce methane as a metabolic byproduct, rendering them ecologically significant in the degradation of organic matter and the carbon cycle. Generally, it is understood that the decomposition rates of organic matter in wetlands are low due to anaerobic conditions, which makes wetlands effective carbon reservoirs [63]. If the Euryarchaeota present in Ag-SU3 are indeed methanogenic, this site might be less efficient at accumulating carbon than the undisturbed zone. Prior research in pasture systems has identified Actinobacteria, Chloroflexi, Firmicutes, and Bacteroidetes as the phyla with the highest differential abundance [64]. Verrucomicrobiota is frequently detected in metagenomic studies of forest and jungle soils [65]. In our analysis, the most significant variation regarding this phylum was related to the years of sampling. It was not observed in any of the sites in 2017, yet it was present in all three sites in 2018. However, by 2019, it was only found on the Ag-UD1 site.
Entothonellaeota, Halobacteriota, and NB1-j are phyla with site-specific abundance, typically inhabiting specialized environments. These include polluted areas rich in copper and chromium or anaerobic settings with high methanogenic activity [66,67,68]. Associating these phyla with specific physicochemical characteristics of the waterholes where they are found could yield intriguing insights.
Our observations reveal that while Ag-UD1, one of the sites in the undisturbed zone of the reserve, has the lowest bacterial diversity, the second site within this protected zone, Ag-UD2, appears to be more similar in terms of alpha diversity to the Ag-SU3 site, which is located in the semi-urbanized zone. These observations align with a previous study comparing urban forest soils and national parks, which found that the alpha diversity of bacteria and fungi increased with higher levels of anthropogenic disturbance [69]. The diversity in the Calakmul wetlands studied over the 2017–2019 period appears stable, potentially due to the adequate protection of macrodiversity in the area, which is reflected in the conservation of microdiversity. The current conservation status of CBR is likely a consequence of the region’s low population density, attributable to limited water availability for human consumption and scarce arable land. Variations between sites could be influenced by differing levels of sunlight access, which may vary depending on the tree canopy at each site. Previous studies have demonstrated how the soil microbiome varies with changes in vegetation coverage [70,71,72].
The shotgun sequencing of metagenomes from the studied sites revealed not only taxonomic redundancy, irrespective of being in the undisturbed or semi-urbanized zones, but also functional redundancy. This finding is consistent with previous reports [47, 48, 73]. These studies also suggest that although the microbial composition is sensitive to disturbances, the community may exhibit resilience and rapidly revert to its pre-disturbance state. This resilience could be attributed to the fast growth rates of bacteria and the metabolic flexibility of prokaryotes in general. Besides cross-site analysis, we also compare our shotgun sequencing results with a metagenomic study carried out in a pond at Cuatro Ciénegas [74]. This oligotrophic, phosphorus-deficient environment has been extensively studied and presents a stark contrast to the Calakmul aguadas. Despite the distinct environmental conditions, we discovered that the functional categories in both studies were very similar, with no notable differences at a global level in the communities on a functional basis.
Analysis using the shotgun sequencing strategy revealed that Proteobacteria and Actinobacteria are the predominant phyla present, consistent with findings from other wetland studies. These bacterial groups are essential for performing numerous functions critical to the biogeochemical cycles within wetland ecosystems.
The Multigenomic Entropy Based Score (MEBS) analysis revealed nitrogen pathways, notably nitrate reduction and ammonia oxidation, were prevalent across all three sites. Alphaproteobacteria, particularly the Rizhobiales, emerged as potential executors of these functions, with negligible taxonomic disparities observed between sites. Additionally, dissimilatory nitrate reduction and denitrification pathways, expressed in Proteobacteria and Bacilli classes, contributed to the nitrogen cycle.
Furthermore, sulfur-related pathways, including sulfite oxidation and dimethylsulfoniopropionate (DMSP) oxidation, were well represented, predominantly by Delta and Gamma class proteobacteria and the Cyanobacteriota phylum. The presence of DMSP in Ag-UD1 suggests its significance as a carbon and energy source, with Cyanobacteriota potentially playing a pivotal role in this function.
Nonetheless, carbon cycle pathways exhibited substantial variation between sites, suggesting a potential bias in bacterial composition influenced by alternative energy sources. In summary, these findings underscore the critical role of wetlands in biogeochemical cycling and nutrient removal processes.
As previously observed, shotgun and amplicon sequencing methods reveal significantly different community structures for nearly all the bacterial communities collected from various locations and sites [75]. In concordance with other studies, we observed that 16S rRNA metabarcoding profiles bacterial communities in greater detail compared to shotgun metagenomics. Consequently, these methods should not be directly compared but can complement each other to address different aspects of the microbial communities under study. However, it is essential to expand the number of studies focussing on the dynamics of microbial communities and metabolic processes in Calakmul aguadas. Characterizing the microbiota in its various microenvironments, identifying its metabolic capacities, and analyzing its potential associations with the diversity of flora and soil types present in this tropical ecosystem are highly desirable objectives.
Conclusions
The composition of microbial communities is often overlooked in ecosystem studies. To the best of our knowledge, this is the first instance of exploratory monitoring of bacterial diversity in wetlands, locally known as aguadas, within Calakmul Biosphere Reserve - the second most important tropical forest after the Amazon - using NExt-Generation Sequencing (NGS). Studies of bacterial communities through metabarcoding and metagenomics provide an essential foundation for initiating high-resolution, random sequencing of large metagenomic fragments. This approach is critical to gaining deeper insights into how populations organize, interact, and evolve within communities and discovering new metabolisms in pristine sites of this vital lung of Mexico and the Yucatan Peninsula. This report presents a survey of soil microbial communities across three sites in undisturbed and semi-urbanized zones. Changes in the soil microbial community structure, diversity, and function were anticipated due to comparing undisturbed and semi-urbanized zones within the Calakmul Biosphere Reserve. Overall, the composition of the microbial communities is strikingly similar across the three sites. While anthropogenic activities have led to a noticeable reduction in plant and animal presence at site Ag-SU3, the impact on the soil microbiota also reflects these changes, increasing microbial diversity, as far as our findings indicate. Our study also presents results regarding spatial changes in bacterial communities. Shotgun sequencing revealed Proteobacteria and Actinobacteria as dominant phyla, essential for performing numerous functions involved in the biogeochemical cycles within wetland ecosystems. Function annotation analysis highlighted nitrogen, sulfur, carbon, and iron cycles, emphasizing wetlands’ importance in nutrient cycling and environmental sustainability. The shotgun data did not permit the assembly of MAGs due to low coverage, suggesting that more extensive sequencing data are required to adequately characterize the functional content of the soil in the aguadas of Calakmul. This study represents an important point of comparison for our further sampling in the same areas once the newly constructed Maya Train is operational, enabling us to assess the changes in microbial communities in response to a project of this magnitude.
Methods
Samples recollection and sediment analysis
The authorization for sampling in the studied areas was regulated by the local office of the National Commission of Natural Protected Areas (CONANP) in Xpujil, Calakmul, which issued a sampling permit for the team led by Dr. Karina Verdel Aranda. Three sites were sampled: two in the undisturbed zone of the Biosphere Reserve, designated as Ag-UD1 and Ag-UD2, and one in the semi-urbanized zone near agricultural areas, referred to as Ag-SU3. The locations of these sites are shown in Fig. 1. Calakmul experiences a seasonal climate.
Ten samples underwent 16 S rRNA sequencing. Of these, nine were collected in September across the years 2017 to 2019, and one sample from the Ag-SU3 site was taken in June 2019. This sampling strategy was employed to investigate how these communities vary over three years. The water level in these reservoirs fluctuates significantly, ranging from 1.5 to 3 m during the rainy season (July-November) to complete dryness in the period preceding the rainy season (February-June).
Approximately 500 g of surface sediments were randomly collected in triplicate. This involved using a tube inserted to a depth of 10 cm, followed by the sediment extraction with a plunger. The collection instruments were sterilized with 70% ethanol. The samples were then placed in sterile plastic bags, kept on ice, and transported back to the laboratory. The triplicate samples were combined and utilized for all experiments in this study. They were stored at − 80 °C until DNA extraction. Soil pH, organic matter content, total nitrogen, and available phosphorus concentrations were measured at the Soil Analysis Laboratory of the Postgraduate College campus Campeche using standard methods [76, 77].
DNA extraction and Illumina sequencing
Genomic DNA was extracted from 1 g of the sediment samples using the PowerSoil DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA, USA). The sequencing service was performed by Zymo Research Microbiomics Services (Zymo Research, Irvine, CA). Bacterial 16 S ribosomal RNA gene-targeted sequencing was performed using the Quick-16S™ NGS Library Prep Kit (Zymo Research, Irvine, CA). The bacterial 16S primers, custom-designed by Zymo Research, amplified the V3-V4 region of the 16S rRNA gene. The final library was sequenced on theIllumina® MiSeq™ platform using a v3 reagent kit (600 cycles), with more than 10% PhiX spike-in. Library preparation and shotgun sequencing were carried out at Langebio, Cinvestav (Irapuato, Mexico) using the NextSeq mid-output 2 × 150 paired-end read format for all samples.
Bioinformatic analyses
The scripts for these analyses are available in the Github repository: https://davidalberto.github.io/Calakmul/. Below, we provide a summary of the steps that were followed.
Amplicon quality control and taxonomic assignation
The DADA2 package, version 1.26.1 [78], is designed to infer accurate amplicon sequence variants from high-throughput amplicon sequencing data [78]. We used demultiplexed fastq files as input for DADA2 to remove substitution and chimera errors, subsequently producing sequence variants and their abundances in the samples. For the sequence’s taxonomic classification, we employed the naive Bayesian classifier RDP and species-level assignment of 16S rRNA gene fragments by exact match. Reads were organized by separating forward (R1) and reverse (R2) copies. Quality control included filtering and trimming the raw readings to keep a quality > 30. We trim the forward reads to 250 bp, and the reverse reads to 230 bp. We then analyzed amplicon sequence variants (ASVs) with the resulting reads, where DADA2 inferred unique sequences. Subsequently, the new R1 and R2 sequences were merged to yield complete, noise-free sequences. A table of ASVs was created, from which chimeras resulting from substitution or insertion/deletion errors (Indels) were removed. Finally, we assigned taxonomy to each ASV, using SILVA version 138 as the reference database for sequences with known taxonomy, and generated exact match assignments.
Amplicon phylogenetic assignment
The phylogenetic analysis was performed using the Phangorn package, version 2.10.0: Phylogenetic Reconstruction and Analysis. This package enables the estimation of phylogenetic trees and networks using methods such as Maximum Likelihood, Maximum Parsimony, distance, and Hadamard conjugation. It also provides tools for tree comparison, model selection, and the visualization of phylogenetic networks [79]. This phylogenetic reconstruction is essential for calculating Beta diversity with UniFrac distances.
Shotgun metagenomic quality control, binning and functional annotation
The sequence quality control (QC) analysis for shotgun metagenomic data was conducted using the FastQC program [80], which facilitates the identification of low-quality regions and sequences and the presence of adapters. The FastQ files underwent trimming and adapter removal using Trimmomatic [81]. The quality clipping threshold was set at a Q score of 30, corresponding to an error rate of 0.001. After quality control, we recovered the following reads: Ag-UD1:7.6 × 10^6, Ag-UD2:6.8 × 10^6, and Ag-SU3 6.5 × 10^0.6. Then, contigs were assembled with MegaHit and classified in bins with MaxBin2 [40] version 2.2.7. The maximum bin completeness was 78.5%, Supplementary Table S1. CheckM [41], Table S2 also evaluated Bin completeness and contamination. Regardless of their contamination percentage, bins with completeness above 70% were compared against genomes in the Genome Taxonomy Database (GTDB) with its tool kit GTDBtk [39]. Shotgun data are available at mgRAST with IDs Ag1:mgm4972426.3, Ag2:mgm4972425.3, and Ag3 mgm4972427. 16 S rRNA sequencing data are available in the NIH Sequence Read Archive (SRA) repository, BioProject ID: PRJNA983657 (Release date: 2024-06-14). Four tools, namely MG-RAST [45], DiTing [44], SUPER-FOCUS [46], and MEBS [43], were employed for the functional annotation of shotgun contigs.
Shotgun metagenomic taxonomic assignment
Kraken2 was used for taxonomic assignment of prokaryotes reads [35]. This program enhances sensitivity and speed compared to other classifiers by using innovative algorithms that employ exact alignments of k-mers. Abundance tables from DADA2 [78] for the 16S rRNA gene and Kraken2 [35] from shotgun metagenomes were analyzed and visualized with Phyloseq [82] 1.44.0 and Pavian [36]. Phyloseq objects were created, including the OTUs table, metadata table, taxonomic table, and the phylogenetic tree for the 16 S rRNA and Shotgun data.
Analysis of alpha and beta diversity
To standardize for differences in library sizes, we utilized rarefaction with the rarefy_even_depth function from the Phyloseq package, setting the sample size to the number of reads from the smallest library. For the 16 S rRNA data, the smallest library contained 10 thousand reads, while the largest contained 39 thousand. In the case of the shotgun sequencing data, the smallest library contained 1.61 million reads, while the largest contained 1.84 million. Alpha diversity estimation was conducted at various taxonomic levels, including OTU defined by the default value and using Phylum and Genus agglomeration as taxonomic units.
Agglomeration by Phylum facilitates comparison with other studies, Table S3. It provides a first approximation to diversity, allowing a general view of the OTU distribution, even if they are not assigned to lower levels. At the same time, while genus-level classification may be less precise in a taxonomical sense than phylum-level, it offers a more refined diversity resolution. Genus level allows for differentiating between closely related microbial genera within a Phylum. Such an approach can be instrumental in identifying the predominant genera within a microbial community and suggesting relevant genera for microbiome analysis. Two phyloseq objects were generated from the tax_glom function to store the Phylum and Genus agglomeration tables. We measure alpha diversity by calculating three metrics using the plot_richness function. These metrics include Observed (the raw count of taxa, or richness), Simpson (reflecting the probability that two randomly selected bacterial species in a community are different), and Fisher (richness metric considering abundance).
Beta diversity represents the ratio of regional to local species diversity. We measure beta indices using the ordination function of Phyloseq 1.42.0. The distance matrix used for 16 S rRNA metabarcoding data was UniFrac, as it considers the phylogeny generated with Phangorn. The resulting distance matrix is used in conjunction with some method of ordination or multidimensional scaling. Again, with the ordination function of Phyloseq, we used the Principal Coordinates Analysis (PCoA) method.
The existence of significant differences in alpha and beta diversity was assessed with R packages: stats v4.3.1, vegan v2.6-4, phyloseq v1.44.0, and pairwiseAdonis v0.4. To establish differences between sites’ alpha diversity indexes, an Analysis of Variance (ANOVA) was implemented, followed by a Tukey post hoc test. The threshold for significant p-values was set to less than 0.05. For beta diversity, a Permutated Multivariate Analysis of Variance (PERMANOVA) was applied with a paired multilevel comparison, also considering p-values less than 0.05 as significant.
Data availability
The datasets supporting the conclusions of this article are available in the NIH Sequence Read Archive (SRA) repository, BioProject ID: PRJNA983657 https://www.ncbi.nlm.nih.gov/bioproject/983657. (Release date: 2024-06-14) and shotgun sequencing data are available at mgRAST with IDs Ag1:mgm4972426.3, Ag2:mgm4972425.3, and Ag3 mgm4972427 https://www.mg-rast.org/.
Abbreviations
- CBR:
-
Calakmul Biosphere Reserve
- Ag-UD1:
-
Wetland 1 in the undisturbed zone from the CBR
- Ag-UD2:
-
Wetland 2 in the undisturbed zone from the CBR
- Ag-SU3:
-
Wetland 3 in the semi-urbanized zone from the CBR
References
IEG (Independent Evaluation Group). The Mesoamerican Biological Corridor. 2011. https://web.archive.org/web/20141208155438/http://ieg.worldbankgroup.org/Data/reports/mbc_rpr.pdf. Accessed 13 Mar 2024.
García Gil G, Palacio Prieto JL, Ortiz Pérez MA. Reconocimiento geomorfológico e hidrográfico de la Reserva de la Biosfera Calakmul, México. Investig Geográficas. 2002;:7–23.
DOF - Diario Oficial de la Federación. https://www.dof.gob.mx/nota_detalle.php?codigo=5700588&fecha=31/08/2023#gsc.tab=0. Accessed 13 Mar 2024.
Reyna Hurtado RA, García Anleu R, Garcia Vetorazzi M, Sanchez Pinzón K, Slater K, Barão Nobrega J, et al. Aguadas De La Selva Maya: Santuarios De Vida Silvestre que unen esfuerzos de conservación internacional. Cienc Nicolaita. 2022. https://doi.org/10.35830/cn.vi84.610.
Dobler-Morales C. Between subsidies and Parks: the impact of Agrarian and Conservation Policy on Smallholder territories of Calakmul, Mexico. In: McCall MK, Boni Noguez A, Napoletano B, Rico-Rodríguez T, editors. Territorialising Space in Latin America: processes and perceptions. Cham: Springer International Publishing; 2021. pp. 57–73.
Špirić J, Vallejo M, Ramírez MI. Impact of productive activities on Forest Cover Change in the Calakmul Biosphere Reserve Region: evidence and research gaps. Trop Conserv Sci. 2022;15:19400829221105712.
Hurtado RR, O’Farrill G, Sima D, Andrade M, Padilla A, Sosa YL. RESERVORIOS DE VIDA SILVESTRE Y DE LA RIQUEZA NATURAL DE MÉXICO.
Slater K. Calakmul Biodiversity Monitoring Report 2014–2019. 2021.
Chuvochina M, Adame MF, Guyot A, Lovelock C, Lockington D, Gamboa-Cutz JN, et al. Drivers of bacterial diversity along a natural transect from freshwater to saline subtropical wetlands. Sci Total Environ. 2021;759:143455.
Li W, Lv X, Ruan J, Yu M, Song Y-B, Yu J, et al. Variations in soil bacterial composition and diversity in newly formed Coastal wetlands. Front Microbiol. 2019;9:3256.
Thomson T, Fusi M, Bennett-Smith MF, Prinz N, Aylagas E, Carvalho S, et al. Contrasting effects of Local Environmental and Biogeographic factors on the composition and structure of bacterial communities in Arid Monospecific Mangrove soils. Microbiol Spectr. 2022;10:e0090321.
Doolittle WF. Phylogenetic classification and the universal tree. Science. 1999;284:2124–9.
Torsvik V, Daae FL, Sandaa RA, Ovreås L. Novel techniques for analysing microbial diversity in natural and perturbed environments. J Biotechnol. 1998;64:53–62.
Torsvik V, Øvreås L. Microbial diversity and function in soil: from genes to ecosystems. Curr Opin Microbiol. 2002;5:240–5.
Lm D, Dn M, Bj W. Distribution and quantification of antibiotic resistant genes and bacteria across agricultural and non-agricultural metagenomes. PLoS ONE. 2012;7.
van Elsas JD, Costa R, Jansson J, Sjöling S, Bailey M, Nalin R, et al. The metagenomics of disease-suppressive soils – experiences from the METACONTROL project. Trends Biotechnol. 2008;26:591–601.
Castelle CJ, Hug LA, Wrighton KC, Thomas BC, Williams KH, Wu D, et al. Extraordinary phylogenetic diversity and metabolic versatility in aquifer sediment. Nat Commun. 2013;4:2120.
Howe AC, Jansson JK, Malfatti SA, Tringe SG, Tiedje JM, Brown CT. Tackling soil diversity with the assembly of large, complex metagenomes. Proc Natl Acad Sci U S A. 2014;111:4904–9.
Kroeger ME, Delmont TO, Eren AM, Meyer KM, Guo J, Khan K, et al. New Biological insights into how Deforestation in Amazonia affects Soil Microbial communities using metagenomics and metagenome-assembled genomes. Front Microbiol. 2018;9:1635.
Navarrete-Euan H, Rodríguez-Escamilla Z, Pérez-Rueda E, Escalante-Herrera K, Martínez-Núñez MA. Comparing sediment microbiomes in contaminated and pristine wetlands along the Coast of Yucatan. Microorganisms. 2021;9:877.
Suárez-Moo P, Remes-Rodríguez CA, Márquez-Velázquez NA, Falcón LI, García-Maldonado JQ, Prieto-Davó A. Changes in the sediment microbial community structure of coastal and inland sinkholes of a karst ecosystem from the Yucatan Peninsula. Sci Rep. 2022;12:1110.
Becerra-Lucio AA, Labrín-Sotomayor NY, Becerra-Lucio PA, Trujillo-Elisea FI, Chávez-Bárcenas AT, Machkour-M’Rabet S, et al. Diversity and Interactomics of Bacterial Communities Associated with Dominant Trees during Tropical Forest Recovery. Curr Microbiol. 2021;78:3417–29.
Pajares S, Campo J, Bohannan BJM, Etchevers JD. Environmental controls on Soil Microbial communities in a seasonally dry Tropical Forest. Appl Environ Microbiol. 2018;84:e00342–18.
Instituto Mexicano de Recursos Renovables. Los recursos naturales del sureste y su aprovechamiento: Situación, problemas, perspectivas, Por E. Beltrán. Instituto Mexicano de Recursos Naturales Renovables; 1959.
Bharti R, Grimm DG. Current challenges and best-practice protocols for microbiome analysis. Brief Bioinform. 2019;22:178–93.
Escobar-Zepeda A, Rosas-Escobar P, Marquez Valdelamar L, de la Torre P, Partida-Martinez LP, Remegaldo R, et al. Distinctive prokaryotic microbiomes in sympatric plant roots from a Yucatan cenote. BMC Res Notes. 2021;14:333.
Yanez-Montalvo A, Gómez-Acata S, Águila B, Hernández-Arana H, Falcón LI. The microbiome of modern microbialites in Bacalar Lagoon, Mexico. PLoS ONE. 2020;15:e0230071.
Mr L, C A, N F. G S, J A, E A, Microbialite response to an anthropogenic salinity gradient in Great Salt Lake. Utah Geobiology. 2017;15.
Instituto Nacional de Ecología. Programa De manejo de la reserva de la Biosfera Calakmul, México. Instituto Nacional de Ecología; 2000.
Daniel R. The metagenomics of soil. Nat Rev Microbiol. 2005;3:470–8.
Hoffland E, Kuyper TW, Comans RNJ, Creamer RE. Eco-functionality of organic matter in soils. Plant Soil. 2020;455:1–22.
Wu X, Rensing C, Han D, Xiao K-Q, Dai Y, Tang Z, et al. Genome-resolved Metagenomics reveals distinct Phosphorus Acquisition strategies between Soil Microbiomes. mSystems. 2022. https://doi.org/10.1128/msystems.01107-21.
Speirs LBM, Rice DTF, Petrovski S, Seviour RJ. The phylogeny, Biodiversity, and Ecology of the Chloroflexi in activated sludge. Front Microbiol. 2019;10:2015.
Santos EN, Menezes LP, Dolabella SS, Santini A, Severino P, Capasso R, et al. Bacillus thuringiensis: from biopesticides to anticancer agents. Biochimie. 2022;192:83–90.
Wood DE, Salzberg SL. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014;15:R46.
Breitwieser FP, Salzberg SL. Pavian: interactive analysis of metagenomics data for microbiome studies and pathogen identification. Bioinformatics. 2020;36:1303–4.
VanInsberghe D, Maas KR, Cardenas E, Strachan CR, Hallam SJ, Mohn WW. Non-symbiotic Bradyrhizobium ecotypes dominate north American forest soils. ISME J. 2015;9:2435–41.
Pseudolabrys taiwanensis gen. nov., sp. nov., an alphaproteobacterium isolated from soil | Microbiology Society. https://www.microbiologyresearch.org/content/journal/ijsem/10.1099/ijs.0.64124-0#tab2. Accessed 21 Mar 2024.
Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics. 2020;36:1925–7.
Wu Y-W, Simmons BA, Singer SW. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics. 2016;32:605–7.
Dh P, M I, Ct S, Gw PH. T. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25.
Delgado-Baquerizo M, Oliverio AM, Brewer TE, Benavent-González A, Eldridge DJ, Bardgett RD, et al. A global atlas of the dominant bacteria found in soil. Science. 2018;359:320–5.
I Z-P VDA, Ac P-H, Le E, B C-M VS. MEBS, a software platform to evaluate large (meta)genomic collections according to their metabolic machinery: unraveling the sulfur cycle. GigaScience. 2017;6.
Xue C-X, Lin H, Zhu X-Y, Liu J, Zhang Y, Rowley G, et al. DiTing: A Pipeline to infer and compare Biogeochemical pathways from Metagenomic and Metatranscriptomic Data. Front Microbiol. 2021;12:698286.
Keegan KP, Glass EM, Meyer F. MG-RAST, a Metagenomics Service for analysis of Microbial Community structure and function. Microbial Environmental Genomics (MEG). New York, NY: Humana; 2016. pp. 207–33.
Silva GGZ, Green KT, Dutilh BE, Edwards RA. SUPER-FOCUS: a tool for agile functional analysis of shotgun metagenomic data. Bioinformatics. 2016;32:354–61.
Chen H, Ma K, Lu C, Fu Q, Qiu Y, Zhao J, et al. Functional redundancy in Soil Microbial Community based on Metagenomics across the Globe. Front Microbiol. 2022;13:878978.
Chen H, Ma K, Huang Y, Yao Z, Chu C. Stable soil Microbial Functional structure responding to Biodiversity loss based on metagenomic evidences. Front Microbiol. 2021;12:716764.
Dai Z, Zang H, Chen J, Fu Y, Wang X, Liu H, et al. Metagenomic insights into soil microbial communities involved in carbon cycling along an elevation climosequences. Environ Microbiol. 2021;23:4631–45.
Gong B, Cao H, Peng C, Perčulija V, Tong G, Fang H, et al. High-throughput sequencing and analysis of microbial communities in the mangrove swamps along the coast of Beibu Gulf in Guangxi, China. Sci Rep. 2019;9:9377.
Yun J, Deng Y, Zhang H. Anthropogenic protection alters the microbiome in intertidal mangrove wetlands in Hainan Island. Appl Microbiol Biotechnol. 2017;101:6241–52.
Vásquez-Dean J, Maza F, Morel I, Pulgar R, González M. Microbial communities from arid environments on a global scale. A systematic review. Biol Res. 2020;53:29.
Ortiz M, Leung PM, Shelley G, Jirapanjawat T, Nauer PA, Van Goethem MW, et al. Multiple energy sources and metabolic strategies sustain microbial diversity in Antarctic desert soils. Proc Natl Acad Sci U S A. 2021;118:e2025322118.
Kaiser K, Wemheuer B, Korolkow V, Wemheuer F, Nacke H, Schöning I, et al. Driving forces of soil bacterial community structure, diversity, and function in temperate grasslands and forests. Sci Rep. 2016;6:33696.
Hartman WH, Richardson CJ, Vilgalys R, Bruland GL. Environmental and anthropogenic controls over bacterial communities in wetland soils. Proc Natl Acad Sci. 2008;105:17842–7.
Wakelin SA, Macdonald LM, Rogers SL, Gregg AL, Bolger TP, Baldock JA. Habitat selective factors influencing the structural composition and functional capacity of microbial communities in agricultural soils. Soil Biol Biochem. 2008;40:803–13.
Shen C, Xiong J, Zhang H, Feng Y, Lin X, Li X, et al. Soil pH drives the spatial distribution of bacterial communities along elevation on Changbai Mountain. Soil Biol Biochem. 2013;57:204–11.
Lladó S, Žifčáková L, Větrovský T, Eichlerová I, Baldrian P. Functional screening of abundant bacteria from acidic forest soil indicates the metabolic potential of Acidobacteria subdivision 1 for polysaccharide decomposition. Biol Fertil Soils. 2016;52:251–60.
Fazi S, Amalfitano S, Pernthaler J, Puddu A. Bacterial communities associated with benthic organic matter in headwater stream microhabitats. Environ Microbiol. 2005;7:1633–40.
Padmanabhan P, Padmanabhan S, DeRito C, Gray A, Gannon D, Snape JR, et al. Respiration of 13 C-labeled substrates added to soil in the field and subsequent 16S rRNA gene analysis of 13 C-labeled soil DNA. Appl Environ Microbiol. 2003;69:1614–22.
Cupit C, Lomstein BA, Kjeldsen KU. Contrasting community composition of endospores and vegetative Firmicutes in a marine sediment suggests both endogenous and exogenous sources of endospore accumulation. Environ Microbiol Rep. 2019;11:352–60.
Filippidou S, Junier T, Wunderlin T, Lo C-C, Li P-E, Chain PS, et al. Under-detection of endospore-forming Firmicutes in metagenomic data. Comput Struct Biotechnol J. 2015;13:299–306.
Satrio AE, Gandaseca S, Ahm OH, Majid NMA. Effect of precipitation fluctuation on Soil Carbon Storage of a Tropical Peat Swamp Forest. Am J Appl Sci. 2009;6:1484.
Rocha FI, Ribeiro TG, Fontes MA, Schwab S, Coelho MRR, Lumbreras JF, et al. Land-use system and forest floor explain prokaryotic metacommunity structuring and spatial turnover in amazonian forest-to-pasture Conversion Areas. Front Microbiol. 2021;12:657508.
Sun W, Li Z, Lei J, Liu X. Bacterial communities of Forest Soils along different elevations: diversity, structure, and functional composition with potential impacts on CO2 Emission. Microorganisms. 2022;10:766.
Dell’Anno F, Rastelli E, Tangherlini M, Corinaldesi C, Sansone C, Brunet C, et al. Highly contaminated Marine sediments can host rare bacterial Taxa potentially useful for bioremediation. Front Microbiol. 2021;12:584850.
Zhang J, Shi Q, Fan S, Zhang Y, Zhang M, Zhang J. Distinction between Cr and other heavy-metal-resistant bacteria involved in C/N cycling in contaminated soils of copper producing sites. J Hazard Mater. 2021;402:123454.
Bräuer L, Basiliko S, Siljanen NMP, Zinder HH. Methanogenic archaea in peatlands. FEMS Microbiol Lett. 2020;367:fnaa172.
Scholier T, Lavrinienko A, Brila I, Tukalenko E, Hindström R, Vasylenko A et al. Urban forest soils harbour distinct and more diverse communities of bacteria and fungi compared to less disturbed forest soils. Mol Ecol. 2022;:mec16754.
Bona E, Massa N, Toumatia O, Novello G, Cesaro P, Todeschini V, et al. Climatic zone and Soil Properties determine the Biodiversity of the Soil Bacterial communities Associated to native plants from Desert areas of North-Central Algeria. Microorganisms. 2021;9:1359.
Moya D, Fonturbel MT, Lucas-Borja ME, Peña E, Alfaro-Sanchez R, Plaza-Álvarez PA, et al. Burning season and vegetation coverage influenced the community-level physiological profile of Mediterranean mixed-mesogean pine forest soils. J Environ Manage. 2021;277:111405.
Wu Z, Lin W, Li B, Wu L, Fang C, Zhang Z. Terminal restriction fragment length polymorphism analysis of Soil Bacterial communities under different vegetation types in Subtropical Area. PLoS ONE. 2015;10:e0129397.
Allison SD, Martiny JBH. Resistance, resilience, and redundancy in microbial communities. Proc Natl Acad Sci. 2008;105 supplement1:11512–9.
Okie JG, Poret-Peterson AT, Lee ZM, Richter A, Alcaraz LD, Eguiarte LE, et al. Genomic adaptations in information processing underpin trophic strategy in a whole-ecosystem nutrient enrichment experiment. eLife. 2020;9:e49816.
Comparing bacterial communities inferred from 16S rRNA gene sequencing and shotgun metagenomics. In: Biocomputing 2011. World Scientific; 2010. p. 165–76.
Allen SE. Chemical Analysis of Ecological materials. Blackwell Scientific; 1989.
Nelson. Total carbon, organic carbon, and organic matter - Google Scholar. https://scholar.google.com/scholar_lookup?journal=ASA+Monograph&title=Total+carbon,+organic+carbon,+and+organic+matter.+In:+Page,+A.L.+(ed.).+Methods+of+Soil+Analysis,+(2nd+ed.)&author=DW+Nelson&author=LE+Sommers&volume=9&issue=2&publication_year=1982&pages=539-579&. Accessed 26 Jun 2023.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Schliep KP. Phangorn: phylogenetic analysis in R. Bioinformatics. 2011;27:592–3.
Andrews S. FASTQC. A quality control tool for high throughput sequence data | BibSonomy. 2010. https://www.bibsonomy.org/bibtex/f230a919c34360709aa298734d63dca3. Accessed 9 Nov 2022.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinforma Oxf Engl. 2014;30:2114–20.
McMurdie PJ, Holmes S. Shiny-phyloseq: web application for interactive microbiome analysis with provenance tracking. Bioinforma Oxf Engl. 2015;31:282–3.
Acknowledgements
Thanks to the personnel of the local office of the National Commission of Natural Protected Areas in Calakmul, we appreciate the facilities and the access to sampling in the Calakmul Biosphere Reserve. Non-objection document for sample collection: Memorandum No. F.00.9 D-RBC-037-2018. This work was supported by CONACyT, as the experimental part of this project was conducted while Karina Verdel-Aranda was affiliated with the Cátedras CONACyT program under Project No. 1016.” We are grateful to Dr. Bárbara Nova-Franco for her expert editing and critical review of this paper. Her insights and attention to detail have been invaluable in enhancing the quality and clarity of our work.
Funding
The metagenome sequencing was funded by support for academic activities (AAA) granted to Karina Verdel-Aranda by Colegio de Postgraduados Campus Campeche (2017-19).
Author information
Authors and Affiliations
Contributions
JLR and AMH participated in the conception of the study, KVA carried out the design of sampling, the sediment samples manipulation and DNA extraction, the analysis and interpretation of data and wrote the initial draft of the manuscript, KVA, and JLR carried out the collection of samples. NSM, DAGA, and SDR carried out bioinformatic analysis and data interpretation. DAGA, NSM, and KVA prepared figures. All authors reviewed the manuscript critically for important intellectual content and approved the final version to be published. DAGA and NSM contributed equally, and both are first authors.
Corresponding authors
Ethics declarations
Ethical approval
Ethics approval and consent to participate are not applicable.
Consent for publication
Consent for publication is not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
García-Estrada, D.A., Selem-Mojica, N., Martínez-Hernández, A. et al. Diversity of bacterial communities in wetlands of Calakmul Biosphere Reserve: a comparative analysis between conserved and semi-urbanized zones in pre-Mayan Train era. BMC Microbiol 24, 376 (2024). https://doi.org/10.1186/s12866-024-03523-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-024-03523-x