
Abstract
Background
Antibiotic-resistant Klebsiella pneumoniae has emerged as a critical public health threat worldwide. Understanding the antimicrobial resistance mechanisms of multidrug-resistant K. pneumoniae (MDR-Kp) and its prevalence in time and space would provide clinical significance for managing pathogen infection.
Methods
Eighteen clinical MDR-Kp strains were analyzed by whole genome sequencing (WGS), and the antimicrobial resistance genes and associated resistance mechanisms were compared with results obtained from the conventional microbiological test (CMT). The sequence homology across strains in our study and those previously collected over time from a wide geographical region was assessed by phylogenetic analysis.
Results
MDR-Kp strains were collected from eighteen patients who had received empirical treatment before strain collection, with sputum (83.3%, 15/18) being the primary source of clinical samples. The commonly received treatments include β-lactamase inhibitors (55.6%, 10/18) and carbapenems (50%, 9/18). Using CMT, we found that all 18 strains were resistant to aztreonam and ciprofloxacin, while 14 (77.8%) showed resistance to carbapenem. Polymyxin B and tigecycline were the only antibiotics to which MDR-Kp strains were sensitive. A total of 42 antimicrobial resistance mechanisms were identified by WGS, surpassing the 40 detected by the conventional method, with 25 mechanisms shared between the two techniques. Despite a 100% accuracy rate of WGS in detecting penicillin-resistant strains, the accuracy in detecting cephalosporin-resistant strains was only at 60%. Among all resistance genes identified by WGS, Klebsiella pneumoniae carbapenemase-2 (KPC-2) was present in all 14 carbapenem-resistant strains. Phenotypic analysis indicated that sequence type (ST) 11 isolates were the primary cause of these MDR-Kp infections. Additionally, phylogenic clustering analysis, encompassing both the clinical and MDR-Kp strains previously reported in China, revealed four distinct subgroups. No significant difference was observed in the sequence homology between K. pneumoniae strains in our study and those previously collected in East China over time.
Conclusion
The application of WGS in identifying potential antimicrobial-resistant genes of MDR-Kp has demonstrated promising clinical significance. Comprehensive genomic information revealed by WGS holds the promise of guiding treatment decisions, enabling surveillance, and serving as a crucial asset in understanding antibiotic resistance.
Similar content being viewed by others


Introduction
Klebsiella pneumoniae is a gram-negative bacterium that belongs to the family Enterobacteriaceae, which includes the two well-known genera Salmonella and Escherichia [1]. The common manifestations of K. pneumoniae infections are pneumonia, urinary tract infections, bloodstream infections, and wound infections [2]. Though K. pneumoniae is classically considered hospital-acquired and infects immunocompromised hosts as an opportunistic pathogen, hypervirulent and multidrug-resistant (MDR) groups have emerged for the presence of a large accessory genome composed of plasmids and chromosomally encoded genes [1, 3, 4]. As K. pneumoniae strains pose a significant clinical health threat worldwide, understanding factors responsible for pathogenicity and resistance will be crucial for effectively treating infected patients.
The treatment of K. pneumoniae infections has been challenging, and the resistance mechanisms are an active area of investigation. Beta-lactams are commonly used in the treatment of K. pneumoniae infections; however, the production of β-lactamase that hydrolyzes oxyimino-cephalosporins leads to acquired resistance and significantly impacts treatment efficacy [5]. Additionally, while broad-spectrum antibiotics such as carbapenems and fluoroquinolones are typically the drug of choice to treat infections caused by extended-spectrum β-lactamase (ESBL)-producing K. pneumoniae [6], carbapenem-resistant K. pneumoniae (CR-Kp) has been linked to resistance against these drugs, resulting in an escalated global risk of disease [7]. Besides, MDR K. pneumoniae (MDR-Kp) may also arise after acquiring resistance to second-line antibiotics treatment against ESBL-producing and CR-Kp strains [8]. More recently, K. pneumoniae vaccination has emerged as a promising and innovative strategy to effectively defeat this life-threatening pathogen. Ranjbarian et al. conducted a systematic review of different vaccines (i.e., conjugate, protein, polysaccharide, lipopolysaccharide) using animal models from 35 studies [9]. Although these attempts have shown effectiveness, major challenges in vaccination therapy persist, including low vaccine responses in immunocompromised patients, time-consuming and costly processes, and safety concerns. Indeed, there is currently no approved and globally available K. pneumoniae vaccine. Kleb4V, a tetravalent bioconjugate vaccine, is the only unlicensed vaccine candidate in the phase 1/2 clinical trial (NCT04959344), which marks the first-time-in-human evaluation of the vaccine’s safety and immunogenicity against K. pneumoniae [10]. Hence, efforts should be made to optimize treatment decisions and find alternative antigens for K. pneumoniae vaccination, as well as apply new strategies to provoke robust immune responses in animal models.
According to the China Antimicrobial Surveillance Network (CHINET), the resistance rate of K. pneumoniae to carbapenem antimicrobial agents, such as imipenem and meropenem, increased by eight times from 2005 to 2021 (3.0%–23.8%) [11]. The isolation rate of MDR strains also experienced a dramatic increase from 0.3% to 3.5% from 2008 to 2016. Critical geographical differences were observed in K. pneumoniae resistance in China, with East China having higher isolation rates than others. Furthermore, it was worth noting that public health surveillance data for this important pathogen was scarce during the outbreak of COVID-19. Considering the clinical importance of K. pneumoniae infections and their high prevalence in East China, gaining insight into how these emerging MDR strains acquire resistance to antimicrobial treatments and how they vary from one another becomes imperative for better disease control.
In this study, we retrospectively analyzed 18 MDR-Kp strains that were isolated from two tertiary first-class hospitals in Xuzhou, a nine-million population city in East China between December 2021 and April 2022, using conventional microbiological test (CMT) and next-generation whole genome sequencing (WGS). The clonal relationships of K. pneumoniae strains were analyzed by MLST, and the genetic relatedness of strains was compared with those previously identified in the same geographical region to evaluate the epidemiology of K. pneumoniae infections.
Materials and methods
Patients and MDR-Kp strain isolation
This study was conducted in accordance with the declaration of Helsinki and approved by the Ethics Committee of the Affiliated Xuzhou Municipal Hospital of Xuzhou Medical University (No. xyyll[2021]116). All patients provided written consent to participate and publication. All patients were empirically treated before strain culture, and the efficacy of treatment was evaluated after 72 h. Primary considerations for empirical treatment were as follows: (i) the local bacterial epidemiology and the associated antimicrobial resistance; (ii) distinguishing between community- and hospital-acquired infections. Clinical samples, including sputum, urine, and blood, were collected from patients, followed by Klebsiella pneumoniae isolation for antimicrobial susceptibility testing (AST) using the VITEK-2 Compact system (bioMérieux, France).
Whole genome sequencing
The genomic DNA of the isolated MDR-Kp strains was extracted using PureLink Genomic DNA Mini Kit (Invitrogen, United States) according to the manufacturer’s instructions. The quantity and quality of DNA were assessed by Qubit 2.0 (Thermo Fisher Scientific, United States) and NanoDrop 2000 (Thermo Fisher Scientific, United States), respectively. DNA libraries were prepared using the KAPA Hyper Prep kit (KAPA Biosystems, United States) and were paired-end sequenced on the Illumina NovaSeq platform using 150 bp fragments (Dinfectome Inc., China). Low-quality reads, adaptor contaminations, duplications, and short reads of less than 36 bp were removed as previously described [12]. The filtered sequencing reads were aligned to the reference Klebsiella pneumoniae subsp. pneumoniae HS11286 [13] using the Burrows-Wheeler Alignment (BWA, v0.7.5) tool [14]. Genotypes were called for all samples individually with HaplotypeCaller from the Genome Analysis Tool Kit (GATK) (v3.3.0) using the default settings [15]. To identify drug-resistant genes, we conducted a comparison of sequencing reads with the Comprehensive Antibiotic Resistance Database (CARD 2023, https://card.mcmaster.ca/) using ResFinder 2.1 [16]. CARD provides both the reference database and toolsets based on detailed controlled molecular sequences and mutation data of antibiotic resistance genes [17, 18]. Srst2 [19] was used to detect multilocus sequence typing (MLST) using Klebsiella pneumoniae MLST sequences and definitions obtained from the PubMLST (https://pubmlst.org/).
Single nucleotide polymorphism (SNP) discovery and phylogenetic construction
The sequencing reads of the 87 common K. pneumoniae strains were downloaded from the Bacterial and Viral Bioinformatics Resource Center (BV-BRC, https://www.bv-brc.org/). The primary inclusion criteria for the strain’s genome sequence were as follows: (i) the assembly level was complete; (ii) genome contamination was less than 1%; (iii) genome completeness exceeded 99%; (iv) passed the NCBI taxonomy check; (v) strains were collected from various regions across China. We used the available K. pneumoniae whole genome(https://www.ncbi.nlm.nih.gov/genome/815) as a reference for mapping and identifying SNPs. Sequencing reads were aligned to the reference using BWA36 (v0.7.5). Default parameters were used, allowing up to four mismatches and one gap when aligning reads to the genome. Alignments were converted from sequence alignment map (SAM) format to sorted, indexed binary alignment map (BAM) files62 (SAMtools v0.1.18). The Picard tool (v1.103; http://broadinstitute.github.io/picard) was used to remove duplicate reads. Genotypes were called for all samples individually with HaplotypeCaller from the Genome Analysis Tool Kit (GATK)38 (v3.3.0) using the default settings. The variant calls were filtered using a minimum PHRED quality threshold of 20 and a minimum and maximum variant coverage of 10 and 500 reads, respectively. SNPs with a deletion rate greater than 80% were excluded. All filter SNPs of the 87 common strains and 18 clinical strains were used to reconstruct a phylogenetic tree by the RAxML program with a gamma distribution and a general time-reversible model under 500 bootstrap repeats. Principal component analysis was performed to compare regional differences among bacteria strains.
Results
Patient characteristics
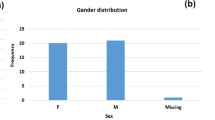
Based on the CMT result, MDR-Kp strains were defined as those resistant to ≥ 3 antimicrobial classes in addition to ampicillin, to which all K. pneumoniae infections are intrinsically resistant. Accordingly, a total of 18 clinical isolates were identified as MDR-Kp and subsequently analyzed by WGS to identify drug-resistant genes. Most patients (72.2%, 13/18) were male and above 65 years of age (Table 1). Sixteen patients have been hospitalized for more than two weeks, and nine underwent treatments in the Intensive Care Unit (ICU) within 30 days before strain culture. The most common comorbidities in these patients were brain disease (94.4%, 17/18) and respiratory disease (94.4%, 17/18), followed by hypertension (55.6%, 10/18) and heart disease (50%, 9/18). Notably, all patients had received empirical treatment before the collection of clinical isolates. The empirical treatment strategy for patients primarily relied on the local bacterial epidemiology and infection types, particularly distinguishing between community- and hospital-acquired infections. For community-acquired infections, antimicrobial drugs that effectively target the most identified bacteria were administered, with a subsequent escalation to carbapenems if significant improvement was not observed. In the case of hospital-acquired infections in critically ill patients, where multiple risk factors for infection with drug-resistant bacteria existed, the empirical use of carbapenems was considered as means to rapidly control the infection and prevent further disease progression. Accordingly, 14 patients (77.8%) were treated with multiple antibiotics, nine patients (50.0%) received carbapenems, and five patients (27.8%) were administered corticosteroids and immunosuppressants as part of their treatment regimen. Besides, it was observed that patients had undergone invasive treatment procedures. The most prevalent type was urinary catheter (66.7%), followed by tracheal intubation (44.4%) and mechanical ventilation (38.9%).
Antimicrobial susceptibility of clinical MDR-Kp strains
The antimicrobial susceptibility of MDR-Kp strains was tested against 40 antibiotics by CMT (Figure S1A and Table S1-S3). Here, we observed a high resistance rate of clinical isolates to β-lactams. Of note, all 18 cultured strains were resistant to aztreonam and ciprofloxacin (Table 2 and Table S1). In great contrast, only one tested strain (10%) was resistant to the cephalosporin antibiotic ceftazidime/avibactam, and all tested strains remained sensitive to tigecycline and polymyxin B (Table 2). Collectively, antimicrobial susceptibility testing (AST) results suggest that tigecycline and polymyxin B might serve as promising treatment strategies for patients infected with K. pneumoniae who have been previously exposed to broad-spectrum antibiotics.
Deciphering the putative drug-resistance genes in MDR-Kp through whole genome sequencing
The optimal treatment option for MDR-Kp infections has not yet been well established. Here, we aimed to identify potential drug-resistance genes in clinical MDR-Kp isolates through WGS. By understanding the specific resistance mechanisms of MDR-Kp clinical isolates, clinicians can tailor the use of appropriate antibiotics, thereby effectively clearing the infection and improving patient clinical outcomes. Among the 42 potential resistance mechanisms detected through WGS, 25 (43.9%) were found to be associated with known drug-resistance mechanisms, which can be readily evaluated by CMT (Figure S1B; Table S4 and S5). Notably, Klebsiella pneumoniae carbapenemase-2 (KPC-2) and parC had the highest detection rate (77.8%, 14/18), followed by rmtB and sul2 (66.7%, 12/18) (Table 3). Next, we assessed the performance of WGS for detecting known mechanisms conferring resistance in MDR-Kp strains. For β-lactams, while WGS achieved a 100% sensitivity and accuracy in identifying penicillin-resistant strains, the performance dropped by 40% in predicting the sensitivity of K. pneumoniae to cephalothin and cefuroxime (Table 4). Regarding other frequently employed antibiotics, WGS demonstrated a moderate performance, exhibiting an accuracy ranging from 77.8 to 100%.
Exploring the genetic divergence of MDR-Kp strains in temporal and spatial scales
The diversity and complexity of MDR-Kp strains in our study were examined using the multilocus sequence typing (MLST) test [20]. The MLST is a nucleotide sequence-based approach developed for K. pneumoniae to characterize the genetic relationships among isolates. Four sequence types (ST) were identified, with ST11 being the predominant type (Table S6). This finding was consistent with others, suggesting that ST11 is the dominant cause of K. pneumoniae infections in China.
Next, we performed a single nucleotide polymorphism (SNP)-based phylogenic analysis to compare the genetic similarities and evolutionary relationships among the 18 MDR-Kp isolates in our study and 87 strains previously identified in China. As a result, MDR-Kp strains investigated in our study were primarily classified into four clusters. Notably, half of the strains were found clustered together with Hunan and Shanghai strains in clusters A, while six strains (33.3%) formed a distinct grouping in cluster B (Figure S2A). However, our analysis did not show any significant spatial or temporal differences in the distribution pattern of these strains (Figure S2B, C). This finding suggests a widespread prevalence of MDR-Kp strains in China, indicating a significant public health threat that necessitates immediate attention.
Discussion
In this study, we utilized WGS to identify potential resistance mechanisms of MDR-Kp strains and compared that with CMT results. The sequence homology of K. pneumoniae strains isolated in local hospitals was also compared with those previously collected in a wider geographical region over time to uncover the diversity and complexity of this pathogen.
K. pneumoniae is one of the most common nosocomial pathogens in the world [21]. It is also a leading cause of ventilator-associated pneumonia among patients in the ICU [22, 23]. Consistently, we found that half of the patients had previous records of being treated in the ICU within 30 days before strain culture, including 2 and 4 patients who were later transferred from the ICU to the respiratory department and the neurology department, respectively (Table 1). This finding suggests that the ICU is the primary source of MDR-Kp infections in the two local hospitals, and the clinical use of antibiotics in the ICU should be more standardized. In addition, we found that 12 out of 18 patients (66.7%) were invaded by urinary tract-associated catheters within three months before strain culture. This finding supports the idea that the urinary tract is a common site of K. pneumoniae infection, with catheter-associated procedures being the primary cause of infection, possibly due to the ability of K. pneumoniae to form biofilms and adhere to catheters. Besides, we report that the primary underlying diseases associated with MDR-Kp infection were respiratory and brain diseases (17/18, 94.4%). However, given that K. pneumoniae is also the second leading cause of bloodstream infections, MDR-Kp strains may have originated from secondary infections that trace back to a known source.
MDR-Kp infections are typically associated with a high mortality rate; however, there is currently no standardized treatment for effectively controlling such diseases. Here, we aim to elucidate the mechanisms underlying antibiotic resistance in clinical MDR-Kp isolates within local hospitals using both conventional methods and WGS. Aztreonam is a vital treatment scheme for severe infection caused by Enterobacter species producing metallo-β-lactamases [24]. The CMT results in our study revealed that all MDR-Kp strains were resistant to aztreonam and ciprofloxacin, suggesting that these two antibiotics are likely ineffective against K. pneumoniae in the Xuzhou region. In addition, we noticed that the resistance rate of K. pneumoniae to ciprofloxacin was significantly higher than previously reported by CHINET (100% vs. 33%). We reasoned that this could be due to regional differences and limited sample size. Moreover, 77.8% of strains were identified to be CR-Kp. Since half of the patients had been previously treated with carbapenem antibiotics, we reasoned that K. pneumoniae might be susceptible to carbapenem resistance. Therefore, clinicians should be more cautious when prescribing such antibiotics. Lastly, we found that all examined MDR-Kp strains were sensitive to tigecycline and polymyxin B. Indeed, there have been previous reports showing that tigecycline effectively treats complex abdominal infections and community-acquired pneumonia caused by refractory bacterial infections [25]. Combining polymyxin B in antimicrobial treatment may provide better efficacy as it reduces the antibiotic dosage and eliminates the generation of drug-resistant strains [26]. Despite this promising result, given that polymyxin B is considered the last resort for treating CR-Kp infections, it is of clinical importance to develop other molecular/genetic tools to identify potential resistance mechanisms of the specific strain for optimal treatment decision-making and avoid misconduct of therapies.
In this regard, WGS is an unbiased approach that enables the broad identification of known and unexpected pathogens and provides auxiliary genomic information to predict drug resistance [27, 28]. In this present study, all WGS results were returned within 48 h, much shorter than the turnaround time of using traditional microbiological tests. Notably, we identified 42 potential resistance mechanisms of locally isolated clinical MDR-Kp strains using WGS, with KPC-2 and parC being the most commonly identified drug-resistant genes among all these strains. While 25 resistance mechanisms were identified by both techniques, 15 were exclusively identified by CMT but not by WGS. Additionally, WGS showed false negative results in 11 strains, most of which were cephalosporin-resistant (5 strains) or quinolone-resistant strains (4 strains). Indeed, several factors may affect the number of mechanisms identified by both techniques, including the scope of CMT (available antibiotics), the reference database used against WGS data, and even treatment strategies implemented for patients before the collection of clinical samples. Therefore, these results comparing the identified resistance mechanisms between CMT and WGS need to be interpreted with caution. Nonetheless, the NGS-facilitated resistant gene detection holds great promise in supplementing the conventional testing approach for prompt treatment decision-making and clearance of pathogen infection.
A deeper understanding of K. pneumoniae diversity will be important for interpreting public health surveillance data and the implementation of novel control strategies against this pathogen. Here, we compared MDR-Kp strains to those previously identified in other regions of China and Xuzhou strains isolated between 2016 and 2021. MDR-Kp strains in this study were mainly ST11 and clustered in four categories. No significant spatial or temporal differences were observed, suggesting a wide geographical spread of clinically relevant MDR-Kp strains in East China. Therefore, close surveillance should be carried out in local hospitals to prevent the outbreak of MDR-Kp strains.
Despite these promising results, there are limitations to this study. First, our study has a relatively small sample size. Expanding the sample size is warranted to provide a more comprehensive view of the potential resistance mechanism of K. pneumoniae to antibiotics. Second, although WGS identified a broad range of antibiotic-resistant genes and promising resistance mechanisms, whether these drug-resistant genes are expressed in vivo awaits to be tested. Phenotypic tests might be considered to validate whether the underlying drug-resistant gene expresses the corresponding enzyme.
Conclusion
To summarize, WGS demonstrated significant clinical implications to identify potential antimicrobial-resistant genes in MDR-Kp strains, which enables prompt treatment strategy adjustment and a better understanding of the resistance mechanism in MDR-Kp strains.
Data Availability
All sequence reads were deposited into the NCBI Sequence Read Archive (SRA) database under the accession number PRJNA956822. All the other relevant data of the study are available from the corresponding authors upon reasonable request.
References
Wyres KL, Lam MMC, Holt KE. Population genomics of Klebsiella pneumoniae. Nat Rev Microbiol. 2020;18(6):344–59.
Podschun R, Ullmann U. Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin Microbiol Rev. 1998;11(4):589–603.
Martin RM, Bachman MA. Colonization, infection, and the Accessory Genome of Klebsiella pneumoniae. Front Cell Infect Microbiol. 2018;8:4.
Holt KE, Wertheim H, Zadoks RN, Baker S, Whitehouse CA, Dance D, et al. Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc Natl Acad Sci U S A. 2015;112(27):E3574–81.
Bush K, Jacoby GA, Medeiros AA. A functional classification scheme for beta-lactamases and its correlation with molecular structure. Antimicrob Agents Chemother. 1995;39(6):1211–33.
Long SW, Olsen RJ, Eagar TN, Beres SB, Zhao P, Davis JJ et al. Population Genomic Analysis of 1,777 Extended-Spectrum Beta-Lactamase-Producing Klebsiella pneumoniae Isolates, Houston, Texas: Unexpected Abundance of Clonal Group 307 mBio, 2017. 8(3).
Xu L, Sun X, Ma X. Systematic review and meta-analysis of mortality of patients infected with carbapenem-resistant Klebsiella pneumoniae. Ann Clin Microbiol Antimicrob. 2017;16(1):18.
van Duin D, Doi Y. Outbreak of Colistin-Resistant, carbapenemase-producing Klebsiella pneumoniae: are we at the end of the Road? J Clin Microbiol. 2015;53(10):3116–7.
Ranjbarian P, Sobhi Amjad Z, Chegene Lorestani R, Shojaeian A, Rostamian M. Klebsiella pneumoniae vaccine studies in animal models. Biologicals. 2023;82:101678.
Organization WH. Bacterial vaccines in clinical and preclinical development 2021: an overview and analysis World Health Organization, License: CC BY-NC-SA 3.0 IGO, 2022.
Fupin GYHU, Demei ZHU, Fu WANG, Xiaofei JIANG, Yingchun XU et al. CHINET surveillance of antimicrobial resistance among the bacterial isolates in 2021. Chin J Infect Chemother, 2022.
Xiao-Wei Z-DM, Yin Q, Zhang Q-X, Ou J, Liu Y, Shao. Zhi-Guang Liu, Clinical metagenomic sequencing for rapid diagnosis of pneumonia and meningitis caused by Chlamydia psittaci. World J Clin Cases; 2021.
Liu P, Li P, Jiang X, Bi D, Xie Y, Tai C, et al. Complete genome sequence of Klebsiella pneumoniae subsp. pneumoniae HS11286, a multidrug-resistant strain isolated from human sputum. J Bacteriol. 2012;194(7):1841–2.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–8.
Bortolaia V, Kaas RS, Ruppe E, Roberts MC, Schwarz S, Cattoir V, et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J Antimicrob Chemother. 2020;75(12):3491–500.
Alcock BP, Huynh W, Chalil R, Smith KW, Raphenya AR, Wlodarski MA, et al. CARD 2023: expanded curation, support for machine learning, and resistome prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2023;51(D1):D690–9.
McArthur AG, Waglechner N, Nizam F, Yan A, Azad MA, Baylay AJ, et al. The comprehensive antibiotic resistance database. Antimicrob Agents Chemother. 2013;57(7):3348–57.
Inouye M, Dashnow H, Raven LA, Schultz MB, Pope BJ, Tomita T, et al. SRST2: Rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 2014;6(11):90.
Urwin R, Maiden MC. Multi-locus sequence typing: a tool for global epidemiology. Trends Microbiol. 2003;11(10):479–87.
Pendleton JN, Gorman SP, Gilmore BF. Clinical relevance of the ESKAPE pathogens. Expert Rev Anti Infect Ther. 2013;11(3):297–308.
Kalanuria AA, Ziai W, Mirski M. Ventilator-associated pneumonia in the ICU. Crit Care. 2014;18(2):208.
Talha KA, Hasan Z, Selina F, Palash MI. Organisms associated with ventilator associated pneumonia in intensive care unit. Mymensingh Med J. 2009;18(1 Suppl):S93–97.
Yasmin M, Fouts DE, Jacobs MR, Haydar H, Marshall SH, White R, et al. Monitoring ceftazidime-avibactam and aztreonam concentrations in the treatment of a bloodstream infection caused by a multidrug-resistant Enterobacter sp. Carrying both Klebsiella pneumoniae Carbapenemase-4 and New Delhi Metallo-beta-Lactamase-1. Clin Infect Dis. 2020;71(4):1095–8.
Zhang Y, Zhao C, Wang Q, Wang X, Chen H, Li H, et al. Evaluation of the in vitro activity of new polymyxin B analogue SPR206 against clinical MDR, colistin-resistant and tigecycline-resistant Gram-negative bacilli. J Antimicrob Chemother. 2020;75(9):2609–15.
Ezadi F, Ardebili A, Mirnejad R. Antimicrobial susceptibility testing for polymyxins: Challenges, Issues, and recommendations. J Clin Microbiol, 2019. 57(4).
Wei Gu SM, Chiu CY. Clinical metagenomic next-generation sequencing for Pathogen Detection. Annu Rev Pathol Mech Dis, 2019.
Malaya MIL, Sahoo K, Yamamoto F, Waggoner JJ, Chou S, Holmes SP, Anderson MW, Pinsky BA. Detection of Cytomegalovirus Drug Resistance mutations by Next-Generation sequencing. J Clin Microbiol, 2013.
Villegas MV, Lolans K, Correa A, Kattan JN, Lopez JA, Quinn JP. Colombian nosocomial resistance study, First identification of Pseudomonas aeruginosa isolates producing a KPC-type carbapenem-hydrolyzing beta-lactamase. Antimicrob Agents Chemother. 2007;51(4):1553–5.
Nawaz M, Sung K, Kweon O, Khan S, Nawaz S, Steele R. Characterisation of novel mutations involved in quinolone resistance in Escherichia coli isolated from imported shrimp. Int J Antimicrob Agents. 2015;45(5):471–6.
Zhou Y, Yu H, Guo Q, Xu X, Ye X, Wu S, et al. Distribution of 16S rRNA methylases among different species of Gram-negative bacilli with high-level resistance to aminoglycosides. Eur J Clin Microbiol Infect Dis. 2010;29(11):1349–53.
Daly M, Villa L, Pezzella C, Fanning S, Carattoli A. Comparison of multidrug resistance gene regions between two geographically unrelated Salmonella serotypes. J Antimicrob Chemother. 2005;55(4):558–61.
Sanchez-Romero I, Asensio A, Oteo J, Munoz-Algarra M, Isidoro B, Vindel A, et al. Nosocomial outbreak of VIM-1-producing Klebsiella pneumoniae isolates of multilocus sequence type 15: molecular basis, clinical risk factors, and outcome. Antimicrob Agents Chemother. 2012;56(1):420–7.
Aldema ML, McMurry LM, Walmsley AR, Levy SB. Purification of the Tn10-specified tetracycline efflux antiporter TetA in a native state as a polyhistidine fusion protein. Mol Microbiol. 1996;19(1):187–95.
Huang Z, Mi Z, Wang C. A novel beta-lactamase gene, LAP-2, produced by an Enterobacter cloacae clinical isolate in China. J Hosp Infect. 2008;70(1):95–6.
Ho PL, Wong RC, Lo SW, Chow KH, Wong SS, Que TL. Genetic identity of aminoglycoside-resistance genes in Escherichia coli isolates from human and animal sources. J Med Microbiol. 2010;59(Pt 6):702–7.
Nuesch-Inderbinen MT, Kayser FH, Hachler H. Survey and molecular genetics of SHV beta-lactamases in Enterobacteriaceae in Switzerland: two novel enzymes, SHV-11 and SHV-12. Antimicrob Agents Chemother. 1997;41(5):943–9.
Scholz P, Haring V, Wittmann-Liebold B, Ashman K, Bagdasarian M, Scherzinger E. Complete nucleotide sequence and gene organization of the broad-host-range plasmid RSF1010. Gene. 1989;75(2):271–88.
Marquez C, Labbate M, Ingold AJ, Roy Chowdhury P, Ramirez MS, Centron D, et al. Recovery of a functional class 2 integron from an Escherichia coli strain mediating a urinary tract infection. Antimicrob Agents Chemother. 2008;52(11):4153–4.
Krizova L, Dijkshoorn L, Nemec A. Diversity and evolution of AbaR genomic resistance islands in Acinetobacter baumannii strains of european clone I. Antimicrob Agents Chemother. 2011;55(7):3201–6.
Datta N, Kontomichalou P. Penicillinase synthesis controlled by infectious R factors in Enterobacteriaceae. Nature. 1965;208(5007):239–41.
Shen P, Jiang Y, Zhou Z, Zhang J, Yu Y, Li L. Complete nucleotide sequence of pKP96, a 67 850 bp multiresistance plasmid encoding qnrA1, aac(6’)-Ib-cr and blaCTX-M-24 from Klebsiella pneumoniae J Antimicrob Chemother, 2008. 62(6): p. 1252-6.
Wu J, Xie L, Zhang F, Ni Y, Sun J. Molecular characterization of ISCR1-mediated blaPER-1 in a non-O1, non-O139 Vibrio cholerae strain from China. Antimicrob Agents Chemother. 2015;59(7):4293–5.
Kadlec K, Kehrenberg C, Schwarz S. Efflux-mediated resistance to florfenicol and/or chloramphenicol in Bordetella bronchiseptica: identification of a novel chloramphenicol exporter. J Antimicrob Chemother. 2007;59(2):191–6.
Ambler RP, Scott GK. Partial amino acid sequence of penicillinase coded by Escherichia coli plasmid R6K. Proc Natl Acad Sci U S A. 1978;75(8):3732–6.
Mugnier P, Casin I, Bouthors AT, Collatz E. Novel OXA-10-derived extended-spectrum beta-lactamases selected in vivo or in vitro. Antimicrob Agents Chemother. 1998;42(12):3113–6.
Hollingshead S, Vapnek D. Nucleotide sequence analysis of a gene encoding a streptomycin/spectinomycin adenylyltransferase. Plasmid. 1985;13(1):17–30.
Chen YT, Lauderdale TL, Liao TL, Shiau YR, Shu HY, Wu KM, et al. Sequencing and comparative genomic analysis of pK29, a 269-kilobase conjugative plasmid encoding CMY-8 and CTX-M-3 beta-lactamases in Klebsiella pneumoniae. Antimicrob Agents Chemother. 2007;51(8):3004–7.
Poirel L, Gniadkowski M, Nordmann P. Biochemical analysis of the ceftazidime-hydrolysing extended-spectrum beta-lactamase CTX-M-15 and of its structurally related beta-lactamase CTX-M-3. J Antimicrob Chemother. 2002;50(6):1031–4.
Gniadkowski M, Schneider I, Palucha A, Jungwirth R, Mikiewicz B, Bauernfeind A. Cefotaxime-resistant Enterobacteriaceae isolates from a hospital in Warsaw, Poland: identification of a new CTX-M-3 cefotaxime-hydrolyzing beta-lactamase that is closely related to the CTX-M-1/MEN-1 enzyme. Antimicrob Agents Chemother. 1998;42(4):827–32.
Gopal M, Elumalai S, Arumugam S, Durairajpandian V, Kannan MA, Selvam E, Seetharaman S. GyrA ser83 and ParC trp106 mutations in Salmonella enterica Serovar Typhi isolated from typhoid fever patients in Tertiary Care Hospital. J Clin Diagn Res. 2016;10(7):DC14–8.
Bonnin RA, Nordmann P, Carattoli A, Poirel L. Comparative genomics of IncL/M-type plasmids: evolution by acquisition of resistance genes and insertion sequences. Antimicrob Agents Chemother. 2013;57(1):674–6.
Chesneau O, Tsvetkova K, Courvalin P. Resistance phenotypes conferred by macrolide phosphotransferases. FEMS Microbiol Lett. 2007;269(2):317–22.
Ali A, Azam MW, Khan AU. Non-active site mutation (Q123A) in New Delhi metallo-beta-lactamase (NDM-1) enhanced its enzyme activity. Int J Biol Macromol. 2018;112:1272–7.
Ouellette M, Bissonnette L, Roy PH. Precise insertion of antibiotic resistance determinants into Tn21-like transposons: nucleotide sequence of the OXA-1 beta-lactamase gene. Proc Natl Acad Sci U S A. 1987;84(21):7378–82.
De Pascale G, Wright GD. Antibiotic resistance by enzyme inactivation: from mechanisms to solutions. ChemBioChem. 2010;11(10):1325–34.
Sanchez-Cespedes J, Marti S, Soto SM, Alba V, Melcion C, Almela M, et al. Two chromosomally located qnrB variants, qnrB6 and the new qnrB16, in Citrobacter spp. isolates causing bacteraemia. Clin Microbiol Infect. 2009;15(12):1132–8.
Acknowledgements
The authors thank all the patients and their families, the investigators who participated in this study.
Tables.
Funding
This work was supported by the Key R&D Program of the Xuzhou Science and Technology Project (Agricultural and Social Development, No. KC19169). The funder had no role in study design, data collection, data analysis, the decision to publish, or manuscript preparation.
Author information
Authors and Affiliations
Contributions
JY designed this study. JY, KZ and CD performed the data acquisition. JY, KZ, SW, and WWW performed data analysis. JY, KZ and SW edited the manuscript. XQL supervised the present study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
SW and WWW are employees of Dinfectome Inc. The remaining authors declare no competing interests.
Abbreviations
MDR, multidrug-resistant; MDR-Kp, multidrug-resistant Klebsiella pneumoniae; ESBL, extended-spectrum β-lactamase; CR-Kp, carbapenem-resistant K. pneumoniae; WGS, whole genome sequencing; ICU, intensive care units; MLST, multilocus sequence typing; ST, sequence type.
Ethics approval and consent to participate
This study was conducted in accordance with the declaration of Helsinki and approved by the Ethics Committee of the Affiliated Xuzhou Municipal Hospital of Xuzhou Medical University (No. xyyll[2021]116). Written informed consent was obtained from each patient before sample collection.
Consent for publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, J., Zhang, K., Ding, C. et al. Exploring multidrug-resistant Klebsiella pneumoniae antimicrobial resistance mechanisms through whole genome sequencing analysis. BMC Microbiol 23, 245 (2023). https://doi.org/10.1186/s12866-023-02974-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-023-02974-y