
Abstract
Background
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by disturbance of pro-inflammatory and anti-inflammatory lymphocytes. Growing evidence shown that gut microbiota participated in the occurrence and development of SLE by affecting the differentiation and function of intestinal immune cells. The purpose of this study was to investigate the changes of gut microbiota in SLE and judge its associations with peripheral T lymphocytes.
Methods
A total of 19 SLE patients and 16 HCs were enrolled in this study. Flow cytometry was used to detect the number of peripheral T lymphocyte subsets, and 16 s rRNA was used to detect the relative abundance of gut microbiota. Analyzed the correlation between gut microbiota with SLEDAI, ESR, ds-DNA and complement. SPSS26.0 software was used to analyze the experimental data. Mann–Whitney U test was applied to compare T lymphocyte subsets. Spearman analysis was used for calculating correlation.
Results
Compared with HCs, the proportions of Tregs (P = 0.001), Tfh cells (P = 0.018) and Naïve CD4 + T cells (P = 0.004) significantly decreased in SLE patients, and proportions of Th17 cells (P = 0.020) and γδT cells (P = 0.018) increased in SLE. The diversity of SLE patients were significantly decreased. Addition, there were 11 species of flora were discovered to be distinctly different in SLE group (P < 0.05). In the correlation analysis of SLE, Tregs were positively correlated with Ruminococcus2 (P = 0.042), Th17 cells were positively correlated with Megamonas (P = 0.009), γδT cells were positively correlated with Megamonas (P = 0.003) and Streptococcus (P = 0.004), Tfh cells were positively correlated with Bacteroides (P = 0.040), and Th1 cells were negatively correlated with Bifidobacterium (P = 0.005). As for clinical indicators, the level of Tregs was negatively correlated with ESR (P = 0.031), but not with C3 and C4, and the remaining cells were not significantly correlated with ESR, C3 and C4.
Conclusion
Gut microbiota and T lymphocyte subsets of SLE changed and related to each other, which may break the immune balance and affect the occurrence and development of SLE. Therefore, it is necessary to pay attention to the changes of gut microbiota and provide new ideas for the treatment of SLE.
Similar content being viewed by others

Introduction
Systemic lupus erythematosus (SLE) is a common chronic autoimmune disease involving multiple organs and systems, which leads to diverse clinical manifestations. The pathogenesis of SLE is not entirely clear, and may be related to various factors such as genetic, immune, hormones, and environment [1]. Pathogenic factors stimulate the generation of autoantibodies and activation of autoreactive T cells, resulting in abnormal immune environment [2]. To a certain extent, immunosuppressive drugs could correct immune disorder and reduce disease activity. However, there are still some SLE patients who are not in remission, and long-term medication may increase the risk of severe infection and death [3]. Therefore, we need to better understand the immune-related pathogenesis of SLE in order to optimize the treatment strategies.
In recent years, numerous studies have highlighted the imbalance of T lymphocyte subsets in the onset of both lupus murine and SLE patients [4, 5]. In addition, the regulation of T subpopulations by gut microbiota and their metabolites in SLE sufferers are also widely recognized [6]. Oral antibiotics could suppress the growth of bacteria and induce autoimmune diseases such as SLE, which suggested the critical role of bacteria balance in SLE [7]. It has been shown that changes in gut microbiota, particularly those of Clostridium and segmented filamentous bacteria (SFB) in mice and Bacteroides fragilis in humans can disrupt the homeostasis between Type 17 T helper (Th17) and T regulatory (Treg) cells (Tregs) in autoimmune diseases [8]. Moreover, a study has increased the proportion of Lactobacillales in gut microbiota, thus stimulating differentiation of Tregs and inhibiting differentiation of pathogenic Th17 cells to alleviate renal inflammation in lupus-prone mice, which suggests another potential treatment for SLE [9].
The development of metagenomic sequencing (16S rRNA and whole-genome sequencing) has made it possible to study the mechanisms of the gut microbiota in many diseases. Here, we report changes of gut microbiota and T lymphocyte subpopulations in SLE patients as well as the correlation between them to explore the pathogenesis for more effective treatment.
Materials and methods
Participants
A total of 19 patients with SLE (mean age of 42.79 ± 14.36 years old) according to the 2019 American college of Rheumatology (ACR) and European League Against Rheumatism (EULAR) classification criteria were included in the study. At the same time, 16 healthy controls (HCs) (mean age of 33.62 ± 12.63 years old) were also enrolled in this research. All participants donated samples of their peripheral blood (PB) and feces for the test of T lymphocyte subsets and intestinal microflora respectively. Clinical data like erythrocyte sedimentation rate (ESR), complement 3 (C3) and complement 4 (C4) were recorded in detail. None of the selected members had serious heart, liver, kidney diseases, tumor, infection, use of antibiotics within two months or gastrointestinal surgery. In addition, none of the selected members used probiotic supplements. Each participant signed the informed consent and our study was approved by Ethics Committee of Second Hospital of Shanxi Medical University (Ethics Number: 2023YX246).
Flow cytometry
Flow cytometry was used to value the plasma levels of T lymphocyte subsets. Surface antibodies, intracellular antibodies were added to label the T cells, including CD4+T (CD3+CD4+), CD8+T (CD3+CD8+), Naïve CD4+T (CD3+CD4+CD45RA+), Memory CD4+T (CD3+CD4+CD45RO+), Naïve CD8+T (CD3+CD8+CD45RA+), Memory CD8+T (CD3+CD8+CD45RO+), Tfh (CD4+CXCR5+PD1+), Tfr (CD4+CXCR5+CD25+Foxp3+), Th1 (CD4+IFN-γ+), Th17 (CD4+IL-17+), Tregs (CD4+CD25+FoxP3+) and γδ T (CD3+TCR γδ +).
DNA extraction from fecal specimens
500 mg fresh stool samples without urine or blood stains from all participants were collected. The samples should be put into pre-prepared storage tubes quickly and transported to laboratory to be frozen and stored at -80 °C for later use. Total DNA were extracted from thawed specimens using the TIANamp Stool DNA Kit according to the manufacturer protocols. The V3-V4 regions of the bacterial 16S rRNA gene sequences were amplified from the diluted DNA extracts with the primers (F-ACTCCTACGGGAGGCAGCAG) and (R-GGACTACHVGGGTWTCTAAT).
PCR amplification and sequencing
PCR amplification was performed in a mixture containing 2 μl of 10 × Buffer, 5 μl of target DNA sample, 1.5 μl of forward primer, 1.5 μl of reverse primer, 0.5 μl of dNTPs, 0.5 μl of magnesium ion, 0.5 μl of Taq DNA Polymerase and added dH2O to 20 μl. The reactions were hot-started at 95˚C for 3 min, followed by 27 cycles at 95˚C for 30 s, 55˚C for 30 s and 72˚C for 60 s, with a final extension step at 72˚C for 7 min. Target PCR products were detected by electrophoresis in 2% agarose gel. The products were purified and recovered by FC magic beans Kit (enlighten) and quantify by Qubit 4.0 (Thermo Fisher Scientific). The Illumina PE250 library was constructed by mixing the samples of equal volume and denatured them with NaOH. Each sample was sequenced on Miseq PE250 (Illumina).
Bioinformatics analysis: species diversity and difference analysis
Sequence reads were filtered to remove low-quality reads, including those that lack exact match with primers, contain ambiguous characters or average quality score of less than 25. High-quality reads are spliced into tags through overlap between reads. The tags with 97% similarity or more were grouped into the same operational taxonomic units (OTUs). Alpha diversity indices (richness, Chao1, Simpson, Shannon) and Beta diversity indices were analyzed using QIIME2 and results are displayed using R software.
Statistical analysis
The collected data were examined using SPSS 26.0. For measurement data such as proportions of T subsets and age, Kolmogorov–Smirnov test and Levene's T test were used to test the normality and homogeneity of variance of all variables respectively. Data conforming to normal distribution were described as mean ± standard deviation (\(\overline{x} \pm s\)) and analyzed by independent-samples t-test. Meanwhile, values that didn’t meet the normal distribution were represented as quartile and analyzed by Mann–Whitney U test. For enumeration data like gender, the Chi-square test was applied for statistical analysis. Moreover, Spearman test was used for testing the correlation between two variables. All P-values reported herein are two-tailed and P-value < 0.05 was taken as statistically significant.
Results
Demographic and clinical characteristics of participants
The mean age of 19 SLE patients was 42.79 ± 14.36 years old (female/male:15/4) and that of 16 HCs was 33.62 ± 12.63 years old (female/male:6/10). There were no statistically differences in age and gender between SLE patients and HCs (P > 0.05) (Table 1). Furthermore, clinical characteristics like erythrocyte sedimentation rate (ESR), complement 3 (C3), complement 4(C4) have been shown in Table 1.
The distribution of T lymphocyte subsets in peripheral blood is unbalanced in SLE patients
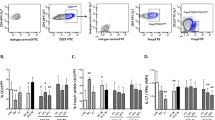
We tested the distribution of T lymphocyte subsets in peripheral blood using flow cytometry and flow chart is detailed in Fig. 1, the specific gating strategy of T lymphocyte subsets are shown in Figure S1-Figure S10 of the supplementary material. And the statistically differences of results were summarized in Fig. 2.

Flow graphs of T subsets with statistically significant differences between SLE patients and HC. A, B Flow graph of Tregs. C, D Flow graph of Tfh cells. E, F Flow graph of Naïve CD4 + T cells. G, H Flow graph of Th17 cells. I, J Flow graph of γδ T cells

Comparison of 5 T subsets with statistically significant differences between SLE patients and HCs
In particular, the percentages of Th17 cells [2.19 (0.83–4.10) vs. 1.18 (0.62–1.40), respectively, P = 0.020] and γδ T cells [1.66 (0.86–2.59) vs. 0.69 (0.50–1.75), respectively, P = 0.018] were higher in SLE patients than those in HCs group (Fig. 2a, b). In contrast, compared with HCs, SLE sufferers had lower proportions of Tregs [0.75 (0.25–1.24) vs. 2.08 (1.37–2.51), respectively, P = 0.001], Tfh cells [0.39 (0.27–0.57) vs. 0.57 (0.42–0.75), respectively, P = 0.018] and Naïve CD4+T cells [19.35 (12.12–33.50) vs. 35.24 (25.38–43.41), respectively, P = 0.004] (Fig. 2 c-e).
However, there were no significant statistical differences in proportions of CD4+T [19.88 (15.39–31.00) vs. 23.05 (18.84–33.48), respectively, P = 0.635], memory CD4+T [49.27 (34.37–60.78) vs. 35.57 (32.48–47.88), respectively, P = 0.109], CD8+T [27.35 (20.87–31.88) vs. 23.89 (21.29–27.44), respectively, P = 0.367], Naïve CD8+T [67.22 (37.89–74.34) vs. 68.94 (60.76–75.04), respectively, P = 0.403], memory CD8+T [17.37 (13.79–22.52) vs. 13.99 (11.33–25.44), respectively, P = 0.350], Tfr [0.13 (0.05–0.16) vs. 0.06 (0.04–0.14), respectively, P = 0.109] and Th1 [11.40 (8.07–26.50) vs. 16.25 (11.68–19.05), respectively, P = 0.961] between SLE patients and HCs.
ESR was negatively correlated with the proportion of Tregs
There was a significant negative correlation between Tregs and ESR (P = 0.031) in SLE patients, suggesting that ESR decreased with the increase of Tregs. And the values of other peripheral T lymphocyte subsets with statistically differences had no correlation with ESR, C3 and C4 (Table 2).
Variation of gut microbiota in SLE patients compared with HCs
We examined the alpha diversity of each sample to reflect the species richness and evenness of gut microbiota. The results showed that SLE patients had reduced community richness and obvious quantity differences in every species based on the richness index, Chao1 index and Simpson index (Fig. 3a-d). The Venn diagram that reflecting distribution of OTU clusters indicated 342 OTUs were shared, 97 OTUs were specific to HCs and only 33 were specific to SLE group (Fig. 3e). Compared with HC, 11 species of flora were discovered to be distinctly different (P < 0.05) from the boxplot diagram (Fig. 3f). These data clearly indicated a decreased richness of gut microbiota in SLE sufferers and suggested that certain microbiological changes may be associated with the pathogenesis of SLE.

The species richness and evenness of gut microbiota
The relevance between highly expressed differential flora and T peripheral lymphocyte subpopulations in SLE group
As is shown in Table 3, there were significant positive correlations between levels of Tregs and Ruminococcus2 (P = 0.042), Th17 cells and Megamonas (P = 0.009), γδT cells and Streptococcus (P = 0.004) as well as Megamonas (P = 0.003), Tfh cells and Bacteroides (P = 0.040). However, Th1 cells and Bifidobacterium were negatively correlated in these participants (P = 0.005).
The relevance between highly expressed differential flora and T peripheral lymphocyte subpopulations in HC group
As is shown in Table 4, there were significant negative correlations between Treg levels and Bacteroides (P = 0.033), Th1 cells and Streptococcus (P = 0.006), γδT cells and Ruminococcus2 (P = 0.008).
C4 was negatively correlated with the level of Unclassified_Ruminococcaceae
There was a negative correlation between Unclassified_Ruminococcaceae and C4 level but no significant correlation with ESR and C3 level. Other highly expressed differential floras had no relevance to ESR, C3 and C4 level (Table 5).
Discussion
SLE, a common immune-mediated inflammatory syndrome, is characterized by over-activity of lymphocytes, production of autoantibodies and effects on multiple organs [10]. The abnormality of T cell subsets is the main pathogenesis of SLE [5]. Five major helper T cell subsets have been identified: Th1, Th2, Th17, Treg and Tfh cells, which coordinate the immune response via providing costimulatory signals and producing cytokines [11]. Th1 cells produce cytokines such as interferon (IFN)-γ, interleukin (IL)-2 and tumor necrosis factor (TNF) to prevent intracellular bacterial infection and delayed hypersensitivity. Th2 cells produce cytokines such as IL-4, IL-5, and IL-13 to enhance humoral immunity and eliminate worms and other extracellular pathogens, which are the drivers of pathological allergic reactions [12]. Th17 cells control the transcription of IL-17 to promote the recruitment and activation of neutrophils and eliminate extracellular bacteria [13]. Compared with healthy individuals, the levels of Th1, Th2 and Th17 cytokines in SLE patients are mostly increased and positively correlated with disease activity [12]. In contrast, Tregs produces cytokines such as transforming growth factor-β (TGF-β), IL-10 and IL-35 to inhibit antigen presentation and the activation and proliferation of T and B lymphocytes [14], and the number and functional defects of Tregs are closely related to SLE. Tfh cells are identified as a special subgroup of CD4+ T cells, which regulate the differentiation of B cells and the production of autoantibodies, and have been proved to be involved in the pathogenesis of SLE [15]. According to the expression of T cell receptor (TCR), T cells can be divided into two subsets (γδ T and αβ T cells). γδ T cells are a small number of T cells composed of γ and δ chains, which can recognize non-peptide antigens. It is widely recognized that γδ T cells may shift from typical Th1-like phenotype to Th2, Th17 and Tregs in a specific microenvironment [16]. In short, imbalance of T cell subsets leads to deregulated levels of cytokine and antibody, which may be one of the multiple factors in the pathogenesis and severity of SLE.
Numerous studies have emphasized the key role of gut microbiota in the pathogenesis of SLE, including the damage of intestinal mucosal barrier, molecular imitation of autoantigens and immune imbalance [17]. Gut pathogens with proinflammatory ability can reshape the immune environment by aberrant activation of innate immune and adaptive immune system [18]. Microbial antigens lead to the differentiation of inflammatory T cell subtypes such as Th17 cells, resulting in the upregulation of pro-inflammatory cytokines such as IL-17, IL-12 and IFN. Meanwhile, it inhibits Tregs differentiation and reduced transformation of anti-inflammatory cytokines such as TGF-β and IL-10. Recent findings have shown that the imbalance of Th17 cells and Tregs is a key feature of SLE disease activity [19, 20]. Gut pathogens can also trigger over-activation of B lymphocytes with the help of Tfh cells and produce pathogenic autoantibodies, which may affect the pathogenesis of SLE [21]. Interestingly, the imbalance of gut microbiota is not only the cause of autoimmunity, but also the result of autoimmunity [17]. Genetic mutations of TCR signal can initiate autoimmunity by promoting the positive selection of auto-reactive T cells and Tfh cells as well as the production of IgG. At the same time, these mutations reduce the positive selection of microbial reactive T cells and intestinal Tfh cells as well as intestinal IgA, leading to intestinal biological disorders, which further promotes the development of SLE. Therefore, gut microbiota imbalance, immune response, and inflammation are interrelated and jointly affect the occurrence and development of SLE.
According to recent research, it is increasingly clear that altered gut microbiome composition in SLE patients and murine models are closely related to disease activity [22, 23]. To better determine the role and mechanisms of gut microbiota in SLE, we compared 19 SLE patients and 16 HCs to characterize the gut microbiota pathophysiological changes in the progression of SLE. Compared with the HCs, the gut microbiome of SLE patients showed the following characteristics. (1) Microbial composition was changed. The distribution of OTU clusters exhibited that 342 OTUs were shared, 97 OTUs were specific to HCs and only 33 were specific to SLE group. (2) Immune disorders. The main upregulated T lymphocyte in SLE patients were Th17 cells and γδ T cells, and the main downregulated T lymphocyte were Tregs and Tfh cells. (3) The gut microbiome and lymphocyte were interrelated, and Megamonas may promote the progression of SLE, and Bifidobacterium and Ruminococcus2 may improve the condition of SLE.
In this study, we observed that genus Megamonas was positively correlated with Th17 cells in SLE patients, and had no significant correlation with C3 and C4. Balmant et al. explored the difference of gut microbiome between inactive SLE women using hydroxychloroquine (HCQ) and healthy women. Megamonas is abundant in SLE females and negatively regulates C3 complement levels [24]. Additionally, a study had evaluated the association between bladder microbiome and urine metabolism, cytokines as well as disease phenotype in active patients SLE, and reported a negative correlation between the abundance of Megamonas and C3 complement levels [25]. At the same time, the abundance of Megamonas in primary Sjogren's syndrome patients was significantly more than HCs, and positively correlated with clinical disease indexes [26]. In addition, in patients with non-rheumatic diseases, Megamonas is associated with pro-inflammatory cytokines [27]. These findings suggest that Megamonas plays an important role in the disease activity of SLE.
Our results showed that Ruminococcus2 was positively correlated with the absolute number of Tregs, while Bifidobacterium was negatively correlated with Th1 cells. Some studies have also demonstrated that Ruminococcus2 was deficient in autoimmune diseases [28]. The insufficiency of Ruminococcus2 may impair immune tolerance of Tregs, which indicated Ruminococcus2 may be one of the microbes that can elicit a protective response to SLE. It is reported that Bifidobacterium decreased in the feces of SLE, which was negatively correlated with disease activity. Supplementation of Bifidobacterium can prevent over-activation of CD4 + lymphocytes and reconstruct the Treg/Th17/Th1 imbalance associated with gut microbiome [29]. Coincidentally, a study has demonstrated that some anti-inflammatory bacteria such as Bifidobacterium reduced in SLE patients, resulting in the release of inflammatory factors and increased level of systemic inflammation [30]. Not only that, short-term intervention of gut microbiota before onset can affect the treatment of prednisone in SLE mice, and the decrease of Bifidobacterium abundance may reduce therapeutic efficiency of prednisone [31]. Therefore, the regulation of gut microbiota may affect the severity, progress, and treatment of SLE, which can be used as important reference for the choice of clinical treatment of SLE.
Compared with the analysis of single microbiome dataset, our study provides additional insights by immune cell subpopulation analysis, which demonstrated that the gut microbiome and cell subsets were changed and interconnected in SLE patients compared with HCs. These associated gut microbiota of SLE are worthy to further research and development, which may provide a new idea for SLE treatment.
Availability of data and materials
Our SRA records will be accessible with the following link: https://www.ncbi.nlm.nih.gov/sra/PRJNA1085098 Or PRJNA1085098.
Abbreviations
- SLE:
-
Systemic Lupus Erythematosus
- SFB:
-
Segmented Filamentous Bacteria
- Th17:
-
Type 17 T helper
- Tregs:
-
T regulatory cells
- ACR:
-
American college of Rheumatology
- EULAR:
-
European League Against Rheumatism
- HCs:
-
Healthy Controls
- ESR:
-
Erythrocyte Sedimentation Rate
- C3:
-
Complement 3
- C4:
-
Complement 4
- IFN-γ:
-
Interferon-γ
- IL-2:
-
Interleukin-2
- TNF:
-
Tumor Necrosis Factor
- TGF-β:
-
Transforming Growth Factor-β
- TCR:
-
T Cell Receptor
- HCQ:
-
Hydroxychloroquine
References
Kiriakidou M, Ching CL. Systemic lupus erythematosus. Ann Intern Med. 2020;172(11):ITC81–96.
Herrada AA, Escobedo N, Iruretagoyena M, et al. Innate immune cells’ contribution to systemic lupus erythematosus. Front Immunol. 2019;10:772.
Al-Rayes H, Al-Swailem R, Arfin M, Sobki S, Rizvi S, Tariq M. Systemic lupus erythematosus and infections: a retrospective study in Saudis. Lupus. 2007;16(9):755–63.
Takeshima Y, Iwasaki Y, Fujio K, Yamamoto K. Metabolism as a key regulator in the pathogenesis of systemic lupus erythematosus. Semin Arthritis Rheum. 2019;48(6):1142–5.
Sharabi A, Tsokos GC. T cell metabolism: new insights in systemic lupus erythematosus pathogenesis and therapy. Nat Rev Rheumatol. 2020;16(2):100–12.
Silverman GJ, Azzouz DF, Alekseyenko AV. Systemic Lupus Erythematosus and dysbiosis in the microbiome: cause or effect or both? Curr Opin Immunol. 2019;61:80–5.
Margolis DJ, Hoffstad O, Bilker W. Association or lack of association between tetracycline class antibiotics used for acne vulgaris and lupus erythematosus. Br J Dermatol. 2007;157(3):540–6.
Kosiewicz MM, Dryden GW, Chhabra A, Alard P. Relationship between gut microbiota and development of T cell associated disease. FEBS Lett. 2014;588(22):4195–206.
Mu Q, Zhang H, Liao X, et al. Control of lupus nephritis by changes of gut microbiota. Microbiome. 2017;5(1):73.
Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365(22):2110–21.
Li H, Boulougoura A, Endo Y, Tsokos GC. Abnormalities of T cells in systemic lupus erythematosus: new insights in pathogenesis and therapeutic strategies. J Autoimmun. 2022;132:102870.
Muhammad Yusoff F, Wong KK, Mohd RN. Th1, Th2, and Th17 cytokines in systemic lupus erythematosus. Autoimmunity. 2020;53(1):8–20.
Craft JE. Follicular helper T cells in immunity and systemic autoimmunity. Nat Rev Rheumatol. 2012;8(6):337–47.
Zhang F, Cheng T, Zhang SX. Mechanistic target of rapamycin (mTOR): a potential new therapeutic target for rheumatoid arthritis. Arthritis Res Ther. 2023;25(1):187.
Jin X, Chen J, Wu J, et al. Aberrant expansion of follicular helper T cell subsets in patients with systemic lupus erythematosus. Front Immunol. 2022;13:928359.
Wu M, Yang J, Li X, Chen J. The Role of gammadelta T Cells in Systemic Lupus Erythematosus. J Immunol Res. 2016;2016:2932531.
Christovich A, Luo XM. Gut Microbiota, Leaky Gut, and Autoimmune Diseases. Front Immunol. 2022;13:946248.
Zhao T, Wei Y, Zhu Y, et al. Gut microbiota and rheumatoid arthritis: From pathogenesis to novel therapeutic opportunities. Front Immunol. 2022;13:1007165.
Talaat RM, Mohamed SF, Bassyouni IH, Raouf AA. Th1/Th2/Th17/Treg cytokine imbalance in systemic lupus erythematosus (SLE) patients: correlation with disease activity. Cytokine. 2015;72(2):146–53.
Zhao C, Chu Y, Liang Z, et al. Low dose of IL-2 combined with rapamycin restores and maintains the long-term balance of Th17/Treg cells in refractory SLE patients. BMC Immunol. 2019;20(1):32.
Cristofori F, Dargenio VN, Dargenio C, Miniello VL, Barone M, Francavilla R. Anti-inflammatory and immunomodulatory effects of probiotics in gut inflammation: a door to the body. Front Immunol. 2021;12:578386.
Ma Y, Xu X, Li M, Cai J, Wei Q, Niu H. Gut microbiota promote the inflammatory response in the pathogenesis of systemic lupus erythematosus. Mol Med. 2019;25(1):35.
Luo XM, Edwards MR, Mu Q, et al. Gut microbiota in human systemic lupus erythematosus and a mouse model of lupus. Appl Environ Microbiol. 2018;84(4):e02288–17.
Balmant BD, Fonseca DC, Prudencio APA, et al. Megamonas funiformis, plasma zonulin, and sodium intake affect C3 complement levels in inactive systemic lupus erythematosus. Nutrients. 2023;15(8):1999.
Liu F, Du J, Zhai Q, et al. The bladder microbiome, metabolome, cytokines, and phenotypes in patients with systemic lupus erythematosus. Microbiol Spectr. 2022;10(5):e0021222.
Li Y, Li Z, Sun W, Wang M, Li M. Characteristics of gut microbiota in patients with primary Sjogren’s syndrome in Northern China. PLoS ONE. 2022;17(11):e0277270.
Ling Z, Jin C, Xie T, Cheng Y, Li L, Wu N. Alterations in the fecal microbiota of patients with HIV-1 infection: an observational study in a Chinese population. Sci Rep. 2016;6:30673.
Wang T, Sternes PR, Guo XK, Zhao H, Xu C, Xu H. Autoimmune diseases exhibit shared alterations in the gut microbiota. Rheumatology (Oxford). 2024;63(3):856–65.
Lopez P, de Paz B, Rodriguez-Carrio J, et al. Th17 responses and natural IgM antibodies are related to gut microbiota composition in systemic lupus erythematosus patients. Sci Rep. 2016;6:24072.
Li Y, Wang HF, Li X, et al. Disordered intestinal microbes are associated with the activity of Systemic Lupus Erythematosus. Clin Sci (Lond). 2019;133(7):821–38.
Zhang Y, Liu Q, Yu Y, Wang M, Wen C, He Z. Early and short-term interventions in the gut microbiota affects lupus severity, progression, and treatment in MRL/lpr mice. Front Microbiol. 2020;11:628.
Acknowledgements
Not applicable.
Funding
This study was supported by the Natural Science Research Project of Shanxi Province, Grand/Award Number: 20210302123275.
Author information
Authors and Affiliations
Contributions
Methodology and experiments: Fen-Ping Lian, Fen Zhang, Chun-Miao Zhao, Xu-Xia Wang; Data curation: Yu-Jie Bu, Xing Cen, Gui-Fang Zhao; Project administration: Chun-Miao Zhao, Jun-Wei Chen; Resources: Jun-Wei Chen; Writing–original draft: Fen-Ping Lian, Fen Zhang; Writing–review & editing: Fen-Ping Lian, Fen Zhang, Sheng-Xiao Zhang, Jun-Wei Chen. All authors read and approved the final manuscript. Fen-Ping Lian and Fen Zhang contribute equally to the article.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Each participant signed the informed consent and our study was approved by Ethics Committee of Second Hospital of Shanxi Medical University (Ethics Number: 2023YX246).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lian, FP., Zhang, F., Zhao, CM. et al. Gut microbiota regulation of T lymphocyte subsets during systemic lupus erythematosus. BMC Immunol 25, 41 (2024). https://doi.org/10.1186/s12865-024-00632-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12865-024-00632-0