
Abstract
Acinetobacter baumannii is a gram-negative bacillus prevalent in nature, capable of thriving under various environmental conditions. As an opportunistic pathogen, it frequently causes nosocomial infections such as urinary tract infections, bacteremia, and pneumonia, contributing to increased morbidity and mortality in clinical settings. Consequently, developing novel vaccines against Acinetobacter baumannii is of utmost importance. In our study, we identified 10 highly conserved antigenic proteins from the NCBI and UniProt databases for epitope mapping. We subsequently screened and selected 8 CTL, HTL, and LBL epitopes, integrating them into three distinct vaccines constructed with adjuvants. Following comprehensive evaluations of immunological and physicochemical parameters, we conducted molecular docking and molecular dynamics simulations to assess the efficacy and stability of these vaccines. Our findings indicate that all three multi-epitope mRNA vaccines designed against Acinetobacter baumannii are promising; however, further animal studies are required to confirm their reliability and effectiveness.
Similar content being viewed by others

Introduction
First identified as a new species in 1986 [1], Acinetobacter baumannii is a gram-negative bacillus implicated in various infections, including soft tissue and urinary tract infections, septicopyemia, bacteremia, pneumonia, and meningitis [2]. Improper treatment of these infections can result in elevated mortality rates. Despite being recognized for decades, Acinetobacter baumannii remains a significant member of the ESKAPE group [3], consistently presenting a formidable challenge as a major pathogen causing difficult-to-treat nosocomial infections worldwide [4].
The increasing resistance to carbapenems in Acinetobacter baumannii is partly attributed to its highly variable capsular polysaccharide, which enhances its ability to evade immune system attacks and resist certain antibacterial drugs and disinfectants [5]. Due to its escalating threat, the World Health Organization has designated Acinetobacter baumannii as a critical research priority, emphasizing the urgent need for new drug development [6].
Research has shown that early immune responses in individuals infected with Acinetobacter baumannii involve key factors such as neutrophils, macrophages, antimicrobial peptides, and the complement system [7]. Neutrophils play a crucial role in pathogen elimination through phagocytosis, degranulation, or NETosis, leading to the generation of reactive oxygen species (ROS). Macrophages, on the other hand, produce nitric oxide and secrete inflammatory cytokines and chemokines [8]. Despite these immune defenses, Acinetobacter baumannii can evade host defenses through various virulence factors and mechanisms. For instance, its capsular polysaccharide impedes the interaction between neutrophils and the negatively charged surfaces of macrophages, preventing effective phagocytosis. Additionally, outer membrane protein A interacts with factor H, inhibiting complement-mediated killing. Consequently, targeting these components with drugs is not only feasible but imperative for effective intervention.
In recent years, numerous studies have focused on developing vaccines against Acinetobacter baumannii. Many of these studies have concentrated on whole-cell antigens of Acinetobacter baumannii [9], while others have explored vaccines targeting its outer membrane vesicles [10, 11]. Additionally, various studies have designed vaccines for important surface antigens such as OmpA [12], Bap [13], FilF, and QnrA. These new vaccines have demonstrated the ability to elicit specific immune responses against these key targets, thereby preventing Acinetobacter baumannii infections. However, none of these vaccines have yet shown the capability to induce a sufficient immune response for use in clinical trials. Therefore, the development of novel vaccines against Acinetobacter baumannii remains an urgent priority.
Vaccines represent a powerful strategy for preventing bacterial infections; however, a comprehensive and effective vaccine targeting Acinetobacter baumannii is currently unavailable. Traditional vaccine development typically involves whole organisms or large proteins, which can sometimes trigger unnecessary allergic reactions or excessively strong immune responses [14]. Furthermore, due to the diversity of Acinetobacter baumannii strains, extensive antigen variation, and strong resistance to most antibiotics, in vitro culture is relatively challenging [15]. As a result, traditional biochemical, serological, and microbial methods for directly identifying relevant antigen targets are inefficient, time-consuming, and require significant resources and funding. Furthermore, the traditional approach is time-consuming and its therapeutic effects can be uncertain. Recently, a novel type of vaccine, known as multi-epitope vaccines, has emerged. These vaccines have shown promising immune responses and fewer side effects in various animal studies and early clinical trials. Incorporating informatics into vaccine research and development, as well as immunization processes, can reduce costs, improve efficiency, save time, and facilitate the design of more effective vaccines [16]. However, despite their potential, synthetic peptide and DNA vaccines often suffer from low immunogenicity, and DNA vaccines may pose risks of mutagenic alterations [17].
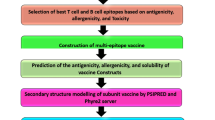
Fortunately, mRNA vaccines can overcome these issues and be produced economically and rapidly at scale. Therefore, the primary objective of our research was to develop multi-epitope mRNA vaccines against Acinetobacter baumannii, incorporating elements such as CTL, HTL, LBL, and others. Initially, we retrieved the amino acid sequences of target proteins from the NCBI and UniProt databases and selected epitopes based on characteristics such as antigenicity and toxicity. Suitable adjuvants were then integrated with the identified T cell and B cell epitopes using various linkers to formulate the vaccines. The structure and physicochemical properties of the constructs were validated using various network tools. Molecular docking and molecular dynamics simulations were performed to elucidate the docking patterns and assess stability. Finally, we optimized the codons, constructed the mRNA vaccines, and evaluated the immunogenicity of the constructs using different analytical tools.
Materials and methods
Target protein sequence screening
Protein sequence retrieval and screening
The protein sequences of AmpD (WP_000810002.1), OmpA (WP_000777878.1), Pal (EEX03100.1, 2009, GenBank), BauA (WP_001073039.1), Omp34 (EEX02037.1), BamA (EEX04588.1), Omp22 (WP_001202415.1), CsuA/B (EEX04344.1), OmpK (WP_001176275.1) and Dcap (EEX05238.1) of Acinetobacter baumannii strain ATCC19606 were retrieved in FASTA format from the National Center for Biotechnology Information Database (NCBI, https://www.ncbi.nlm.nih.gov/).
Prediction and selection of epitope
Prediction and selection of Cytotoxic T lymphocyte epitope
Cytotoxic lymphocytes (CTLs), a subset of white blood cells, are specialized T cells that secrete various cytokines and play a crucial role in immune functions. They are essential for eliminating viruses, cancer cells, and other antigenic substances, serving as a key defense mechanism in antiviral and anti-tumor immunity, alongside natural killer cells.
The NetCTL 1.2 server (https://services.healthtech.dtu.dk/services/NetCTL-1.2/) was used to predict CTL epitopes within protein sequences. The latest version, 1.2, can predict epitopes for 12 major MHC-I supertypes. Epitopes with combined scores greater than 0.75 were selected for further analysis, including allergenicity, antigenicity, and immunogenicity tests.
The VaxiJen v2.0 server (https://ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html/) is a precise tool for predicting protective antigens and subunit vaccines without requiring sequence alignment. We set the bacterial threshold value at 0.4 to reflect the capacity of sequences to bind with specific antibodies and CTLs. Recognizing that antigenicity alone does not ensure efficacy, we also considered safety aspects, such as minimizing toxicity and allergenicity. To address these concerns, we used AllerTOP v2.0 (https://ddg-pharmfac.net/AllerTOP/) to predict allergenicity and ToxinPred (https://webs.iiitd.edu.in/raghava/toxinpred/index.html) to predict virulence. Sequences that were non-allergic, non-toxic, and had an antigenicity score of ≥ 0.4 were selected for further assessment of immunogenicity.
Unlike antigenicity, which refers to the ability of an antigen to be recognized by the immune system, immunogenicity pertains to the antigen's ability to induce an immune response in the host. To predict immunogenicity, we utilized the Class I Immunogenicity tool (http://tools.iedb.org/immunogenicity/) from the Immune Epitope Database & Tools (IEDB, https://iedb.org/).
To prevent epitopes from being cleaved by signal peptidase, we used SignalP-6.0 (https://services.healthtech.dtu.dk/services/SignalP-6.0/), which predicts the presence of signal peptides and their cleavage sites. Additionally, we employed the DeepTMHMM server (https://dtu.biolib.com/DeepTMHMM) for deep learning-based prediction of the membrane topology of transmembrane proteins. This approach allowed us to recheck epitope locations, predict transmembrane helix domains, and select epitopes that are free from predicted transmembrane regions and signal peptides.
Recognizing that conserved epitopes typically offer greater protective effects, we used Epitope Analysis Tools in IEDB (http://tools.iedb.org/conservancy/) to assess the conservation or variation of specific peptides within a given protein sequence set. Sequences with an identity threshold of ≥ 95% were selected to identify highly conserved epitopes.
Prediction and selection of helper T lymphocyte epitope
Helper T lymphocytes (HTLs) play a crucial role in stimulating antibody production by activating complement pathways and producing cytokines and chemokines, thereby significantly contributing to immune responses.
The MHC II Binding Tool (http://tools.iedb.org/mhcii/) from the Immune Epitope Database (IEDB) is used for HTL epitope prediction, utilizing the NetMHCIIpan-4.1 server, which includes separate predictors for binding and elution. We selected a comprehensive HLA reference set covering various alleles such as HLA-DRB101:01, HLA-DRB103:01, HLA-DRB104:01, HLA-DRB104:05, and others. Default values for peptide lengths were used in the predictions.
Predicted peptides were then evaluated for antigenicity, immunogenicity, toxicity, and allergenicity using VaxiJen v2.0 (https://ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html/), ToxinPred (https://webs.iiitd.edu.in/raghava/toxinpred/index.html), IEDB Class I Immunogenicity (https://webs.iiitd.edu.in/raghava/toxinpred/index.html), and AllerTOP v2.0 (https://ddg-pharmfac.net/AllerTOP/).
Interferon plays a vital role in the maturation and differentiation of antigen-presenting cells, thus enhancing the immune response. Therefore, it is essential to confirm the potential of our epitopes to induce interferon. The IFNepitope server (http://crdd.osdd.net/raghava/ifnepitope/) predicts the potential of peptides to induce IFN-γ. Additionally, IL-4 is critical for regulating immune cell formation and differentiation, and its effects can be counteracted by interleukin-10 (IL-10). The potential of peptides to induce IL-4 and IL-10 was predicted using IL4pred (http://crdd.osdd.net/raghava/il4pred/) and IL-10pred (http://crdd.osdd.net/raghava/IL-10pred/). Transmembrane helix domains and signal peptides were predicted using the DeepTMHMM server (https://dtu.biolib.com/DeepTMHMM), and peptide conservativeness was assessed through Epitope Analysis Tools in IEDB (http://tools.iedb.org/conservancy/).
Prediction and selection of B cell epitope
B cell epitopes, located on the surface of antigen protein molecules, are capable of binding to B cell receptors (BCR) and antibodies. Linear B cell (LBL) epitopes are characterized by their sequential arrangement in the amino acid sequence.
To identify LBL epitopes, we used the ABCpred network server (https://webs.iiitd.edu.in/raghava/abcpred/ABC_submission.html/), which employs a trained recurrent neural network (RNN) with a window length of 16 amino acids and a threshold value of 0.51. The predicted peptides were then subjected to a comprehensive analysis, including assessments of antigenicity, toxicity, immunogenicity, and allergenicity using VaxiJen v2.0 (https://ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html/), ToxinPred (https://webs.iiitd.edu.in/raghava/toxinpred/index.html), IEDB Class I Immunogenicity (https://webs.iiitd.edu.in/raghava/toxinpred/index.html), and AllerTOP v2.0 (https://ddg-pharmfac.net/AllerTOP/).
Additionally, we predicted transmembrane helix domains and signal peptides using the DeepTMHMM server (https://dtu.biolib.com/DeepTMHMM). The conservativeness of the epitopes was assessed using IEDB (http://tools.iedb.org/conservancy/).
Population coverage
Predicting the population coverage of MHC alleles is crucial for assessing the effectiveness of the constructed vaccines, as it depends on the distribution and expression frequency of HLA alleles that interact with the vaccine constructs. To achieve this, we used the population coverage tool from IEDB (http://tools.iedb.org/population/) to forecast coverage. Peptides that provided coverage of at least 5% of the global population were then selected for further analysis.
Molecular docking of peptides and HLA alleles
We utilized the SWISS-MODEL server (https://swissmodel.expasy.org/interactive) for homologous modeling of our selected MHC molecules, employing ProMod3, an in-house comparative modeling engine based on Open Structure. Additionally, PEP-FOLD3 (https://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD3/) was used to predict peptide structures from amino acid sequences, utilizing a structural alphabet (SA letters) to describe the conformations of four consecutive residues. The MDockPep server (https://zougrouptoolkit.missouri.edu/MDockPeP/) was employed to predict HLA allele-peptide complexes, starting with the HLA allele protein structure and peptide sequence. This server ranked the predicted models using the knowledge-based scoring function, ITScorePeP. Finally, we used LigPlus, a virtual screening software, to screen compounds against potential drug targets.
Design of multi-epitope vaccine constructs
β-Defensin 3 is a molecule featuring three β-strands and a short helix in its N-terminal region [18]. It is known for its antibacterial activity against both Gram-negative and Gram-positive bacteria and its immunomodulatory effects, which include recruiting naive T cells and immature dendritic cells to the site of infection [19]. RS09, a widely utilized synthetic TLR-4 agonist, is recognized for inducing strong immune activation and enhancing antibody production when used as an adjuvant [20]. Additionally, the Cholera Toxin B subunit (CTB) acts as an effective adjuvant by binding to antigen-presenting cells (APCs), improving stability, extending half-life, and boosting immune activation [21]. These three adjuvants have been extensively applied in multi-epitope vaccine design and have shown significant activity in eliciting immune responses.
In constructing the vaccine sequences, the existing signal-peptide sequences in these adjuvants were removed. The treated peptides were then combined with the PADRE peptide [22], a universal synthetic peptide that activates antigen-specific CD4 + T cells and initiates innate immune responses. The PADRE peptide has been shown to enhance therapeutic responses to MHC I-restricted epitopes by increasing IFN-γ production. The PADRE sequence was linked to the three adjuvants using an EAAAK linker. After removing the signal-peptide sequence, methionine was reintroduced at the N-terminus, as methionine is typically the first amino acid in polypeptide chains synthesized by eukaryotic or prokaryotic ribosomes.
The protein and CTB sequences were fused using EAAAK, GPGPG, and AAY linkers to create a chimeric protein, which significantly boosts the vaccine's immunogenicity and enhances immune responses. Subsequently, CPGPG linkers were used to connect HTL epitopes, AAY linkers for CTLs, and KK linkers for LBLs. The last HTL epitopes were linked to the first CTL epitopes via AAY linkers, while the final CTL epitopes were connected to the first LBL epitopes using KK linkers. Finally, a "6xHis tag" was appended to the C-terminal of the vaccine construct to facilitate protein purification without affecting the functionality of the fusion proteins. And we named the vaccine construct, which is bound with CTB, ABV1. Vaccine bound with RS09 named ABV2, and vaccine bound with β-defensin 3 named ABV3.
Prediction of biophysical properties and biochemical characteristics of the designed vaccine constructs
In the design of these three constructs, we assessed their antigenicity using both Vaxijen and ANTIGENpro (http://scratch.proteomics.ics.uci.edu/). Then, the allergenicity and toxicity of the vaccine was evaluated via AllerTop v.2.0 and ToxinPred2 server, respectively. And various physicochemical properties such as molecular weight, theoretical isoelectric point (pI), half-value period, instability index, aliphatic index, and grand average of hydropathicity (GRAVY) index of the vaccine was predicted by ProtParam (https://web.expasy.org/protparam/). Subsequently, the SOLpro was employed to predict the protein's solubility (https://scratch.proteomics.ics.uci.edu/).
Secondary structure prediction, tertiary structure modeling, refinement and validation
The PSIPRED4.0 server (http://bioinf.cs.ucl.ac.uk/psipred/) was employed to establish the secondary structure of vaccines using position-specific scoring matrices, ensuring high precision. Subsequently, Colabfold v1.53 (https://alphafold.com/) was utilized for the prediction and optimization of the tertiary structures of the vaccines. The most superior model for each vaccine was selected and then committed to the ERRAT and PROCHECK tools on the structure validation services SAVS v6.0 (https://saves.mbi.ucla.edu/). Additionally, the models were submitted to the ProSA-Web (https://prosa.services.came.sbg.ac.at/prosa.php) server to evaluate the Z-score, ensuring the quality and validation of the predicted structures.
Molecular docking between designed vaccine and receptors
Molecular docking of the vaccine construct and the receptors facilitated understanding of the binding mode between the two molecules. Toll-like receptor 4 (TLR4), a member of the Toll-like receptor family, plays a role in innate immunity and mediates inflammatory responses by recognizing PAMPs such as lipopolysaccharide, bacterial endotoxins, or lipoteichoic acid [23]. In addition to TLR4, we explored the molecular docking of our designed vaccine with HLA-A02:01 and HLA-DRB101:01. Afterwards, we utilized the ClusPro2.0 server [24] to evaluate the binding affinity and employed the HADDOCK2.4 server to refine the complexes.
Molecular dynamics simulation and analysis
MD simulation
Utilizing Gromacs v2023.3 software, we conducted MD simulations to predict the dynamics and stability of receptors-vaccine docking complexes [25]. At the initial stage, the AMBER14SB_ parmbsc1force field was applied to generate the coordinate file and topology of the complexes. Subsequently, these complexes underwent solvation by introducing Transferable Intermolecular Potential 3P (TIP3P) water molecules and counter ions (Na + and Cl-) to neutralize the charge of the simulated system [26]. Following this, we minimized the energy. To maintain a constant temperature isopiestic pressure environment, we set NVT simulations in 200 ps and NPT simulations in 1 ns, ensuring equilibration of temperature and pressure to 310 K and 1 atm. The MD simulation was then conducted for 100 ns in an isothermal and isopiestic environment. Lastly, the LINCS algorithm was employed to constrain hydrogen bonds, and the Particle-Mesh Ewald summation scheme was utilized for the calculation of long-range electrostatic interactions.
MM-PBSA
The final 10 ns of the trajectory were isolated and then input into the gmx_MMPBSA v1.62 tool [27]. This tool was used to compute estimates of binding free energy between the vaccine and the receptors.
Immune simulation
We utilized the C-IMMSIM v10.1 tool (https://kraken.iac.rm.cnr.it/C-IMMSIM/) to evaluate the potential immune efficacy of our constructed vaccine. It employs Position Specific Scoring Matrix (PSSM) methods to recognize epitopes and can synchronously simulate the immune reactions in 3 compartments representing keest, thymus gland and lymphedema of mammals to epitopes [28]. Our simulation time steps were configured at 1050 (with 1 time stage equivalent to 8 h), and each injection was scheduled at time step-1, step-84, and step-168. The default injection dose for each administration was set to 1,000, with a four-week time interval between each dose, following the most recommended settings.
Multi-epitope mRNA construction and secondary structure prediction
To construct an effective, conclusive and verifiable mRNA vaccine, we attached signal peptide tissue plasminogen activator(tPA) [29] to the 5’end of DNA sequence with the connection of EAAAK, and MHC I targeting domain(MITD) sequence [30], which was contributed to induce the presentation of CTL epitope, to the 3’end of the DNA sequence with EAAAK as well. Then used the EMBOSS (http://emboss.open-bio.org/) server to process reverse translation and optimize this construct. After that, added Kozak sequence [31], which helped to facilitate extracellular secretion, to the completed verified DNA sequence. And attached 5’ untranslated region and 3’ untranslated region to the beginning of the sequence and the ending of the sequence, with a TAA codon ends the translation in front of 3’UTR [32, 33]. And finally, added the restriction site GGATCC to the beginning and CTCCAG to the end. Afterwards, we applied the Biochen (https://www.biochen.org/cn/tools/transform_sequence) server to translate the DNA sequence to mRNA sequence and utilized the RNAfold server(http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) to predict the secondary structure.
Codon optimization and in silico cloning
In order to enhance the expression of the designed vaccines in the chosen host, we subjected our construct to codon optimization by using EMBOSS (http://emboss.open-bio.org/), choosing E. coli (Strain K12) as the expression host. We assessed the CAI-Value and GC-Content of the optimized sequence. Following this, we introduced XhoI and BamHI restriction endonuclease sites at the C-terminal and N-terminal of the vaccines, respectively. Ultimately, the refined sequence was incorporated into the pET-28a ( +) vector using GenSmart(https://www.genscript.com/gensmart-design/), completing the modification process (Fig. 1).

Graphical abstract
Results
Protein sequence retrieval
Previous investigations into Acinetobacter baumannii have identified numerous protein targets, including AmpD, OmpA, Pal, BauA, Omp34, BamA, Omp22, CsuA/B, OmpK, and Dcap, which have been recognized as promising therapeutic targets for vaccine design (Supplementary Table 1) [9, 34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71]. We retrieved their sequences from the reference standard Acinetobacter baumannii strain ATCC19606, and their UniProtKB entries and amino acid sequences are presented in Table 1.
Epitopes selections
The immune system comprises diverse cell types working collectively for health maintenance. In our study, we considered CTL, HTL, and LBL. CTL primarily mediates cellular immunity, inducing target cell death and clearing circulating antigens. Upon activation, B cells undergo differentiation into plasma cells, a pivotal process contributing to the initiation of humoral immune responses and the production of antibodies targeted against pathogens. Another category of immune cells known as Helper T cells (HTLs) plays a significant role by bolstering both humoral and cellular immunity through the secretion of diverse cytokines. In order to construct a great effective multi-epitope design, considerations encompass CTLs, HTLs and LBLs. Following predictions of antigenicity, immunogenicity, toxicity, allergenicity, and the ability to induce IFN-γ, IL-4, and IL-10, while ensuring signal peptide and transmembrane helix presence, we identified 8 optimal HTL epitopes (Table 2), 8 LBL epitopes (Table 3), and 8 CTL epitopes (Table 4).
Population coverage prediction
In accordance with the HLA alleles that bind to T cell epitopes (Supplementary Table 2, Supplementary Table 3), we conducted predictions of population coverage to assess the proportion of the population in various regions worldwide. As illustrated in Fig. 2 and Table 5, Table 6, the formulated vaccines were identified as effective in these areas for combating Acinetobacter baumannii.

The population coverage of vaccine
Structure and characteristics of multi-epitope vaccine constructs
The final sequences of our vaccines are presented in Fig. 3, with their characteristics detailed in Table 7. According to the predictions from VaxiJen 2.0 and ANTIGENpro, all three vaccines exhibit significant antigenicity. Results from ToxinPred2 and AllerTop 2.0 indicate that they are non-toxic and non-allergenic. With instability indices of 11.79, 16.24, and 14.61, these vaccines demonstrate stability. The aliphatic indices of 66.2, 70.33, and 66.07 indicate dynamic thermal stability. Furthermore, these vaccines boast prolonged half-time periods, and SOLpro predictions confirm their excellent solubility. And the plot about the signal peptide prediction of the vaccine construct was showed in Supplementary Fig. 1.

A, B, C, D, E The structure and characteristics of ABV1. F, G, H, I, J The structure and characteristics of ABV2. K, L, M, N O The structure and characteristics of ABV3. A, F, K The 3D structure of vaccine. B, G, L The amino acid sequences of the vaccine construct. The adjuvants are tagged in red, the Pan HLA DR-binding epitope (PADRE) tagged in yellow, the CTL epitopes are tagged in green, the HTL epitopes are tagged in orange, the LBL epitopes tagged in blue and the linkers in black. C, H, M The secondary structure of the vaccine construct. D, I, N The Ramachandran plot depicting the refined 3D model, as generated through the PROCHECK server, highlights various regions. In this representation, the red-colored areas denote the most favored regions, while the dark yellow and light–yellow regions represent additional allowed and generously allowed regions, respectively. Meanwhile, the white regions signify disallowed regions. E, J, O The Z-score plot of the refined 3D model generated by the ProSA-web server
Secondary structure prediction, tertiary structure modeling, refinement and validation
The PSIPRED4.0 server analysis indicated that the ABV1 consists of 52.93% coil, 27.62% α-helix, and 19.45% β-strand (Fig. 3C). Similarly, the ABV2 comprises 58.98% coil, 24.35% α-helix, and 16.49% β-strand (Fig. 3H), while the ABV3 exhibits 60.71% coil, 20.48% α-helix, and 18.81% β-strand (Fig. 3M). Subsequently, the structural integrity of these three vaccines was validated using the SAVESv6.0 and ProSA-web servers.
In the Ramachandran plot, the ABV1 demonstrated that 94.2% of its residues occupied the most favored regions, with 4.8% in additional allowed regions and a mere 1.0% in generously allowed regions (Fig. 3D). The ERRAT score for ABV1 achieved a perfect 100, and the ProSA-web Z-score was -5.32 (Fig. 3E). Meanwhile, for the ABV2, the Ramachandran plot illustrated that 93.7% of residues were in the most favored regions, 5.3% in additional allowed regions, and only 0.3% in generously allowed regions (Fig. 3I). The ERRAT score for ABV2 was 94.3182, accompanied by a ProSA-web Z-score of -3.69 (Fig. 3J). Furthermore, the ABV3 showcased a composition of 97.0% residues in the most favored regions, 2.7% in additional allowed regions, and only 0.3% in generously allowed regions (Fig. 3N). The ERRAT score for vaccine-β-defensin3 was 97.4359, with a ProSA-web Z-score of -3.59 (Fig. 3O).
The fact that over 90% of residues are situated in the most favored regions serves as a strong indicator of the exceptional quality of the vaccine models. The ERRAT score, differentiating correct and incorrect regions, and the ProSA-web Z-score, indicating overall model quality, both confirm the reliable and stable 3D structure of the developed vaccine.
Molecular docking
Molecular docking of the T-cell epitopes with MHC molecules
Except for a few HTL epitopes such as (NHIAGHSDIAPGRKT), (FEAEYNQVKGDVDGA), (DGVTRGVNVSTAVGI), (VGATFVGNDGEADIK), and (GAAAGYGISKSNANS), other epitopes exhibited diverse bindings. Some epitopes even demonstrated a remarkably large number, reaching up to 19 alleles (AAANAGVTV). In our investigation, we conducted docking analyses for each T cell epitope with all corresponding alleles. The ITScorePeP, a statistical potential-based scoring function tailored for protein-peptide dockings, considers both protein-peptide interactions (inter-score) and interactions among non-neighboring residues within the peptide (intra-score) [72]. The scores obtained indicated a strong affinity between T cell epitopes and HLA alleles. (Table 8, Supplementary Table 4, Supplementary 5).
Lepus predictions further illustrated the presence of abundant hydrogen bonds and salt bridges between T cell epitopes and HLA alleles (Fig. 4, Fig. 5). These results convincingly support the notion that T cell epitopes effectively bind with MHC molecules, emphasizing their potential in eliciting immune responses (Supplementary Table 6, Supplementary Table 7).

The plot of visualized analysis of the docking complex: the CTL epitopes docking with HLA alleles, (A, C, E, G) The plot of visualized analysis of the docking complex: the CTL epitopes docking wit) the 3D structure of CTL-HLA docking complexes, (B, D, F, H) the interaction forces between CTL-HLA complexes

The plot of visualized analysis of the docking complex:the HTL epitopes docking with HLA alleles, (I, K) the 3D structure of HTL-HLA docking complexes, (J, L) the interaction forces between HTL-HLA complex
Molecular docking of the vaccines with receptor molecules
We utilized the Cluspro2.0 server to evaluate the binding affinity between the vaccine construct and receptor molecules (TLR4, HLA-A02:01, and HLA-DRB101:01). The server generated numbers of candidate models for each complex, and these complexes showed individual binding energies. The complex whose binding energy score was the lowest would be selected for further refinement. Subsequently, we employed the HADDOCK 2.4 server to improve the complexes.
The HADDOCK scores for the ABV1-A2 complex (Fig. 6A), ABV1-DRB1 complex (Fig. 6G), and ABV1-R4 complex (Fig. 6M) were -477.6 ± 5.8, -718.4 ± 9.0, and -301.2 ± 0.6, respectively. For the ABV2-A2 complex (Fig. 6C), ABV2-DRB1 complex (Fig. 6I), and ABV2-R4 complex (Fig. 6O), the HADDOCK scores were -443.8 ± 4.9, -721.5 ± 2.6, and -358.9 ± 2.9. Lastly, the HADDOCK scores for the ABV3-A2 complex (Fig. 6E), ABV3-DRB1 complex (Fig. 6K), and ABV3-R4 complex (Fig. 6Q) were -490.0 ± 8.0, -692.5 ± 4.2, and -310.0 ± 2.1. The consistently low HADDOCK scores indicated the stable binding between vaccines and receptors (Table 9 Table 10).

Docking analysis of Vaccine-receptors docking complex (A, C, E, G, I, K, M, O, Q) The 3D structure of vaccine-receptors docking complex. B, D, F, H, J, L, N, P, R) In the analysis of the interactions inside the complex, the connections between the amino acids colored in red refer to salt bridges, yellow refers to disulphide bonds, blue refers to hydrogen bonds and orange dotted line refers to non-bonded contacts
Moreover, the hydrogen bonds and salt bridges formed between the constructs and receptors were documented in Table 11. This comprehensive data affirms the robust interactions between the designed construct and the receptors (TLR4, HLA-A02:01, and HLA-DRB101:01).
MD simulation
Molecular Dynamics (MD) simulation, employing Gromacs v2022.1, was carried out to evaluate the atomic-level stability of complexes. A thorough examination of trajectories included the computation of Backbone root-mean-square deviation (RMSD, Fig. 7C, F, I), radius of gyration (Rg, Fig. 7A, D, G), root mean square fluctuation (RMSF, Supplementary Fig. 2), and the determination of hydrogen bond quantities (Fig. 7B, E, H).

The analysis of MD simulation of complexes. The red curve refers to ABV1, the blue refers to ABV2 and the black ones refers to ABV3 A, D, G The radius of gyration of the complexes. B, E, H) Hydrogen bonds of the complexes. C, F, I Root mean square deviation plots of the complexes
RMSD, reflecting the conformational fluctuations of complexes on account of the primary structure, demonstrated a gradual increase in value between 1–20 ns, followed by stabilization. This leveling off of the RMSD curve indicated equilibration of the protein structure across all vaccine-receptor complexes(22).
Rg, characterizing the compactness of protein structure, exhibited relatively constant values after 30 ns for all nine curves in our study. The Rg value of β-HLA-DRB1*01:01 stabilized later, at 50 ns, suggesting a consistent distribution of atoms around the axis [73].
The vaccine-receptor complexes initially formed about 10–20 hydrogen bonds, increasing after 20 s and subsequently maintaining stability in all these simulations. The consistent presence of hydrogen bonds indicated a strong affinity between the vaccine and receptors.
RMSF, measuring individual residue flexibility, revealed higher values for residues 250–350 of the β-receptor complex in the first simulation, signifying increased flexibility. Notably, residues 250–360, which did not interact with the receptor, displayed higher flexibility during the first simulation due to the less compact structure of the vaccine.
MM-PBSA calculation
We performed MM-PBSA calculations on trajectories extracted at 100 ps intervals over the last 10 ns of the simulation period. These calculations evaluated diverse binding interaction energies, including van der Waals, electrostatic, polar solubility, and the overall binding energy (ΔGbinding) of the complex. The detailed energy analysis, as outlined in Table 12, underscored the vaccine's substantial affinity for all three receptor molecules.
Immune simulation
The immune stimulation process was conducted using the C-ImmSim web server, revealing the vaccine's potential to induce robust adaptive immunity in the host. Following the initial injection, a decline in lgM antibody titers indicated a predominant B-cell isotype lgM population, accompanied by the detection of a modest B-cell isotype lgG population. Subsequent injections resulted in heightened levels of IgM antibodies, along with IgG1, IgG1 + IgG2, and lgG + lgM (Fig. 8B, E, H). Additionally, a substantial increase in memory B cells was observed (Fig. 7C, F, I). The immune simulation also noted the activation of TC cells, NK cells, and macrophages upon exposure to the vaccine. The vaccine exhibited the capacity to stimulate various cytokines (CKs, Fig. 8A, D, G), including IFN-γ, IL-4, and IL-1. In summary, our vaccine demonstrated the ability to induce the production of a significant quantity of immune cells and a diverse array of CKs, signifying its successful elicitation of both adaptive immunity and memory immunity in the host. The whole immune simulation results can be found in Supplementary Fig. 3, Supplementary Fig. 4 and Supplementary Fig. 5.

In silico immune simulation spectrum. A, D, G The activation of cytokines including IFN-γ, IL-4, TGF-β. B, E, H Variations in antibody titers (C, F, I) Variations in B cell population
Multi-epitope mRNA vaccine construction and in silico cloning
We performed reverse translation and codon optimization utilizing EMBOSS, after optimization. Our three vaccines run the length of 2262 bp(ABV2), 2253 bp(ABV1) and 2379(ABV3), including full length as well as tPA, MITD, TTA codon and UTR sequences. And their CAI values were 0.99365002437207(ABV2), 0.9944592969051331(ABV1), 0.9940022424132773(ABV3) and the GC contents were 51.181498240321766(ABV2), 50.526315789473685(ABV1), 50.902184235517566(ABV3). Notably, both the Codon Adaptation Index (CAI) value and the GC content for each vaccine fell within desirable ranges, indicative of a high potential for gene expression and excellent expression capability in E. coli (K12 strain). Then we used RNAfold to predict the free energy of the secondary structure of ABV2 (Supplementary Fig. 6A, D), ABV1 (Supplementary Fig. 6B, E) and ABV3 (Supplementary Fig. 6C, F) were -770.08 kcal/mol, -835.63 kcal/mol and -806.04 kcal/mol. And the plasmid structures’ minimum free energy were -735.00 kcal/mol, -797.30 kcal/mol and -769.90 kcal/mol (Supplementary Fig. 6). Subsequently, the GenSmart tool was employed to insert the cDNA sequence of the vaccine into the pET28a( +) vector (Fig. 9).

In silico cloning of the designed vaccine inserted into pET28a ( +) expression vector; the pale green part was substituted with the vaccine’s codon, (A) ABV1’s codon. B ABV2’s codon. C ABV3’s codon
Discussion
Since its discovery, Acinetobacter baumannii has remained a prominent pathogen responsible for nosocomial infections, particularly in immunocompromised populations like the elderly, HIV-infected individuals, and those with malignant tumors [74]. Given the absence of a specific Acinetobacter baumannii vaccine and the escalating drug resistance [75], there is a pressing need for the development of an innovative Acinetobacter baumannii vaccine. Active vaccination is recognized as one of the most effective strategies for preventing Acinetobacter baumannii infections, yet no vaccines have currently advanced to clinical trials. mRNA-based drugs have demonstrated high efficacy and minimal side effects as an immunotherapy [76]. These vaccines offer strong effectiveness, excellent stability, and low toxicity, while also enabling modular platform development and rapid production [77]. Moreover, mRNA vaccines are efficiently translated into intracellular proteins via cellular mechanisms, leading to quicker and more significant presentation to antigen-presenting cells, which enhances the adaptive immune response [78, 79]. Consequently, our multi-epitope mRNA vaccine against Acinetobacter baumannii holds promise as an ideal candidate for preventing infections caused by this pathogen.
Previous research on Acinetobacter baumannii vaccines has often focused on whole cell antigens [9], outer membrane vesicles [10], or a limited number of outer membrane protein antigens [80]. While these vaccines hold theoretical promise, their protective efficacy and coverage tend to be relatively narrow. This study, however, identifies a range of core conserved targets with high immunogenicity and antigenicity. By designing various mRNA vaccines integrated with different adjuvants, this research provides new insights into developing vaccines with broader protection against Acinetobacter baumannii infections. Acinetobacter baumannii exists in numerous variants, many of which pose significant threats to human health. Consequently, our objective is to create a comprehensive multi-epitope vaccine capable of safeguarding against the majority of Acinetobacter baumannii strains [81]. Traditional vaccines often target a single pathogen, making it challenging to predict their efficacy, especially when aiming for a broad-spectrum multi-epitope vaccine. We identified 12 proteins as potential targets based on extensive vaccine studies, elucidated the sequences of CTL, HTL, and LBL proteins suitable for targeting, and subjected them to thorough analyses of antigenicity, immunogenicity, irritability, toxicity, and conservation. Excluding poorly conserved protein sequences ensures the safety of the vaccine while maintaining its ability to induce a robust immune response. These sequences were interconnected through EAAAK, AAY, GPGPG, KK and other linkers and adjuvants such as CTB, RS09 and β-defensin to design our multi-epitope vaccine, and subsequent verification encompassed assessments of antigenicity, allergenicity, and various physicochemical properties, for instance, adipose index and instability index. Many studies have employed these linkers and adjuvants to enhance immune activation and induce more sustained immune responses.
We assessed the binding capacity of CTL, HTL, and HLA alleles, selecting protein sequences that interact effectively with multiple HLA alleles. Docking results demonstrated strong affinity between our sequences and HLA molecules, facilitating effective presentation by MHC molecules and eliciting robust immune responses. Considering the regulatory roles of IL-4 and IL-10 in helper T cell activation, where IL-4 promotes activation and IL-10 inhibits it, we meticulously screened for sequences that induce IL-4 without triggering IL-10 [82, 83]. For HTL epitopes, we prioritized those capable of inducing IFN-γ, which is crucial for macrophage activation and cytokine secretion. Our diverse epitope selection provides a foundation for a potentially effective multi-epitope vaccine, exhibiting a favorable immune response profile with minimal side effects [84]. These designs contribute to the enhanced stability and applicability of our vaccines.
Augmenting the immunogenicity of our multi-epitope vaccines through adjuvants is essential for inducing a durable and stable immune response. We incorporated three adjuvants—β-defensin 3, cholera toxin B subunit (CTB) [85], and RS09—into our vaccines. β-defensin 3, known for its antibacterial properties, enhances immunomodulation and recruits naive immune cells to infection sites. RS09, a widely used TLR-4 receptor agonist, acts as an adjuvant to enhance immune activation and antibody production [86]. CTB binds to APCs, improving stability and prolonging the half-life to strengthen the immune response. Molecular docking with TLR4 and HLA molecules demonstrated robust affinity and stable interactions, effectively triggering the immune response.
In molecular dynamics simulations, the RMSD stabilized beyond 10–20 ns, indicating structural stability of the complex. The radius of gyration (Rg) reached equilibrium after 50 ns, further confirming complex stability. High RMSF values indicated sensitivity and flexibility, while increased hydrogen bonds and salt bridges underscored stable binding and high affinity between our vaccines and receptors.
MM-PBSA analysis showed that our vaccine-receptor complex required less energy for stabilization and exhibited strong binding to TLR4 and HLA molecules, demonstrating the capability to transition from the natural environment to the human body and induce an effective immune response. Immune simulations revealed proliferation of TC, TH, and B cells, along with production of IgA, IgG, IFN-γ, IL-2, and other cytokines post-vaccine administration. Repeated exposure to the vaccine resulted in memory B cell formation, suggesting potential for both cellular and humoral immunity, as well as long-term protection against Acinetobacter baumannii. Codon optimization and cloning of the vaccine were performed to assess mass production feasibility, and the CAI and GC content indicated successful expression in E. coli K12.
The C-IMMSIM immune simulation results highlighted variations in immune responses induced by our vaccines—CTB, RS09, and β-defensin 3. Notably, the CTB-adjuvanted vaccine demonstrated a faster induction of TH and DC cells compared to RS09 and β-defensin 3. Despite this, all three vaccines showed comparable abilities in inducing differentiation and proliferation of TC cells, NK cells, and various cytokines, including IgM, IgG, and β-defensin 3. These cell and cytokine levels were sustained over time.
Docking analyses using ClusPro2.0 and HADDOCK2.4 showed stable interactions of all three vaccines with HLA-A02:01, HLA-DRB101:01, and TLR4. The stability of interaction with HLA-DRB101:01 was notably higher than with HLA-A02:01 and TLR4. HADDOCK’s scoring revealed that β-defensin 3 and CTB vaccines exhibited more stable binding to HLA-A02:01, while RS09 vaccines showed greater stability with TLR4. Hydrogen bond and salt bridge analyses supported these findings, with the CTB vaccine and HLA-DRB101:01 displaying a higher abundance of hydrogen bonds, reinforcing stability.
Finally, the protein sequence was converted into DNA and engineered into a multi-epitope mRNA vaccine, with the full-length sequence transferred to the pET28(a) plasmid for expression in the E. coli system.
In summary, all three vaccines effectively elicit immune responses, with stable interactions with receptors, establishing them as promising candidates for further investigation. However, the nuanced differences among these vaccines warrant comprehensive follow-up experiments for thorough exploration and validation.
Conclusions
Due to the escalating bacterial resistance and the proliferation of susceptible populations, Acinetobacter baumannii poses a significant threat to global public health. In this study, we employed immunoinformatic technology to craft a multi-epitope mRNA vaccine for Acinetobacter baumannii, characterized by robust antigenicity, immunogenicity, non-sensitivity, and non-toxicity. Molecular docking and MD simulation results underscore the vaccine's robust affinity for innate immune receptors, forming a stable vaccine-receptor complex capable of inducing both humoral and cellular immunity in the human body. Subsequent codon optimization demonstrated the vaccine's potential for mass production. In conclusion, the designed Acinetobacter baumannii vaccine holds promise for effective protection against Acinetobacter baumannii infections. However, further in vitro and in vivo testing is imperative to ascertain its safety and efficacy his section is not mandatory but can be added to the manuscript if the discussion is unusually long or complex.
Availability of data and materials
The protein sequences of AmpD (WP_000810002.1), OmpA (WP_000777878.1), Pal (EEX03100.1, 2009, GenBank), BauA (WP_001073039.1), Omp34 (EEX02037.1, 2009, GenBank), BamA (EEX04588.1, 2009, GenBank), Omp22 (WP_001202415.1), CsuA/B (EEX04344.1, 2009, GenBank), OmpK (WP_001176275.1) and Dcap (EEX05238.1, 2009, GenBank) were retrieved from NCBI (https://www.ncbi.nlm.nih.gov/) and UniProt Database (https://www.uniprot.org/) and their sequences were listed in supplementary materials. The DNA and RNA sequences are predicted in EMBOSS (http://emboss.open-bio.org/) and Biochen server (https://www.biochen.org/cn/tools/transform_sequence) and listed in supplementary materials. The linked genotype and phenotype data were listed in supplementary materials as well, their sequences were caught from IPD-IMGT/HLA Database (https://www.ebi.ac.uk/ipd/imgt/hla/) and most of their structure were predicted via homologous modeling in Swiss-model (https://swissmodel.expasy.org/), and some of the structure was retrieved from RCSB PDB, HLA-A*02:06(PDB ID:3OXR), HLA-B*35:01(PDB ID:4LNR), HLA-B*51:01(PDB ID:4MJI), HLA-DRB1*04:01(PDB ID:5JLZ), HLA-C*06:02(PDB ID:5W67), HLA-B*57:01(PDB ID:6BXP), HLA-E*01:03(PDB ID:6GGM), HLA-A*01:01(PDB ID:6MPP), HLA-C*07:02(PDB ID:6PA1), HLA-A*68:01(PDB ID:6PBH), HLA-B*15:02(PDB ID:6VIU), HLA-A*02:01(PDB ID:7SA2), HLA-B*27:05(PDB ID:7T0L), HLA-A*29:02(PDB ID:7TLT), HLA-B*58:01(PDB ID:7X00), HLA-A*11:01(PDB ID:8I5C). Otherwise, our article isn’t involved with Genetic polymorphisms, macromolecular structure-microarray data and crystallographic data for small molecules.
Abbreviations
- ESKAPE:
-
Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp
- ROS:
-
Reactive oxygen species
- NCBI:
-
National Center for Biotechnology Information Database
- CTL:
-
Cytotoxic lymphocyte
- MHC:
-
Major histocompatibility complex
- IEDB:
-
Immune Epitope Database & Tools
- HTL:
-
Helper T lymphocyte
- BCR:
-
B cell receptor
- LBL:
-
Linear B cell
- RNN:
-
Recurrent neural network
- CTB:
-
Cholera Toxin B subunit
- APC:
-
Antigen Presenting cells
- PADRE:
-
Pan DR epitope
- pI:
-
Isoelectric point
- GRAVY:
-
Grand average of hydropathicity
- TLR4:
-
Toll-like receptor4
- PAMP:
-
Pathogen Associated Molecular Pattern
- MD:
-
Molecular Dynamic
- TIP3P:
-
Transferable Intermolecular Potential 3P
- NVT:
-
Network Virtual Terminal
- PSSM:
-
Position Specific Scoring Matrix
- tPA:
-
Tissue Plasminogen activator
- MITD:
-
MHC I targeting domain
- Rg:
-
Radius of gyration
- RMSD:
-
Root-mean-square deviation
- RMSF:
-
Root mean square fluctuation
References
Giammanco A, Vieu JF, Bouvet PJ, Sarzana A, Sinatra A. A comparative assay of epidemiological markers for Acinetobacter strains isolated in a hospital. Zentralbl Bakteriol. 1989;272(2):231–41.
Sianturi J, Priegue P, Hu J, Yin J, Seeberger PH. Semi-synthetic glycoconjugate vaccine lead against Acinetobacter baumannii 17978. Angew Chem Int Ed Engl. 2022;61(41):e202209556.
Tillery LM, Barrett KF, Dranow DM, Craig J, Shek R, Chun I, Barrett LK, Phan IQ, Subramanian S, Abendroth J, et al. Toward a structome of Acinetobacter baumannii drug targets. Protein Sci. 2020;29(3):789–802.
Kasimova AA, Arbatsky NP, Tickner J, Kenyon JJ, Hall RM, Shneider MM, Dzhaparova AA, Shashkov AS, Chizhov AO, Popova AV, et al. Acinetobacter baumannii K106 and K112: two structurally and genetically related 6-deoxy-l-talose-containing capsular polysaccharides. Int J Mol Sci. 2021;22(11):5641.
Villa EA, Escalante-Semerena JC. Acinetobacter baumannii Catabolizes Ethanolamine in the Absence of a Metabolosome and Converts Cobinamide into Adenosylated Cobamides. Bio. 2022;13(4):e0179322.
Whiteway C, Breine A, Philippe C, Van der Henst C. Acinetobacter baumannii. Trends Microbiol. 2022;30(2):199–200.
Monem S, Furmanek-Blaszk B, Lupkowska A, Kuczynska-Wisnik D, Stojowska-Swedrzynska K, Laskowska E. Mechanisms protecting acinetobacter baumannii against multiple stresses triggered by the host immune response, antibiotics and outside-host environment. Int J Mol Sci. 2020;21(15):5498. https://doi.org/10.3390/ijms21155498.
Qiu H, KuoLee R, Harris G, Van Rooijen N, Patel GB, Chen W. Role of macrophages in early host resistance to respiratory Acinetobacter baumannii infection. PLoS ONE. 2012;7(6): e40019.
Khan MA, Allemailem KS, Maswadeh H, Younus H: Safety and Prophylactic Efficacy of Liposome-Based Vaccine against the Drug-Resistant Acinetobacter baumannii in Mice. Pharmaceutics 2022, 14(7).
Weng Z, Yang N, Shi S, et al. Outer Membrane Vesicles from Acinetobacter baumannii: Biogenesis, Functions, and Vaccine Application. Vaccines (Basel). 2023;12(1):49. https://doi.org/10.3390/vaccines12010049.
Anasagasti JI, Peralta V, Harto A, Chinchilla A, Ledo A. Study of personality in patients with chronic urticaria using the 16-PF questionnaire. Rev Clin Esp. 1986;178(4):177–80.
Lin L, Tan B, Pantapalangkoor P, Ho T, Hujer AM, Taracila MA, Bonomo RA, Spellberg B. Acinetobacter baumannii rOmpA vaccine dose alters immune polarization and immunodominant epitopes. Vaccine. 2013;31(2):313–8.
Mansouri M, Sadeghpoor M, Abdollahi M, Vafaei AJ, Jalali Nadoushan M, Rasooli I. Synergistic immunoprotection by Oma87 and Bap against Acinetobacter baumannii sepsis model. Int Immunopharmacol. 2023;122:110650.
Chauhan V, Rungta T, Goyal K, Singh MP. Designing a multi-epitope based vaccine to combat Kaposi Sarcoma utilizing immunoinformatics approach. Sci Rep. 2019;9(1):2517.
Sheweita SA, Batah AM, Ghazy AA, Hussein A, Amara AA. A new strain of Acinetobacter baumannii and characterization of its ghost as a candidate vaccine. J Infect Public Health. 2019;12(6):831–42.
Tan C, Zhu F, Pan P, Wu A, Li C. Development of multi-epitope vaccines against the monkeypox virus based on envelope proteins using immunoinformatics approaches. Front Immunol. 2023;14:1112816.
Asadinezhad M, Khoshnood S, Asadollahi P, Ghafourian S, Sadeghifard N, Pakzad I, Zeinivand Y, Omidi N, Hematian A, Kalani BS. Development of innovative multi-epitope mRNA vaccine against pseudomonas aeruginosa using in silico approaches. Brief Bioinform. 2023;25(1):bbad502.
Dhople V, Krukemeyer A, Ramamoorthy A. The human beta-defensin-3, an antibacterial peptide with multiple biological functions. Biochim Biophys Acta. 2006;1758(9):1499–512.
Yang D, Chertov O, Bykovskaia SN, Chen Q, Buffo MJ, Shogan J, Anderson M, Schroder JM, Wang JM, Howard OM, et al. Beta-defensins: linking innate and adaptive immunity through dendritic and T cell CCR6. Science. 1999;286(5439):525–8.
Shanmugam A, Rajoria S, George AL, Mittelman A, Suriano R, Tiwari RK. Synthetic Toll like receptor-4 (TLR-4) agonist peptides as a novel class of adjuvants. PLoS ONE. 2012;7(2): e30839.
Antonio-Herrera L, Badillo-Godinez O, Medina-Contreras O, Tepale-Segura A, Garcia-Lozano A, Gutierrez-Xicotencatl L, Soldevila G, Esquivel-Guadarrama FR, Idoyaga J, Bonifaz LC. The nontoxic cholera B subunit is a potent adjuvant for intradermal DC-targeted vaccination. Front Immunol. 2018;9:2212.
Hung CF, Tsai YC, He L, Wu TC. DNA vaccines encoding Ii-PADRE generates potent PADRE-specific CD4+ T-cell immune responses and enhances vaccine potency. Mol Ther. 2007;15(6):1211–9.
Kim HJ, Kim H, Lee JH, Hwangbo C. Toll-like receptor 4 (TLR4): new insight immune and aging. Immun Ageing. 2023;20(1):67.
Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, Beglov D, Vajda S. The ClusPro web server for protein-protein docking. Nat Protoc. 2017;12(2):255–78.
Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–18.
Boonstra S, Onck PR, Giessen E. CHARMM TIP3P water model suppresses peptide folding by solvating the unfolded state. J Phys Chem B. 2016;120(15):3692–8.
Valdes-Tresanco MS, Valdes-Tresanco ME, Valiente PA, Moreno E. gmx_MMPBSA: a new tool to perform end-state free energy calculations with GROMACS. J Chem Theory Comput. 2021;17(10):6281–91.
Liu Y, Gong W, Yang Z, Li C. SNB-PSSM: a spatial neighbor-based PSSM used for protein-RNA binding site prediction. J Mol Recognit. 2021;34(6):e2887.
Kou Y, Xu Y, Zhao Z, Liu J, Wu Y, You Q, Wang L, Gao F, Cai L, Jiang C. Tissue plasminogen activator (tPA) signal sequence enhances immunogenicity of MVA-based vaccine against tuberculosis. Immunol Lett. 2017;190:51–7.
Kreiter S, Selmi A, Diken M, Sebastian M, Osterloh P, Schild H, Huber C, Tureci O, Sahin U. Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. J Immunol. 2008;180(1):309–18.
Kim SC, Sekhon SS, Shin WR, Ahn G, Cho BK, Ahn JY, Kim YH. Modifications of mRNA vaccine structural elements for improving mRNA stability and translation efficiency. Mol Cell Toxicol. 2022;18(1):1–8.
Rcheulishvili N, Mao J, Papukashvili D, Liu C, Wang Z, Zhao J, Xie F, Pan X, Ji Y, He Y, et al. Designing multi-epitope mRNA construct as a universal influenza vaccine candidate for future epidemic/pandemic preparedness. Int J Biol Macromol. 2023;226:885–99.
Rybakova Y, Kowalski PS, Huang Y, Gonzalez JT, Heartlein MW, DeRosa F, Delcassian D, Anderson DG. mRNA Delivery for Therapeutic Anti-HER2 Antibody Expression In Vivo. Mol Ther. 2019;27(8):1415–23.
Lei L, Yang F, Zou J, Jing H, Zhang J, Xu W, Zou Q, Zhang J, Wang X. DNA vaccine encoding OmpA and Pal from Acinetobacter baumannii efficiently protects mice against pulmonary infection. Mol Biol Rep. 2019;46(5):5397–408.
Zeng X, Wang N, Xiang C, Liu Q, Li D, Zhou Y, Zhang X, Xie Y, Zhang W, Yang H, et al. Peptidoglycan-associated lipoprotein contributes to the virulence of Acinetobacter baumannii and serves as a vaccine candidate. Genomics. 2023;115(2):110590.
Beiranvand S, Doosti A, Mirzaei SA. Putative novel B-cell vaccine candidates identified by reverse vaccinology and genomics approaches to control Acinetobacter baumannii serotypes. Infect Genet Evol. 2021;96:105138.
Badmasti F, Ajdary S, Bouzari S, Fooladi AA, Shahcheraghi F, Siadat SD. Immunological evaluation of OMV(PagL)+Bap(1–487aa) and AbOmpA(8–346aa)+Bap(1–487aa) as vaccine candidates against Acinetobacter baumannii sepsis infection. Mol Immunol. 2015;67(2 Pt B):552–8.
Raoufi Z, Abdollahi S, Armand R. DcaP porin and its epitope-based subunit promise effective vaccines against Acinetobacter baumannii; in-silico and in-vivo approaches. Microb Pathog. 2022;162:105346.
Fereshteh S, Abdoli S, Shahcheraghi F, Ajdary S, Nazari M, Badmasti F. New putative vaccine candidates against Acinetobacter baumannii using the reverse vaccinology method. Microb Pathog. 2020;143:104114.
Rahbar MR, Mubarak SMH, Hessami A, Khalesi B, Pourzardosht N, Khalili S, Zanoos KA, Jahangiri A. A unique antigen against SARS-CoV-2, Acinetobacter baumannii, and Pseudomonas aeruginosa. Sci Rep. 2022;12(1):10852.
Akbari Z, Rasooli I, Ghaini MH, Chaudhuri S, Farshchi Andisi V, Jahangiri A, Ramezanalizadeh F, Schryvers AB. BauA and Omp34 surface loops trigger protective antibodies against Acinetobacter baumannii in a murine sepsis model. Int Immunopharmacol. 2022;108:108731.
Ramezanalizadeh F, Rasooli I, Owlia P, Darvish Alipour Astaneh S, Abdolhamidi R. Vaccination with a combination of planktonic and biofilm virulence factors confers protection against carbapenem-resistant Acinetobacter baumannii strains. Sci Rep. 2022;12(1):19909.
Vieira de Araujo AE, Conde LV, da Silva Junior HC, de Almeida Machado L, Lara FA, Chapeaurouge A, Pauer H, Pires Hardoim CC, Martha Antunes LC, D’Alincourt Carvalho-Assef AP, et al. Cross-reactivity and immunotherapeutic potential of BamA recombinant protein from Acinetobacter baumannii. Microbes Infect. 2021;23(4–5):104801.
Ahmad S, Ranaghan KE, Azam SS. Combating tigecycline resistant Acinetobacter baumannii: a leap forward towards multi-epitope based vaccine discovery. Eur J Pharm Sci. 2019;132:1–17.
Solanki V, Tiwari M, Tiwari V. Investigation of peptidoglycan-associated lipoprotein of Acinetobacter baumannii and its interaction with fibronectin to find its therapeutic potential. Infect Immun. 2023;91(5):e0002323.
Abdollahi S, Rasooli I, Mousavi Gargari SL. An in silico structural and physicochemical characterization of TonB-dependent copper receptor in A. baumannii. Microb Pathog. 2018;118:18–31.
Du X, Xue J, Jiang M, Lin S, Huang Y, Deng K, Shu L, Xu H, Li Z, Yao J, et al. A multiepitope peptide, rOmp22, encapsulated in chitosan-PLGA nanoparticles as a candidate vaccine against acinetobacter baumannii Infection. Int J Nanomedicine. 2021;16:1819–36.
Bentancor LV, Routray A, Bozkurt-Guzel C, Camacho-Peiro A, Pier GB, Maira-Litran T. Evaluation of the trimeric autotransporter Ata as a vaccine candidate against Acinetobacter baumannii infections. Infect Immun. 2012;80(10):3381–8.
Luo G, Lin L, Ibrahim AS, Baquir B, Pantapalangkoor P, Bonomo RA, Doi Y, Adams MD, Russo TA, Spellberg B. Active and passive immunization protects against lethal, extreme drug resistant-Acinetobacter baumannii infection. PLoS ONE. 2012;7(1): e29446.
Tajuelo A, Terron MC, Lopez-Siles M, McConnell MJ. Role of peptidoglycan recycling enzymes AmpD and AnmK in Acinetobacter baumannii virulence features. Front Cell Infect Microbiol. 2022;12:1064053.
Ramezanalizadeh F, Owlia P, Rasooli I. Type I pili, CsuA/B and FimA induce a protective immune response against Acinetobacter baumannii. Vaccine. 2020;38(34):5436–46.
Aghajani Z, Rasooli I, Mousavi Gargari SL. Exploitation of two siderophore receptors, BauA and BfnH, for protection against Acinetobacter baumannii infection. APMIS. 2019;127(12):753–63.
Guo SJ, Ren S, Xie YE. Evaluation of the protective efficacy of a fused OmpK/Omp22 protein vaccine candidate against acinetobacter baumannii infection in mice. Biomed Environ Sci. 2018;31(2):155–8.
Mehdinejadiani K, Hashemi A, Bandehpour M, Rahmani H, Ranjbar MM, Yardel V, Jalali SA, Mosaffa N. Evaluationof the new outer membrane protein a epitope-based vaccines for mice model of Acinetobacter baumannii associated pneumonia and sepsis infection. Iran J Allergy Asthma Immunol. 2021;20(5):537–49.
Fereshteh S, Ajdary S, Sepehr A, Bolourchi N, Barzi SM, Haririzadeh Jouriani F, Riazi-Rad F, Shahcheraghi F, Badmasti F. Immunization with recombinant DcaP-like protein and AbOmpA revealed protections against sepsis infection of multi-drug resistant Acinetobacter baumannii ST2(Pas) in a C57BL/6 mouse model. Microb Pathog. 2023;174:105882.
Sun P, Li X, Pan C, Liu Z, Wu J, Wang H, Zhu L. A short peptide of autotransporter ata is a promising protective antigen for vaccination against Acinetobacter baumannii. Front Immunol. 2022;13:884555.
Golestani F, Malekan M, Rasooli I, Jahangiri A, Ramezanalizadeh F, Chaudhuri S, Farshchi Andisi V, Schryvers AB. Immunogenicity of loop 3 of Omp34 from A. Baumannii in loopless C-lobe of TbpB of N. meningitidis. Int Immunopharmacol. 2022;110:109013.
Naghipour Erami A, Rasooli I, Jahangiri A, Darvish Alipour Astaneh S. Anti-Omp34 antibodies protect against Acinetobacter baumannii in a murine sepsis model. Microb Pathog. 2021;161(Pt B):105291.
Viale AM, Evans BA. Microevolution in the major outer membrane protein OmpA of Acinetobacter baumannii. Microb Genom. 2020;6(6):e000381. https://doi.org/10.1099/mgen.0.000381.
Ranjbar A, Rasooli I, Jahangiri A, Ramezanalizadeh F. Specific egg yolk antibody raised to biofilm associated protein (Bap) is protective against murine pneumonia caused by Acinetobacter baumannii. Sci Rep. 2022;12(1):12576.
Jahangiri A, Rasooli I, Owlia P, Imani Fooladi AA, Salimian J. Highly conserved exposed immunogenic peptides of Omp34 against Acinetobacter baumannii: an innovative approach. J Microbiol Methods. 2018;144:79–85.
Chaudhuri S, Rasooli I, Oskouei RH, Pishgahi M, Jahangir A, Andisi VF, Schryvers AB. Hybrid antigens expressing surface loops of BauA from Acinetobacter baumannii are capable of inducing protection against infection. Front Immunol. 2022;13:933445.
Singh R, Capalash N, Sharma P. Immunoprotective potential of BamA, the outer membrane protein assembly factor, against MDR Acinetobacter baumannii. Sci Rep. 2017;7(1):12411.
Sefid F, Rasooli I, Jahangiri A, Bazmara H. Functional exposed amino acids of BauA as potential immunogen against Acinetobacter baumannii. Acta Biotheor. 2015;63(2):129–49.
Huang W, Yao Y, Wang S, Xia Y, Yang X, Long Q, Sun W, Liu C, Li Y, Chu X, et al. Immunization with a 22-kDa outer membrane protein elicits protective immunity to multidrug-resistant Acinetobacter baumannii. Sci Rep. 2016;6:20724.
Fattahian Y, Rasooli I, Mousavi Gargari SL, Rahbar MR, Darvish Alipour Astaneh S, Amani J. Protection against Acinetobacter baumannii infection via its functional deprivation of biofilm associated protein (Bap). Microb Pathog. 2011;51(6):402–6.
Jahangiri A, Owlia P, Rasooli I, Salimian J, Derakhshanifar E, Naghipour Erami A, Darzi Eslam E, Darvish Alipour Astaneh S. Specific egg yolk antibodies (IgY) confer protection against Acinetobacter baumannii in a murine pneumonia model. J Appl Microbiol. 2019;126(2):624–32.
Jahangiri A, Rasooli I, Owlia P, Fooladi AA, Salimian J. In silico design of an immunogen against Acinetobacter baumannii based on a novel model for native structure of outer membrane protein A. Microb Pathog. 2017;105:201–10.
Dey J, Mahapatra SR, Raj TK, Misra N, Suar M. Identification of potential flavonoid compounds as antibacterial therapeutics against Klebsiella pneumoniae infection using structure-based virtual screening and molecular dynamics simulation. Mol Divers. 2023. https://doi.org/10.1007/s11030-023-10738-z.
Dey J, Mahapatra SR, Singh PK, Prabhuswamimath SC, Misra N, Suar M. Designing of multi-epitope peptide vaccine against Acinetobacter baumannii through combined immunoinformatics and protein interaction-based approaches. Immunol Res. 2023;71(4):639–62.
Dey J, Mahapatra SR, Raj TK, Kaur T, Jain P, Tiwari A, Patro S, Misra N, Suar M. Designing a novel multi-epitope vaccine to evoke a robust immune response against pathogenic multidrug-resistant Enterococcus faecium bacterium. Gut Pathog. 2022;14(1):21.
Xu X, Yan C, Zou X. MDockPeP: An ab-initio protein-peptide docking server. J Comput Chem. 2018;39(28):2409–13.
Yanao T, Koon WS, Marsden JE, Kevrekidis IG. Gyration-radius dynamics in structural transitions of atomic clusters. J Chem Phys. 2007;126(12):124102.
Lee CR, Lee JH, Park M, Park KS, Bae IK, Kim YB, Cha CJ, Jeong BC, Lee SH. Biology of Acinetobacter baumannii: pathogenesis, antibiotic resistance mechanisms, and prospective treatment options. Front Cell Infect Microbiol. 2017;7:55.
Zarrilli R, Pournaras S, Giannouli M, Tsakris A. Global evolution of multidrug-resistant Acinetobacter baumannii clonal lineages. Int J Antimicrob Agents. 2013;41(1):11–9.
Xu S, Yang K, Li R, Zhang L. mRNA vaccine era-mechanisms, drug platform and clinical prospection. Int J Mol Sci. 2020;21(18):6582.
Kon E, Elia U, Peer D. Principles for designing an optimal mRNA lipid nanoparticle vaccine. Curr Opin Biotechnol. 2022;73:329–36.
Verbeke R, Hogan MJ, Lore K, Pardi N. Innate immune mechanisms of mRNA vaccines. Immunity. 2022;55(11):1993–2005.
Sureshchandra S, Lewis SA, Doratt BM, Jankeel A, Coimbra Ibraim I, Messaoudi I. Single-cell profiling of T and B cell repertoires following SARS-CoV-2 mRNA vaccine. JCI Insight. 2021;6(24):e153201.
Lau YT, Tan HS. Acinetobacter baumannii subunit vaccines: recent progress and challenges. Crit Rev Microbiol. 2024;50(4):434–49. https://doi.org/10.1080/1040841X.2023.2215303.
Carvalheira A, Silva J, Teixeira P. Acinetobacter spp. in food and drinking water - a review. Food Microbiol. 2021;95:103675.
Sahoo A, Wali S, Nurieva R. T helper 2 and T follicular helper cells: regulation and function of interleukin-4. Cytokine Growth Factor Rev. 2016;30:29–37.
Fang D, Zhu J. Molecular switches for regulating the differentiation of inflammatory and IL-10-producing anti-inflammatory T-helper cells. Cell Mol Life Sci. 2020;77(2):289–303.
Deng Z, Ding W, Li F, Shen S, Huang C, Lai K. Pulmonary IFN-gamma causes lymphocytic inflammation and cough hypersensitivity by increasing the number of IFN-gamma-secreting T lymphocytes. Allergy Asthma Immunol Res. 2022;14(6):653–73.
Lingwood C. Therapeutic uses of bacterial subunit toxins. Toxins (Basel). 2021;13(6):378.
Kim E, Erdos G, Huang S, Kenniston TW, Balmert SC, Carey CD, Raj VS, Epperly MW, Klimstra WB, Haagmans BL, et al. Microneedle array delivered recombinant coronavirus vaccines: immunogenicity and rapid translational development. EBioMedicine. 2020;55: 102743.
Acknowledgements
Authors would like to acknowledge Department of Respiratory Medicine of Xiangya Hospital and the Central South University and Technology for their unconditional support and for providing ambient atmosphere to conduct this research project.
Funding
This research was funded by:
1. The Scientific Research Program of FuRong Laboratory, No.2023SK2101
2. Natural Science Foundation of ChangSha, No.kq2208368
3. Natural Science Foundation of Hunan Province of China, No.2023JJ30930
4. The National Natural Science Foundation of China, No.81770080
5. Supported by the Project Program of National Clinical Research Center for Geriatric Disorders, Xiang ya Hospital, Grant No. 2020LNJJ05
6. The National Natural Science Foundation of China, No.8210012334
7. National Key R&D Program of China, No. 2016YFC1304204
8. Key R&D Program of Hunan Province, No. 2022SK2038
9. Project Program of central south university graduate education teaching reform, No.2022JGB025
10. The national key clinical specialist construction programs of China, Grant Number z047-02
11. Research Project of Teaching Reform in Colleges and Universities in Hunan Province, 2021jy139-2
12. China Postdoctoral Science Foundation, 2022M713520
13. Project on the topic of emergency response to combat the COVID-19 epidemic in Hunan Province, 2022SK2122
Author information
Authors and Affiliations
Contributions
Conceptualization, ZF and XYZ.; Methodology, XYZ.; Validation, ZF, ZZY; Formal analysis, ZPP.; Investigation, TCX.; Resources, PPH.; Data curation, XYZ.; Writing—original draft preparation, XYZ.; Writing—review and editing, ZF, MSY.; Visualization, ZZY, LYY, QRL.; Supervision, CJ.; Project administration, ZF, PPH.; Funding acquisition, PPH, CJ. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participants
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, Y., Zhu, F., Zhou, Z. et al. A novel mRNA multi-epitope vaccine of Acinetobacter baumannii based on multi-target protein design in immunoinformatic approach. BMC Genomics 25, 791 (2024). https://doi.org/10.1186/s12864-024-10691-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10691-7