
Abstract
Background
Mitochondria play crucial roles in the growth, development, and adaptation of plants. Blackcurrant (Ribes nigrum L.) stands out as a significant berry species due to its rich nutritional profile, medicinal properties, and health benefits. Despite its importance, the mitochondrial genome of blackcurrant remains unassembled.
Results
This study presents the first assembly of the mitochondrial genome of R. nigrum in the Grossulariaceae family. The genome spans 450,227 base pairs (bp) and encompasses 39 protein-coding genes (PCGs), 19 transfer RNAs (tRNAs), and three ribosomal RNAs (rRNAs). Protein-coding regions constitute 8.88% of the entire genome. Additionally, we identified 180 simple sequence repeats, 12 tandem repeats, and 432 pairs of dispersed repeats. Notably, the dispersed sequence R1 (cotig3, 1,129 bp) mediated genome recombination, resulting in the formation of two major conformations, namely master and double circles. Furthermore, we identified 731 C-to-U RNA editing sites within the PCGs. Among these, cox1-2, nad1-2, and nad4L-2 were associated with the creation of start codons, whereas atp6-718 and rps10-391 were linked to termination codons. We also detected fourteen plastome fragments within the mitogenome, constituting 1.11% of the total length. Phylogenetic analysis suggests that R. nigrum might have undergone multiple genomic reorganization and/or gene transfer events, resulting in the loss of two PCGs (rps2 and rps11) during its evolutionary history.
Conclusions
This investigation unveils the molecular characteristics of the R. nigrum mitogenome, shedding light on its evolutionary trajectory and phylogenetic implications. Furthermore, it serves as a valuable reference for evolutionary research and germplasm identification within the genus.
Similar content being viewed by others


Introduction
Mitochondria, crucial organelles in eukaryotes, originated from endosymbiotic Alphaproteobacteria within primitive eukaryotic cells approximately 1.5 billion years ago [1]. They gradually evolved into semi-autonomous organelles through gene transfer with the host cell nucleus [2, 3]. In plants, mitochondria are predominantly maternally inherited, with very few species exhibiting patrilineal or biparental inheritance [4]. Beyond their role in providing energy for cellular metabolism, mitochondria are closely associated with biological processes such as plant fertility [5], ecological adaptation [6], programmed cell death [7], and intracellular signal transduction [8]. Furthermore, under stress conditions like drought, salinity, or extreme temperatures, plant mitochondria produce retrograde signals, such as reactive oxygen species, prompting the regulation of stress response-related genes and cellular activities to restore homeostasis [9].
Plant mitochondrial genomes display distinct sequence specificities and complex physical structures. Varying significantly in size across species (ranging from 66 Kb to 11.7 Mb) [10, 11], they exhibit a much greater range than animal mitochondria (14–20 Kb) [12]. Plant mitogenomes also showcase structural diversity, featuring cyclic, linear, and reticular types [13]. The gene-coding region typically constitutes a small portion of the plant mitogenome, around 10%, and harbours numerous repeated sequences that drive evolution and structural diversity in angiosperm genomes [14, 15]. For instance, the “fossilised” mitogenome of Liriodendron tulipifera contains 65 genes [16], whereas extant plants may contain 32–67 genes due to numerous gene transfer or loss events across their evolutionary history [13]. Many RNA-editing events in plant mitogenomes are intricately linked to species evolution [17], phenotypic variation [18], and cytoplasmic male sterility [19]. Additionally, plant mitogenomes can integrate exogenous or migratory DNA sequences through mechanisms like homologous recombination, sequence transfer, or selective domestication, contributing to their rapid evolution [20, 21]. Owing to these characteristics, plant mitogenomes serve as crucial tools for species identification, evolutionary analysis, and trait inheritance research.
Blackcurrant (Ribes nigrum L.), also referred to as black currant, cassis, black cassis, or dry grape, is a perennial deciduous shrub indigenous to northern Europe and northern Asia, belonging to the genus Ribes of the Grossulariaceae family. It is extensively cultivated in temperate and boreal regions, with major producers including Germany, Poland, Norway, China, the United States, and Russia [22, 23]. In 2021, the global planted area and annual production of R. nigrum were approximately 41,860 ha and 118,002 tons of fresh fruit, respectively (International Blackcurrant Association, http://www.blackcurrant-iba.com/agronomy/statistics/). Known for its exceptionally high content of vitamins, γ-linolenic acid, and phenolics, blackcurrant’s bioactive components confer antioxidant and anticancer effects, cardiovascular and cerebrovascular benefits, and immune enhancement [24]. Apart from fresh consumption, R. nigrum is processed into juice, jam, or wine [23, 24], and its seeds are extracted for medicines, oils, etc., often used in cosmetics [25], while the leaves are utilised for tea and functional foods [26, 27]. The Ribes genus boasts rich germplasm resources, with over 160 species worldwide [24], including 59 species and 30 varieties identified in China alone, with significant wild resources distributed in Heilongjiang, Jilin, Liaoning, Nei Mongolia, Xinjiang, and Tibet [28]. A comprehensive understanding of plant genomes, including the mitogenome, is crucial for leveraging these germplasm resources for future development; however, no mitogenome has been reported for the Grossulariaceae family to date.
Integration of second and third-generation high-throughput sequencing technologies facilitates more effective exploration of plant mitogenomes [29]. In this study, we focused on the widely cultivated Chinese blackcurrant cultivar ‘Hanfeng’. Initially, we assembled and characterised the mitogenome features of this cultivar. Subsequently, we predicted and verified the presence of repeated sequences and their involvement in repeat-mediated recombination, besides identifying RNA-editing events in the mitogenome protein-coding genes (PCGs). Additionally, we assembled the plastome and identified homologous DNA sequences using the mitogenome. Finally, we delved into the evolutionary relationship of R. nigrum with related species. This study lays a scientific foundation for a deeper understanding of the genetic characteristics of R. nigrum, with implications for germplasm identification and evolutionary studies.
Results
R. Nigrum mitogenome assembly
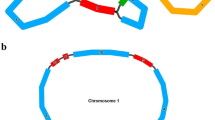
The assembly of the R. nigrum mitogenome utilised 11.81-Gb short reads and 8.73-Gb long reads employing a hybrid assembly strategy. The detailed assembly process for the graphical representation of the mitochondrial genome in R. nigrum is depicted in Fig. S1, while a simplified representation is presented in Fig. 1. The genome sketch comprises three contigs (Fig. 1A), with contig1 being the longest at 302,735 bp length, with a sequencing depth of 129X and 18,325 reads. Subsequently, contig2 spans 145,234 bp with a sequencing depth of 128X and 10,710 reads. Meanwhile, contig3 is the shortest at 1,129 bp in length, with a sequencing depth of 255X and 513 reads. Notably, the genome sketch features a double-bifurcating structure represented by contig3. The utilisation of the Unicycler software to exclude repeated regions identified by long reads led to the establishment of a complete master circle structure encompassing a total length of 450,227 bp (Fig. 1B) with a guanine–cytosine (GC) content of 45.82%. This genome surpasses in size those of Sedum plumbizincicola (212,159 bp), Rhodiola tangutica (257,378 bp), and R. crenulata (194,106 bp) within the same Order, while exhibiting similar GC contents (44.24–45.21%) (Table S1).

A mitogenome sketch for R. nigrum (A) and the master circle structure (B)
Molecular features
Annotation of the R. nigrum mitogenome identified sixty-one unique genes (Fig. 2; Table 1), comprising 24 core, 15 non-core, 19 transfer RNA (tRNA), and three ribosomal RNA (rRNA) genes. The coding sequences for PCG, tRNA, and rRNA measured 32,874 bp, 1,690 bp, and 5,432 bp, respectively, cumulatively accounting for only 8.88% of the entire genome. Notably, the exons of nad1, nad2, and nad5 are dispersed or not co-located on the same strand, representing trans-splicing genes (Table S2). Furthermore, ten genes in the R. nigrum mitogenome PCGs contain introns, surpassing the number of introns observed in closely related species (Table S3 and Table S4).
Relative synonymous codon usage (RSCU) analysis, reflective of gene expression optimization through molecular evolution [30, 31], revealed a general preference for amino acid codon usage in the R. nigrum mitogenome (Fig. S2 and Table S5), with exceptions noted for the start codon (AUG) and tryptophan (UGG) (RSCU = 1.00). For instance, alanine prefers GCU (RSCU = 1.58), while the stop codon favours UAA (RSCU = 1.54). Conversely, histidine exhibits a lesser preference for CAC (RSCU = 0.47), and both tyrosine and phenylalanine demonstrate insignificant usage preferences.

Mitochondrial genome diagram (outer ring) and repetitive sequence distribution (inner ring) of R. nigrum. The colour line on the C1 circle connects two repeated dispersed repeats. The blue, orange and purple lines represent palindromic, forward, and reverse repeats, respectively. The red line on the C2 circle represents tandem repeats. The black line on the C3 circle represents SSRs
Repeat elements and repeat-mediated recombination
A substantial number of repeated sequences were identified in the R. nigrum mitogenome, with their distribution depicted in Fig. 2. Specifically, this genome harbours 180 simple sequence repeats (SSRs) (Table S6), encompassing 70 monomers, 38 dimers, 19 trimers, 45 tetramers, five pentamers, and three hexamers, where monomers and dimers collectively constitute 60.00% of all SSRs. Notably, thymine repeats comprise 48.57% (n = 34) of all monomers. Additionally, twelve tandem repeats with a match of ≥ 85% and lengths ranging from 10 to 39 bp were identified in this mitogenome (Table S7).
Dispersed repeats classified as transposable factors, represent a class of repeated sequences capable of changing their position within the genome. In this investigation, we detected 432 pairs of dispersed repeats ≥ 30 bp in length, including 193 pairs of palindromic repeats, 238 pairs of forward repeats, one pair of reverse repeats, and no complementary repeats (Table S8). The longest forward and palindromic repeats measure 1,129 bp and 337 bp, respectively. The cumulative length of dispersed repeats amount to 43,204 bp, accounting for 9.60% of the mitogenome. This suggests that repeated sequences may serve as crucial drivers of mitogenome size amplification in plants [32].
In general, the plant mitogenome represents a complex, dynamically evolving hybrid state devoid of a singular circular structure [33]. Analysis of the aforementioned repeats unveiled that the forward repeat R1 (contig3) serves as a double-bifurcating structure potentially mediating genomic recombination. Consequently, long reads were employed to validate potential recombinant DNA sequences based on BLASTn. As depicted in Fig. 3A, the recombinant sequence delineates four potentially inferred genomic paths, where ctg1-ctg3-ctg2 and ctg2-ctg3-ctg1 signify the master circular configuration. At the same time, ctg1-ctg3-ctg1 and ctg2-ctg3-ctg2 denote the potential split configuration of the two smaller circles. A total of 35 long reads corroborates the master circle, whereas 170 reads support the split configuration, accounting for proportions of 17.07% and 82.93%, respectively (Table S9). Furthermore, primers were designed to validate the potential structures (Fig. 3A), with the corresponding electrophoresis results depicted in Fig. 3B. Specifically, all borders of the repeated sequences amplified bands of the anticipated size, and the Sanger sequencing comparison results corroborated these findings (Fig. S3). Collectively, both approaches substantiate that the repeated sequence ctg3 can mediate the formation of the two major conformations of the R. nigrum mitogenome: a master circle (Fig. 1B) and two smaller circles (Fig. 3C).

Structural prediction of R. nigrum mitogenome. (A) Primer design, (B) PCR validation, (C) two small circle structures
RNA-editing events
RNA editing, a widespread phenomenon in higher plant mitochondria, constitutes a post-transcriptional modification and regulatory process primarily occurring within coding regions [34]. In the R. nigrum mitogenome, we identified RNA-editing events across all 39 PCGs, totalling 731 editing sites, exclusively manifested as C-to-U edits (Fig. 4A), with the majority occurring at a frequency exceeding 0.80 (614 instances, approximately 83.99%) (Table S10). Notably, nad4 hosts the highest count of editing sites among the mitogenome PCGs, with 53 edits, comprising nearly 7.25% of the total, followed by ccmB, harbouring 47 editing events, accounting for 6.43% of the total. Subsequent analysis revealed 415 RNA edits affecting the second base encoding the amino acid, while 238 edits occurred at the first base, constituting 56.77% and 32.56% of the total edits, respectively.

Number of RNA editing sites (A) and amino acid conversions (B), new start codon and stop codon sites validation (C) and Sanger sequencing sequence comparison (D) in PCGs of the R. nigrum mitogenome
Of all RNA-editing events observed, 653 resulted in non-synonymous codon changes (approximately 89.33%), predominantly involving three types of amino acid alterations: Pro to Leu (164 instances), Ser to Leu (143 instances), and Ser to Phe (97 instances) (Fig. 4B). We predicted three editing sites within these PCGs responsible for creating start codons and two editing sites for generating stop codons (Table 2). Validation via polymerase chain reaction (PCR) yielded results confirming the existence of these five editing sites (Fig. 4C), with Sanger sequencing further validating these findings (Fig. 4D), as detailed in Fig. S4. Notably, the editing efficiency of the site atp6-718 was found to be low, while the site rps10-391 exhibited a larger band in genomic DNA (gDNA) compared to complementary DNA (cDNA) due to the presence of an intron spanning 850 bp.
DNA transfer
Using the same sequencing data, we assembled the R. nigrum plastome, which spans 157,459 bp in size (Fig. 5A). Throughout evolution, sequence migration between plant plastomes and mitogenomes has been extensive, leading to the incorporation of mitochondrial plastid DNAs (MTPTs) - chloroplast-derived DNA fragments - into the mitogenome [13, 35]. We identified fourteen MTPTs exhibiting > 80% sequence similarity (Fig. 5B), with a cumulative length of 4,990 bp within the R. nigrum mitogenome. Notably, two fragments surpassed 1,000 bp in length: MTPT13 (1,755 bp) and MTPT14 (1,278 bp). Additionally, among the MTPTs, twelve chloroplast genes were identified, including six PCGs - petG, petL, psbE, psbF, psbJ, and psbL, and six tRNA genes - trnD-GUC, trnH-GUG, trnM-CAU, trnN-GUU, trnp-UGG, and trnW-CCA. Similarly, eight fragments of chloroplast genes were detected within the MTPTs (Table 3). However, genes within the MTPTs exhibited varying degrees of sequence loss or replacement in the mitogenome, evolving into pseudogenes [13].

R. nigrum plastome map (A) and sequence migration analysis (B). Note: The bule arcs in Fig. 5B represent the mitogenome and the light green arcs represent the plastome. The connecting lines between the arcs are homologous fragments, where the red line represents sequence similarity equal to 100%, yellow represents sequence similarity 90-100%, and dark blue represents sequence similarity 80-90%
Genome evolution
Plant mitogenomes have undergone extensive events of PCG loss or gain throughout their evolutionary history [2, 35]. While the fossilized plant mitogenome boasts 41 PCGs, only 18 PCGs are common to both the mitogenomes of R. nigrum and 29 related species. These shared PCGs include atp1, atp4, atp6, atp8, ccmB, ccmC, ccmFC, cox2, cox3, matR, nad1, nad2, nad4L, nad5, nad6, nad7, nad9, and rpl5. The phylogenetic tree constructed in this study reveals R. nigrum’s closest relation to S. plumbizincicola of the Crassulaceae family, within the Order Saxifragales (Fig. 6A). Moreover, the phylogenetic topology based on mitochondrial DNA aligns with the latest classification of the Angiosperm Phylogeny Group IV system [36].

Phylogenetic relationships (A) and variable gene deletions (B) between R. nigrum and related species. Note: The numbers in Fig. 6A indicate bootstrap support values
Comparisons of variable genes among 11 closely related species within Santalales, Saxifragales, Zygophyllales, and Ranunculales were also conducted. While rpl5 is present across all species, deletions of rps2 and rps11 are more prevalent. Notably, rps11 is exclusive to Pulsatilla chinensis, and rps2 is only found in Tolypanthus maclurei and P. chinensis (Fig. 6B). Intriguingly, rpl2, rps1, rps10, rps19, and sdh3 are absent in S. plumbizincicola, R. tangutica, and R. crenulata but present in R. nigrum.
Furthermore, mitogenome collinearity analysis was performed on eight closely related species within Saxifragales, Zygophyllales, and Ranuncullales (Fig. 7 and Table S11). Despite detecting numerous homologous collinear blocks between R. nigrum and its Saxifragales relatives, these blocks are relatively short in length. Specifically, between R. nigrum and S. plumbizincicola, only 65 collinear blocks exceed 300 bp in length, with the longest measuring 3,241 bp and a total collinear length of 69,888 bp, constituting merely 16% of R. nigrum’s mitogenome. Moreover, the order of these collinear blocks varies significantly, indirectly indicating frequent events of homologous recombination and/or genomic rearrangement between R. nigrum and its evolutionary relatives.

Evolution between R. nigrum and related species. The gray areas and red areas indicate collinear blocks with consistent and inconsistent arrangement orders, respectively. Light green, yellow-gray, orange-red, blue, dark green, purple, indigo, and red in the figure indicate Zygophyllum fabago, Tribulus terrestris, Rhodiola crenulata, Rhodiola tangutica, Sedum plumbizincicola, Ribes nigrum, Pulsatilla chinensis, and Aconitum kusnezoffii, respectively. The proportional numbers on either side indicate the percentage of all homologous sequences between the two species
Discussion
Molecular features and comparison of the R. Nigrum mitogenome among close relatives
The assembly of the complete mitogenome of R. nigrum was successful, revealing a length of 450,227 bp, with a GC content of 45.82%, and adopting a circular structure. This structure mirrors that of 29 related species, yet the genome size significantly differs from those of its counterparts (ranging from 194,106 to 900,031 bp), with slight variations observed in GC content (ranging from 42.48 to 46.86%) (Table S1). Although the genomic GC content has historically remained relatively stable during evolution, recent research suggests that GC-rich species exhibit an enhanced ability to thrive in regions characterized by extremely cold winters or seasonal drought [37]. Additionally, during adaptive evolution, the R. nigrum mitogenome has developed its preferences in amino acid codon usage, such as an elevated preference for GCU in alanine (RSCU = 1.58) and CAU in histidine (RSCU = 1.53) (Table S5).
In plants, complete mitochondrial genes typically encompass 24 core and 17 variable genes [16]. Despite their limited number, mitochondrial genes play crucial roles. For instance, Ayabe et al. [38] demonstrated that defects in the mitochondrial gene nad7 of Arabidopsis thaliana can profoundly affect mitochondrial gene expression by altering copy numbers, resulting in severe growth inhibition and, potentially, plant mortality. Throughout angiosperm evolution, the transfer and loss of ribosomal proteins and succinate dehydrogenase genes to the nucleus have been notably frequent and episodic [2, 13]. The R. nigrum mitogenome encompasses all core genes. However, in comparison to the “fossilised” L. tulipifera [16], R. nigrum lacks two variable genes (rps2 and rps11) while harbouring additional genes (rpl2, rps1, rps4, rps10, rps19, and sdh3) in comparison to S. plumbizincicola, a close relative within the same Order [39]. Furthermore, the mitogenomes of land plants feature diverse numbers of introns, some of which encode open reading frames involved in the evolutionary dissemination and/or splicing of introns [13]. In this investigation, introns were also identified in ccmFC, cox2, nad1, nad2, nad4, nad5, nad7, rpl2, rps3, and rps10, potentially serving as scientific references for species identification [40].
Repeat sequences and genome recombination
Repeated sequences are abundant in mitogenomes and play crucial roles in plant adaptive evolution, phenotypic trait variation, gene expression regulation, and genome size expansion [32]. As mitogenome coding sequences exhibit higher conservation compared to chloroplast or nuclear genes, the development of mitochondrial markers for species identification offers higher accuracy [41]. In this investigation, 180 SSRs were identified in the R. nigrum mitogenome, offering ample references for group classification within Ribes species.
Although the structure of the plant mitogenome is complex and variable, it is commonly depicted as a cyclic molecule [13, 33]. Longer repeated sequences (> 1,000 bp) can facilitate genome recombination, resulting in mitogenome rearrangements and alterations in mitogenome conformation [42]. The frequent occurrence of mitogenome rearrangements is associated with the high incidence of DNA repair through non-homologous DNA end joining at non-coding regions. Such imprecise repairs often incorporate nuclear or plastid DNA fragments during the repair process, significantly contributing to the duplication of non-coding regions and increasing the frequency of genome recombination [43, 44]. In higher plants like sugar beets [45] and rice [5], frequent recombination of repeated sequences can lead to cytoplasmic male sterility, and alterations in promoter positions, thereby affecting gene expression [32]. In this study, 432 pairs of dispersed repeats were identified, offering substantial reference information for genetic evolution and epigenetic traits in Ribes and closely related species.
Roles of RNA editing in plant evolution and ecological adaptation
RNA editing is prevalent in mitochondrial transcripts and is crucial for the production of functional proteins [17, 46]. Particularly, RNA editing involves effective non-synonymous edits that tend to produce codons encoding hydrophobic amino acids, thereby promoting protein folding and functionality [34]. For instance, Jiang et al. [47] identified two editing sites in the conserved region of the soybean maintenance line NJCMS2B that could convert hydrophilic serine into hydrophobic leucine, altering the direction of protein transmembrane spanning; conversely, RNA editing did not occur in the atp9 transcript product of the soybean sterility line NJCMS2A. In this study, 731 C-to-U editing sites were identified in 39 PCGs, most of which cause shifts in amino acids toward hydrophobic ones; however, further exploration is needed to understand their functional roles thoroughly.
Additionally, RNA editing typically restores evolutionarily conserved codons, potentially creating new start and stop codons [48] and enhancing the expression of mitochondrial genes [46]. Despite RNA editing producing new stop codons that shorten the original transcript, these truncated transcripts often remain active. For example, Gallagher et al. [19] demonstrated that maize pollen sterility is linked to truncation of the orf77 chimeric open reading frame due to early termination of mitochondrial editing. Intriguingly, new start codons introduced by RNA editing can initiate gene transcripts from new points. For instance, Kadowaki et al. [49] and Quiñones et al. [50] identified new start codons for cox1 in tomato and potato transcripts, respectively. Notably, three editing sites (cox1-2, nad1-2, and nad4L-2) that create new start codons and two editing sites (atp6-718 and rps10-391) that generate new stop codons were predicted; however, further investigation is warranted to elucidate the roles these editing sites might play in the growth and development of R. nigrum.
Gene transfer and plant evolution
Gene transfer within plant mitochondrial DNA is a common phenomenon. In this investigation, we identified 14 plastome fragments within the R. nigrum mitogenome, constituting 1.11% of its total length. Similar occurrences have been noted in other species such as Mangifera indica (7–10 homologous fragments, comprising 0.51–0.61%) [51], Mentha spicata (No. 17, 3.97%) [52], and Panax notoginseng (No. 12, 3.11%) [53]. Among these, twelve chloroplast genes were discerned in the R. nigrum MTPTs, while most other gene-coding sequences have undergone degradation. Typically, due to base mutations or genomic rearrangements, these genes tend to degenerate into pseudogenes post-integration into the mitogenome [13, 35]. Despite their inability to perform standard functions, these MTPT genes are pivotal in driving adaptive evolution and fostering genetic diversity among terrestrial plants [21].
Evolutionary analysis of R. Nigrum and its relatives
Plant mitogenomes exhibit a propensity for integrating exogenous or migratory DNA sequences, thereby resulting in frequent gain or loss PCG [2, 35]. Our study revealed that only 18 PCGs were shared between R. nigrum and its relatives, underscoring significant divergence during evolution. Collinearity analysis further demonstrated substantial discrepancies in genome sequences between R. nigrum and related species. Even in the case of S. plumbizincicola, which is considered a closer relative, the concordant sequence length accounted for approximately 16% of the total length. However, multiple factors contribute to sequence disparity among mitogenomes, including homologous recombination among species, repeat-mediated recombination within genomes, and horizontal gene transfer, which may be a primary contributing factor [43, 44]. Therefore, understanding the variation and evolution of the R. nigrum mitogenome, along with its evolutionary lineage, necessitates further investigation.
Conclusions
This study marks the successful assembly and comprehensive analysis of the mitogenome of R. nigrum, representing the first complete exploration within the Grossulariaceae family. With a length of 450,227 bp, it harbours 61 unique genes and features a rich presence of repeat sequences, RNA-editing events, and plastome fragments. Notably, it exhibits both master and double circle major conformations. Despite evolutionary conservation, our findings indicate multiple genomic recombination and/or gene transfer events during the species’ evolutionary journey. This study offers valuable insights for deeper investigations into genetic evolution and germplasm identification not only in R. nigrum but also in related species.
Materials and methods
Plant sample preparation and sequencing
Plant material was sourced from a plot cultivating the ‘Hanfeng’ variety of R. nigrum within the National Modern Agricultural Demonstration Area at the Institute of Rural Revitalization Science and Technology (coordinates: 45°50’3’’N; 126°51’14’’E; elevation: 139 m) of the Heilongjiang Academy of Agricultural Sciences, China. Young new shoots were meticulously selected, promptly frozen in liquid nitrogen and preserved at − 86 °C in an ultra-low temperature refrigerator (Thermo Fisher Scientific, Massachusetts, USA). DNA extraction was carried out using the Plant Genomic DNA Extraction Kit (Tiangen Biotech Co., Ltd., Beijing, China), while RNA extraction employed the Plant Total RNA Extraction Kit (Tiangen, Beijing, China), conducted in triplicate to ensure robustness. The quality of the extracted samples was meticulously assessed using a NanoDrop One Microvolume UV-Vis Spectrophotometer (Thermo Fisher Scientific, Massachusetts, USA).
High-quality samples underwent sequencing procedures at Wuhan Benagen Technology Co., Ltd. (Wuhan, China). Second-generation sequencing was executed utilizing a NovaSeq 6000 (Illumina Inc., San Diego, CA, USA), with subsequent evaluation and quality control of the raw short reads achieved through Trimmomatic v0.35. Third-generation sequencing was conducted employing a Nanopore PromethION sequencer (Oxford Nanopore Technologies, Oxford, UK), and quality control filtering of the raw long reads was accomplished using NanoFilt v2.8.0. For long non-coding RNA analysis, an MGISEQ-2000 (Shenzhen Huada Intelligent Technology Co., Ltd., Shenzhen, China) was employed, and SOAPnuke v2.0 facilitated quality control filtering of the raw reads.
Mitogenome assembly
The long-read sequencing data underwent de novo assembly using Flye v2.9.2 (University of California, San Diego, USA) with default settings [54]. The resulting assembly contained sequences from the nuclear, chloroplast, and mitochondrial genomes. Subsequently, a contig library crucial for subsequent analyses was generated using the makeblastdb utility. To pinpoint contigs containing mitochondrial DNA segments, we employed the BLASTn algorithm, referencing the A. thaliana mitogenome (NC_037304) and applying stringent parameters: “-evalue 1e-5 -outfmt 6 -max_hsps 10 -word_size 7 -task blastn-short” [55]. Next, GetOrganelle v1.7.7.0 (Kunming Institute of Botany, Chinese Academy of Sciences, Kunming, China) was utilised to extract short reads specific to the mitochondrial genome, which were then used to construct a graphical representation of the genome via SPAdes v3.15.5 (Saint Petersburg Academic University of the Russian Academy of Sciences, Saint Petersburg, Russia). To validate and consolidate these findings, we employed Unicycler v0.5.0 (The University of Melbourne, Victoria, Australia) and integrated BWA v0.7.11 [56] to align the contigs derived from the graphical genome assembly with the mitochondrial contigs obtained from the long-read assembly. Finally, the assembly results were visualised using Bandage v0.8.1 [57].
Genome annotation and analysis
L. tulipifera (NC_021152.1) and A. thaliana were used as references to annotate R. nigrum mitogenome PCGs using Geseq v2.03 [58]. Exon and intron information of the encoded genes was manually extracted based on the annotation files. Subsequently, rRNA genes were annotated using BLASTN and tRNA genes were annotated using tRNAscan-SE v.2.0.11 [59]. Annotation errors were manually corrected using Apollo v1.11.8 software [60]. The protein-coding sequences of this genome were extracted using PhyloSuite v1.2.2 [61], and Mega v7.0.26 software was used to analyse codon preference and calculate RSCU values [62]. If RSCU > 1, the codon is preferentially used by the amino acids; if RSCU < 1, the opposite is true.
Repeat sequences analysis and repeat-mediated recombination validation
The online tool MISA v2.1 (https://webblast.ipk-gatersleben.de/misa/) [63] was used to identify SSRs with a length of 1–6 bp in the R. nigrum mitogenome using parameters “1–10 2–5 3–4 4 − 3 5 − 3 6 − 3.” Tandem repeat sequences were identified using TRF v4.09 (https://tandem.bu.edu/trf/trf.unix.help.html) [64] with the following threshold requirements: match ≥ 85% and length ≥ 7 bp. The REPuter program (https://bibiserv.cebitec.uni-bielefeld.de/reputer/) was used to identify dispersed repeats [65], and the minimum size was set to 30 bp. The location and details of genome distribution were visualised using Circos v0.69-9 and Excel v2021 [66].
Two approaches were used to validate the possible genomic structures mediated by repeats, as follows. (1) The BLASTN program was applied using long reads to compare repeat sequences and extend 500 bp at both ends to obtain all possible paths (with the parameter -evaluate 1e-10) and calculate the proportion of predicted structures based on the screening comparison results. (2) Based on the R. nigrum mitogenome data, repeat sequences and 500 bp upstream and downstream sequences were extracted, and primers were designed using Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast) for validation. Primers were then synthesised by Sangon Biotech Co., Ltd. (Shanghai, China) (Table S12). The PCR amplification system included 1 µL of template gDNA, 2 µL each of upstream and downstream primers (10 µmol/L), 25 µL of 2× Rapid Taq Master Mix, and 20 µL of ddH2O. The amplification program consisted of pre-denaturation at 95 °C for 3 min, denaturation at 95 °C for 15 s, annealing at 60 °C for 30 s, and extension at 72 °C for 30 s, with a total of 35 cycles, and final extension at 72 °C for 10 min [55]. Electrophoresis was performed using 1% agarose gels, and photographs were obtained by a fully automated gel-imaging analysis system (Peijqing JS-2000, Shanghai, China). PCR products were sent to Sangon Biotech for Sanger sequencing, and PDF files were generated using SeqMan Pro v7.1.0 (44.1) (DNASTAR, Madison, USA).
Prediction and validation of RNA-editing sites
To predict RNA-editing sites in R. nigrum, RNA-seq reads were aligned to the mitogenome PCG coding sequence using TopHat2 [67] with a tolerance of up to seven mismatches. Differences between DNA and RNA sequences were analysed to identify potential RNA-editing sites using REDItools v2.0 [68], with stringent criteria: a minimum coverage depth of 50× and an editing frequency of at least 0.1. To validate the predicted RNA-editing sites, we focused on genes where editing sites corresponded to start and stop codons. Primers were designed based on sequences flanking the predicted editing sites, using Primer-BLAST (Table S13). RNA extracted from the samples was reverse transcribed into cDNA, followed by PCR amplification using both gDNA and cDNA templates. The resulting amplicons were sequenced and compared, and differences in base peaks were analysed using SnapGene Viewer v7.0.1 (GSL Biotech LLC, Chicago, IL, USA).
Plastome assembly and DNA transfer analysis
The plastome of R. nigrum was assembled by integrating short and long reads, utilising GetOrganelle [69] with specific parameters “-R 15 -k 21,45,65,85,105 -F embplant_pt.” Annotation of chloroplast genes was conducted using CPGAVAS2 [70], with subsequent corrections made using CPGView [71]. Homologous fragments shared between the plastome and mitogenome were identified through BLASTN analysis, applying strict filtering criteria: an e-value threshold of 1e-6, a minimum word size of 7, a sequence length of at least 30 bp, and a sequence similarity of 80% or higher. The distribution pattern of homologous sequences was visualised using the Circos package [66].
Phylogenetic and collinearity analysis
For phylogenetic analysis, mitogenome sequences of closely related species were retrieved from NCBI (Table S1), with Pulsatilla chinensis (NC_068017.1) and Aconitum kusnezoffii (NC_053920.1) used as outgroups. Common genes among these genomes were extracted and aligned using MAFFT v7.505 [72]. Phylogenetic reconstruction was performed with IQ-TREE v1.6.12 using the “GTR + F + I + I + R2” model [73], and the resulting tree was visualised using iTOL v6 (https://itol.embl.de/) [74]. Comparison of the mitogenomes of eight closely related species, including A. kusnezoffii, P. chinensis, R. crenulata, R. nigrum, R. tangutica, S. plumbizincicola, T. terrestris, and Z. fabago, was conducted using the BLAST program. Homologous sequences with a length of at least 300 bp were retained for subsequent analysis, and gene synteny was visualised using TBtools v1.09876, based on the MCscanX algorithm [75].
Data availability
The mitogenome sequence data supporting the conclusions of this study are available in GenBank (https://www.ncbi.nlm.nih.gov/) under accession number OR296460. The Illumina, Nanopore, and transcriptome data of Ribes nigrum are deposited in the Sequence Read Archive repository under SRR25211462, SRR25211461, and SRR25211460, respectively. RNA-seq mapping results (BAM files) were uploaded to the Figshare platform: doi:https://doi.org/10.6084/m9.figshare.23912691.
Abbreviations
- Bp:
-
Base pair
- cDNA:
-
Complementary DNA
- GC:
-
Guanine–cytosine
- gDNA:
-
Genomic DNA
- MTPT:
-
Mitochondrial plastid sequence
- PCG:
-
protein coding gene
- rRNA:
-
ribosomal RNA
- RSCU:
-
Relative synonymous codon usage values
- SSR:
-
Simple sequence repeat
- tRNA:
-
transfer RNA
References
Lang BF, Gray MW, Burger G. Mitochondrial genome evolution and the origin of eukaryotes. Annu Rev Genet. 1999;33:351–97.
Adams KL, Palmer JD. Evolution of mitochondrial gene content: gene loss and transfer to the nucleus. Mol Phylogenet Evol. 2003;29(3):380–95.
Roger AJ, Muñoz-Gómez SA, Kamikawa R. The origin and diversification of mitochondria. Curr Biol. 2017;27(21):R1177–92.
Munasinghe M, Agren JA. When and why are mitochondria paternally inherited? Curr Opin Genet Dev. 2023;80:102053.
Fujii S, Kazama T, Yamada M, Toriyama K. Discovery of global genomic re-organization based on comparison of two newly sequenced rice mitochondrial genomes with cytoplasmic male sterility-related genes. BMC Genomics. 2010;11:209.
Liberatore KL, Dukowic-Schulze S, Miller ME, Chen C, Kianian SF. The role of mitochondria in plant development and stress tolerance. Free Radic Biol Med. 2016;100:238–56.
Ravagnan L, Roumier T, Kroemer G. Mitochondria, the killer organelles and their weapons. J Cell Physiol. 2002;192(2):131–37.
Wang J, Xu G, Ning Y, Wang X, Wang GL. Mitochondrial functions in plant immunity. Trends Plant Sci. 2022;27(10):1063–76.
Ng S, De Clercq I, Van Aken O, Law SR, Ivanova A, Willems P, Giraud E, Van Breusegem F, Whelan J. Anterograde and retrograde regulation of nuclear genes encoding mitochondrial proteins during growth, development, and stress. Mol Plant. 2014;7(7):1075–93.
Skippington E, Barkman TJ, Rice DW, Palmer JD. Miniaturized mitogenome of the parasitic plant Viscum Scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc Natl Acad Sci USA. 2015;112(27):E3515–24.
Putintseva YA, Bondar EI, Simonov EP, Sharov VV, Oreshkova NV, Kuzmin DA, Konstantinov YM, Shmakov VN, Belkov VI, Sadovsky MG, Keech O, Krutovsky KV. Siberian larch (Larix sibirica Ledeb.) Mitochondrial genome assembled using both short and long nucleotide sequence reads is currently the largest known mitogenome. BMC Genomics. 2020;21(1):654.
Barr CM, Neiman M, Taylor DR. Inheritance and recombination of mitochondrial genomes in plants, fungi and animals. New Phytol. 2005;168(1):39–50.
Mower JP, Sloan DB, Alverson AJ. Plant mitochondrial genome diversity: the genomics revolution. In: Wendel JF, Greilhuber J, Dolezel J, Leitch IJ, editors. Plant Genome Diversity volume 1: plant genomes, their residents, and their Evolutionary Dynamics. Vienna: Springer Vienna; 2012. 123 – 44.
Oliver KR, McComb JA, Greene WK. Transposable elements: powerful contributors to angiosperm evolution and diversity. Genome Biol Evol. 2013;5(10):1886–901.
Wicker T, Gundlach H, Spannagl M, Uauy C, Borrill P, Ramírez-González RH, De Oliveira R, International Wheat Genome Sequencing Consortium, Mayer KFX, Paux E, Choulet F. Impact of transposable elements on genome structure and evolution in bread wheat. Genome Biol. 2018;19(1):103.
Richardson AO, Rice DW, Young GJ, Alverson AJ, Palmer JD. The fossilized mitochondrial genome of Liriodendron tulipifera: ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate. BMC Biol. 2013;11:29.
Gommans WM, Mullen SP, Maas S. RNA editing: a driving force for adaptive evolution? BioEssays. 2009;31(10):1137–45.
Luo Z, Xiong J, Xia H, Wang L, Hou G, Li Z, Li J, Zhou H, Li T, Luo L. Pentatricopeptide repeat gene-mediated mitochondrial RNA editing impacts on rice drought tolerance. Front Plant Sci. 2022;13:926285.
Gallagher LJ, Betz SK, Chase CD. Mitochondrial RNA editing truncates a chimeric open reading frame associated with S male-sterility in maize. Curr Genet. 2002;42(3):179–84.
Archibald JM, Richards TA. Gene transfer: anything goes in plant mitochondria. BMC Biol. 2010;8:147.
Ma J, Wang S, Zhu X, Sun G, Chang G, Li L, Hu X, Zhang S, Zhou Y, Song CP, Huang J. Major episodes of horizontal gene transfer drove the evolution of land plants. Mol Plant. 2022;15(5):857–71.
Dong Q, Huo J, Wei S, Zhang Z. Black currant production, breeding and processing in China. Acta Hortic. 2012;926:119–22.
Zdunić G, Šavikin K, Pljevljakušić D, Djordjević B. Black (Ribes nigrum L.) and Red Currant (Ribes rubrum L.) cultivars. In: Simmonds MSJ, Preedy VR, editors. Nutritional composition of Fruit cultivars. London: Academic; 2016. 101 – 26.
Sun Q, Wang N, Xu W, Zhou H. Genus Ribes Linn. (Grossulariaceae): a comprehensive review of traditional uses, phytochemistry, pharmacology and clinical applications. J Ethnopharmacol. 2021;276:114166.
Lampi AM, Heinonen M. Berry seed and grapeseed oils. In: Moreau RA, Kamal-Eldin A, editors. Gourmet and Health-promoting Specialty oils. London: Academic Press and AOCS; 2009. 215 – 35.
Vagiri M, Conner S, Stewart D, Andersson SC, Verrall S, Johansson E, Rumpunen K. Phenolic compounds in blackcurrant (Ribes nigrum L.) leaves relative to leaf position and harvest date. Food Chem. 2015;172:135–42.
Redha AA, Siddiqui SA, Zare R, Spadaccini D, Guazzotti S, Feng X, Bahmid NA, Wu Y, Ozeer FZ, Aluko RE. Blackcurrants: a nutrient-rich source for the development of functional foods for improved athletic performance. Food Rev Int. 2024;40(1):135–57.
Lu L. A study on the genus Ribes L. in China. Acta Phytotaxon Sin. 1995;33(1):58–75.
Wang Y, Zhao Y, Bollas A, Wang Y, Au KF. Nanopore sequencing technology, bioinformatics and applications. Nat Biotechnol. 2021;39(11):1348–65.
Sharp PM, Emery LR, Zeng K. Forces that influence the evolution of codon bias. Philos T R Soc B. 2010;365(1544):1203–12.
Song H, Liu J, Chen T, Nan ZB. Synonymous codon usage pattern in model legume Medicago truncatula. J Integr Agr. 2018;17(9):2074–81.
Vicient CM, Casacuberta JM. Impact of transposable elements on polyploid plant genomes. Ann Bot. 2017;120(2):195–207.
Sloan DB. One ring to rule them all? Genome sequencing provides new insights into the ‘master circle’ model of plant mitochondrial DNA structure. New Phytol. 2013;200(4):978–85.
Wu CS, Chaw SM. Evolution of mitochondrial RNA editing in extant gymnosperms. Plant J. 2022;111(6):1676–87.
Garcia LE, Edera AA, Palmer JD, Sato H, Sanchez-Puerta MV. Horizontal gene transfers dominate the functional mitochondrial gene space of a holoparasitic plant. New Phytol. 2021;229(3):1701–14.
The Angiosperm Phylogeny Group, Chase MW, Christenhusz MJM, Fay MF, Byng JW, Judd WS, Soltis DE, Mabberley DJ, Sennikov AN, Soltis PS, Stevens PF. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot J Linn Soc. 2016;181(1):1–20.
Šmarda P, Bureš P, Horová L, Leitch IJ, Mucina L, Pacini E, Tichý L, Grulich V, Rotreklová O. Ecological and evolutionary significance of genomic GC content diversity in monocots. Proc Natl Acad Sci USA. 2014;111(39):E4096–102.
Ayabe H, Toyoda A, Iwamoto A, Tsutsumi N, Arimura SI. Mitochondrial gene defects in Arabidopsis can broadly affect mitochondrial gene expression through copy number. Plant Physiol. 2023;191(4):2256–75.
Ding H, Bi D, Zhang S, Han S, Ye Y, Yi R, Yang J, Liu B, Wu L, Zhuo R, Kan X. The Mitogenome of Sedum Plumbizincicola (Crassulaceae): insights into RNA editing, lateral gene transfer, and phylogenetic implications. Biology (Basel). 2022;11(11):1661.
Grosser MR, Sites SK, Murata MM, Lopez Y, Chamusco KC, Love Harriage K, Grosser JW, Graham JH, Gmitter FG Jr, Chase CD. Plant mitochondrial introns as genetic markers - conservation and variation. Front Plant Sci. 2023;14:1116851.
Khera P, Saxena R, Sameerkumar CV, Saxena K, Varshney RK. Mitochondrial SSRs and their utility in distinguishing wild species, CMS lines and maintainer lines in pigeonpea (Cajanus cajan L). Euphytica. 2015;206:737–46.
Kozik A, Rowan BA, Lavelle D, Berke L, Schranz ME, Michelmore RW, Christensen AC. The alternative reality of plant mitochondrial DNA: one ring does not rule them all. PLoS Genet. 2019;15(8):e1008373.
Gualberto JM, Newton KJ. Plant mitochondrial genomes: dynamics and mechanisms of mutation. Annu Rev Plant Biol. 2017;68:225–52.
Cole LW, Guo W, Mower JP, Palmer JD. High and variable rates of repeat-mediated mitochondrial genome rearrangement in a genus of plants. Mol Biol Evol. 2018;35(11):2773–85.
Satoh M, Kubo T, Mikami T. The Owen mitochondrial genome in sugar beet (Beta vulgaris L.): possible mechanisms of extensive rearrangements and the origin of the mitotype-unique regions. Theor Appl Genet. 2006;113(3):477–84.
Edera AA, Gandini CL, Sanchez-Puerta MV. Towards a comprehensive picture of C-to-U RNA editing sites in angiosperm mitochondria. Plant Mol Biol. 2018;97(3):215–31.
Jiang W, Yang S, Yu D, Gai J. A comparative study of ATPase subunit 9 (Atp9) gene between cytoplasmic male sterile line and its maintainer line in soybeans. Afr J Biotechnol. 2011;10:10387–92.
Knoop V, C-to-U. U-to-C: RNA editing in plant organelles and beyond. J Exp Bot. 2023;74(7):2273–94.
Kadowaki K, Ozawa K, Kazama S, Kubo N, Akihama T. Creation of an initiation codon by RNA editing in the coxI transcript from tomato mitochondria. Curr Genet. 1995;28(5):415–22.
Quiñones V, Zanlungo S, Holuigue L, Litvak S, Jordana X. The cox1 initiation codon is created by RNA editing in potato mitochondria. Plant Physiol. 1995;108(3):1327–8.
Niu Y, Gao C, Liu J. Complete mitochondrial genomes of three Mangifera species, their genomic structure and gene transfer from chloroplast genomes. BMC Genomics. 2022;23(1):147.
Jiang M, Ni Y, Zhang J, Li J, Liu C. Complete mitochondrial genome of Mentha spicata L. reveals multiple chromosomal configurations and RNA editing events. Int J Biol Macromol. 2023;251:126257.
Yang H, Ni Y, Zhang X, Li J, Chen H, Liu C. The mitochondrial genomes of Panax notoginseng reveal recombination mediated by repeats associated with DNA replication. Int J Biol Macromol. 2023;252:126359.
Kolmogorov M, Yuan J, Lin Y, Pevzner PA. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol. 2019;37(5):540–46.
Lu G, Wang W, Mao J, Li Q, Que Y. Complete mitogenome assembly of Selenicereus monacanthus revealed its molecular features, genome evolution, and phylogenetic implications. BMC Plant Biol. 2023;23:541.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60.
Wick RR, Judd LM, Gorrie CL, Holt KE. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. 2017;13(6):e1005595.
Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017;45(W1):W6–11.
Lowe TM, Chan PP. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016;44(W1):W54–57.
Misra S, Harris N. Using Apollo to browse and edit genome annotations. Curr Protoc Bioinf. 2005;12(1):951–9528.
Xiang CY, Gao F, Jakovlić I, Lei HP, Hu Y, Zhang H, Zou H, Wang GT, Zhang D. Using PhyloSuite for molecular phylogeny and tree-based analyses. iMeta. 2023;2(1):e87.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–29.
Beier S, Thiel T, Münch T, Scholz U, Mascher M. MISA-web: a web server for microsatellite prediction. Bioinformatics. 2017;33(16):2583–85.
Behboudi R, Nouri-Baygi M, Naghibzadeh M. RPTRF: a rapid perfect tandem repeat finder tool for DNA sequences. BioSystems. 2023;226:104869.
Kurtz S, Schleiermacher C. REPuter: fast computation of maximal repeats in complete genomes. Bioinformatics. 1999;15(5):426–27.
Cui Y, Cui Z, Xu J, Hao D, Shi J, Wang D, Xiao H, Duan X, Chen R, Li W. NG-Circos: next-generation Circos for data visualization and interpretation. NAR Genom Bioinform. 2020;2(3):lqaa069.
Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–11.
Lo Giudice C, Tangaro MA, Pesole G, Picardi E. Investigating RNA editing in deep transcriptome datasets with REDItools and REDIportal. Nat Protoc. 2020;15(3):1098–131.
Jin JJ, Yu WB, Yang JB, Song Y, Yi TS, Li DZ. GetOrganelle: a simple and fast pipeline for de novo assembly of a complete circular chloroplast genome using genome skimming data. bioRxiv. 2018;4:256479.
Liu C, Shi L, Zhu Y, Chen H, Zhang J, Lin X, Guan X. CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genomics. 2012;13:715.
Liu S, Ni Y, Li J, Zhang X, Yang H, Chen H, Liu C. CPGView: a package for visualizing detailed chloroplast genome structures. Mol Ecol Resour. 2023;23(3):694–704.
Nakamura T, Yamada KD, Tomii K, Katoh K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics. 2018;34(14):2490–92.
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32(1):268–74.
Letunic I, Bork P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49(W1):W293–96.
Wang Y, Li J, Paterson AH. MCScanX-transposed: detecting transposed gene duplications based on multiple colinearity scans. Bioinformatics. 2013;29(11):1458–60.
Acknowledgements
The authors sincerely thank the experimental personnel and bioinformatics analysis team at Wuhan Benagen Technology Co., Ltd (www.benagen.com), as well as the MitoRun research group that participated in this project.
Funding
This research was supported by the Science and Technology Program of Tibet Autonomous Region, China (XZ202301ZY0020N) and the Scientific Research Start-up Fund for High-level Introduced Talents of Henan Institute of Science and Technology (103020224001/073).
Author information
Authors and Affiliations
Contributions
Guilong Lu, Youxiong Que, and Lan Deng conceived and designed the research. Guilong Lu, Wenhua Wang, and Shanshan Zhang performed the experiments and bioinformatic analysis. Guilong Lu performed wrote the initial draft. Lan Deng and Youxiong Que reviewed and edited the manuscript. Guang Yang and Kun Zhang provided the materials. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethical approval and consent to participate
This study’s material collections and experimental research complied with relevant institutional, national, and international guidelines and legislation. No specific permissions or licenses were required.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it.The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lu, G., Wang, W., Zhang, S. et al. The first complete mitochondrial genome of Grossulariaceae: Molecular features, structure recombination, and genetic evolution. BMC Genomics 25, 744 (2024). https://doi.org/10.1186/s12864-024-10654-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10654-y