
Abstract
Background
Biological mechanisms affecting gametogenesis, embryo development and postnatal viability have the potential to alter Mendelian inheritance expectations resulting in observable transmission ratio distortion (TRD). Although the discovery of TRD cases have been around for a long time, the current widespread and growing use of DNA technologies in the livestock industry provides a valuable resource of large genomic data with parent–offspring genotyped trios, enabling the implementation of TRD approach. In this research, the objective is to investigate TRD using SNP-by-SNP and sliding windows approaches on 441,802 genotyped Holstein cattle and 132,991 (or 47,910 phased) autosomal SNPs.
Results
The TRD was characterized using allelic and genotypic parameterizations. Across the whole genome a total of 604 chromosomal regions showed strong significant TRD. Most (85%) of the regions presented an allelic TRD pattern with an under-representation (reduced viability) of carrier (heterozygous) offspring or with the complete or quasi-complete absence (lethality) for homozygous individuals. On the other hand, the remaining regions with genotypic TRD patterns exhibited the classical recessive inheritance or either an excess or deficiency of heterozygote offspring. Among them, the number of most relevant novel regions with strong allelic and recessive TRD patterns were 10 and 5, respectively. In addition, functional analyses revealed candidate genes regulating key biological processes associated with embryonic development and survival, DNA repair and meiotic processes, among others, providing additional biological evidence of TRD findings.
Conclusions
Our results revealed the importance of implementing different TRD parameterizations to capture all types of distortions and to determine the corresponding inheritance pattern. Novel candidate genomic regions containing lethal alleles and genes with functional and biological consequences on fertility and pre- and post-natal viability were also identified, providing opportunities for improving breeding success in cattle.
Similar content being viewed by others

Background
Exceptions to ordinary Mendelian principles, as the non-random transmission of alleles from parents to offspring, have been described in both model and non-model organisms (e.g., [1,2,3,4]. This phenomenon, where Mendelian inheritance expectations are altered (regardless of the cause), is known as transmission ratio distortion (TRD, [5]. From a biological point of view, a wide variety of mechanisms that affect germ cells (e.g., meiotic drive, germline selection, gametic competition, [6], embryo lethality [7], and even differential postnatal viability [8] have the potential to violate Mendel’s law of segregation. Although TRD remains an unclear and ambiguous phenomenon in the scientific literature [9, 10], it can be considered the ultimate outcome of several genetic factors that arise at different stages of the reproductive process and early neonatal life. In fact, particular cases of TRD, such as the absence of homozygosity, are already being used to target recessive defects that affect reproduction (e.g., decreased fertility, embryo loss) in different livestock species [11, 12]. Indeed, the absence of homozygous offspring may also be attributed to inbreeding depression, which is a potential source of TRD [13]. Nowadays, the rapid development of high-throughput genotyping technologies and the increasing number of genotyped animals, with over 3,000,000 Holstein genotypes in North America alone [14], has opened alternative genomic strategies to improve reproductive efficiency. This is crucial because reproductive defects and genetic abnormalities are prevalent and have a significant impact on animal productivity. The TRD approach, which requires only genomic information without phenotypic records, aims to be an applicable strategy with potential outcomes to improve reproductive success in livestock [15]. Within this context, the aim of this study is to investigate TRD in Holstein cattle with the hypothesis that novel chromosomal regions containing lethal alleles or potential genes affecting reproduction can be discovered. This will provide valuable insights into the genetic background of the reproduction.
In this study, we focused on implementing different TRD models based on tracing allele inheritance from parents to offspring using parent–offspring genotype trios of Holstein cattle. The TRD models derived from two parameterizations: (i) allelic parameterization with specific sire- and dam-TRD, or merging both into one overall TRD [15, 16], and (ii) genotypic parameterization modeling interaction between alleles in offspring genotypes, including additive and dominance components of TRD [17]. Therefore, the main objectives of this study were (1) to describe, interpret and compare TRD parameterizations and to highlight their differences and potential applications, (2) to present a comprehensive characterization of TRD across the entire genome of Holstein cattle, (3) to assess the inheritance pattern of regions with known lethal alleles in order to determine the reliability of the TRD approach, (4) to identify novel genomic regions with moderate to high TRD penetrance potential to harbor deleterious mutations, and (5) to annotate genes and to evaluate the functional consequences of TRD regions using functional analyses.
Results and discussion
Prevalence of TRD across Holstein genome
Decisive evidence (Bayes factor (BF) ≥ 100) according to Jeffreys’ scale [18] was identified for TRD in both individual SNPs and haplotypes across the Holstein genome. All the post TRD analyses criteria were implemented to minimize and discard TRD artifacts coming from genotyping errors and random TRD given the sample size of informative offspring. Particularly, the specific approximate empirical null distributions of additive- and dominance-TRD used to discard random TRD from the genotypic model were developed following [19] and summarized in the supplementary Material 1. After applying the previously given filtering criteria, 271 and 700 SNPs were identified with distorted segregation from SNP-by-SNP analyses in raw- and imputed-data, respectively. Using imputed data for haplotype analyses, the total numbers of allele-regions with TRD were 3,115, 8,566, 17,558, 25,638 and 39,433 for 2-, 4-, 7-, 10- and 20-SNP haplotypes, respectively. The potential of capturing more signals of TRD with haplotype-based method is given its ability to exploit the available SNPs and provide additional range of allele frequencies across the whole genome as reported by Id-Lahoucine et al. [19] in more details. Among these findings, it is worth highlighting that the majority of regions were detected with more than one of the models applied (i.e., parent-unspecific model, parent-specific model and genotypic model). Only 3.06% and 2.49% regions were identified uniquely on parent-specific or genotypic TRD models, respectively. Despite this overlap, different statistical significance values were obtained for TRD estimates, suggesting different degrees of fit among the models. After exclusively keeping the allele-region (i.e., a SNP or a haplotype from a window) with the highest BF within a region, 51,364 regions were identified (including totally or partially overlapped windows). This reported initial number of regions can be considered reasonable for TRD phenomenon in comparison to results in Hoff et al. [12], who reported 12,020 haplotypes of 20-SNP with absence of homozygosity in 3,993 genotyped animals and a minor allele frequency (MAF) of 0.02.
Rare variants identified by TRD approach
Most of the regions detected with TRD presented low frequencies and were supported by the large dataset. Among them, 47,839, 44,424, 40,358, 36,864, 26,419, 18,516 and 5 regions had a MAF < 0.05, 0.02, 0.01, 0.005, 0.001, 0.0005 and 0.0001, respectively. These findings, in contrast to other methodologies, revealed the TRD approach as a very powerful method to detect rare variants. In fact, it has been suggested that targeting rare variants is more power to detect causal mutations than common variants [20]. Moreover, Ghanem and Nishibori [21] reported that most of the recessive variants associated with a decline in fertility and pregnancy losses in cattle are difficult to discover and may not be detected.
Integration of TRD mapping
A total of 51,364 regions were identified across the whole genome. This high number of regions is a result of different features, including the sliding window approach used, the different window sizes implemented, the level of linkage disequilibrium (LD) and the patterns of TRD observed for individual SNPs (or short haplotypes), which are also displayed across the haplotypes that include them. We assumed that the different patterns observed across adjacent regions were generated from one single mutation that underlies the observed TRD. The allele-region with the highest BF was assumed to be the best candidate region harboring the causal variant or in strong LD with it. This assumption was made taking into consideration that the BF simultaneously combines both the magnitude of TRD and the number of informative offspring [19]. After the integration of LD with the smoothing process in order to obtain clear highlighted peaks of TRD across the whole genome, 797 core regions were differentially identified. This result was based on bandwidth of 500,000 bp for the smoothing process, which was assumed as a sensible distance to obtain a considerable initial number of candidate regions. Notice that the choice of the bandwidth for smoothing is an important component of its implementation. Figure 1 shows the different patterns of kernel smoothing for several fixed bandwidths across a single chromosome with the rescaled smoothed BF. Here, with bandwidth of 500,000 bp a specific region was assumed to show significant TRD at 95, 99 and 99.9% up to ± 980,000, ± 1,290,000 and ± 1,645,000 bp, respectively. Among the obtained 797 core regions after the smoothing process, 193 regions were excluded as they were plausibly explained by genotyping errors after individually checking the parameters estimated and the corresponding distribution of the offspring across matings. Supplementary Material 2 summarizes the information for the final 604 chromosomal regions deemed with TRD.

Bayes factor for transmission ratio distortion in BTA1 with Kernel smoothing using different bandwidth (i.e., 500,000, 2,000,000 and 5,000,000 bp)
Goodness-of-fit and pattern of TRD
Multiple mechanisms from gamete formation in the parental generation to offspring viability can cause TRD. In order to differentiate among the various potential causes, Pardo-Manuel de Villena et al. [5] developed a test to discriminate between meiotic and postmeiotic mechanisms of TRD based on the recombinational status between the centromere and the distorted locus. Leppälä et al. [22] used a likelihood ratio test to distinguish between gametic or zygotic TRD in classical experimental designs of F2 crosses. The different models developed by Casellas et al. 2012, 16, 15, 18, arising from different complementary parameterizations, can determine the genetic mechanism (or inheritance pattern) involved in a region and even provide additional evidence on the level where the TRD is likely to be [23].
When comparing different TRD models across the Holstein genome, different goodness-of-fit in terms of deviance information criterion (DIC) were observed. Models with smaller DIC values indicate a better fit, and differences between models greater than 3 DIC units are considered statistically relevant [24]. For allelic parameterization, in the absence of parent-specific TRD, similar magnitudes of TRD effects (i.e., overall, sire- and dam-TRD) and similar fit with minimal differences in DIC units between the parent-unspecific and -specific models, were observed, supporting the overall TRD pattern. However, under a parent-specific TRD pattern, differences from 3 up to 599 DIC units were observed between the allelic models (i.e., parent-unspecific and -specific TRD). On the other hand, the genotypic model was favored in certain genomic regions reaching reductions up to 6,442.85 DIC units in relation to the allelic model.
Different TRD models were clearly favored by DIC among TRD regions suggesting the corresponding inheritance pattern of the observed TRD. Within this context, among 604 identified regions, 195 fit better to the allelic model with overall TRD. When the parent-specific model displayed minimal DIC units, 91 regions exhibited sire-TRD (null via dam), 6 showed dam-TRD (null via sire) whereas 220 regions presented both sire- and dam-TRD with different magnitudes (i.e., different penetrance or LD among them). Finally, the genotypic model fit better for 92 regions highlighting the corresponding additive and dominance components of TRD.
Allelic versus genotypic patterns
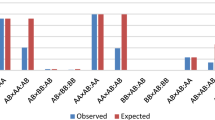
From a biological point of view, the allelic parameterization, which focuses on a single-allele basis, is more likely to be associated with TRD occurring prior to fertilization (i.e., haploid phase), whereas the genotypic parameterization is more appropriate for events occurring after fertilization given its basis on the (combined) genotype itself (i.e., during the diploid phase; [23]. However, the peculiarity of TRD findings on real data, such as the low frequency of alleles on TRD regions, must be taken into consideration. Most of the identified TRD regions showed a lack of some types of matings and an imbalanced number of trios. Thus, a relatively few (or none) heterozygous-by-heterozygous mating and the absence or near absence of homozygous individuals in both parental and offspring generations further supports deleterious effects of the identified TRD regions. Nevertheless, at the biological level, the imbalanced number of trios among matings or the absence of specific matings could make it difficult to differentiate between allelic and genotypic related TRD phenomenon [23]. Therefore, although most regions detected showed allelic patterns (~ 85%), this does not ensure their occurrence before fertilization. In fact, functional and positional evidence for regions with the allelic pattern suggested that biological related events act both pre-and post-fertilization. Further, the presence or absence of a specific allele, independently of the homologous one, may be sufficient to induce lethality and consequently generate TRD. Moreover, taking into consideration the moderate-to-high correlation between the overall TRD and additive-TRD, the allelic patterns could also be viewed as an additive effect in the offspring genotype, where the presence of the allele gives a dosage effect, reducing the viability of carrier offspring. It must be noted that an important part of regions with allelic pattern showed complete or quasi-complete absence (lethality) for homozygous individuals, but also an under-representation (reduced viability) of heterozygous offspring. Indeed, Khatib et al. [25] described a similar pattern of distortion in a fertility candidate gene study. These observed patterns across the Holstein genome emphasize the importance of the allelic parameterization. Table 1 and Fig. 2 illustrates the distribution of offspring for each type of mating showing different patterns of TRD with the corresponding TRD estimates for the fitted model.

Distribution of offspring across matings (Sire × Dam:Offpsring) from different regions with transmission ratio distortion
On the other hand, the genotypic model was sensitive to the lack of some type of matings and the imbalance in number of trios on TRD estimations. Under these conditions, TRD estimates must be interpreted with caution, as they could be less accurate due to the ability of the model to converge to a combination of additive- and dominance-TRD that maximize the imbalance of matings. Nevertheless, when a clear interaction between alleles is observed and supported by the distribution of the offspring genotypes, this could substantiate the post fertilization events of the observed TRD. For example, the genotypic model highlighted regions with the classical recessive patterns, in these cases, homozygous offspring were not observed from heterozygous-by-heterozygous matings, whereas the theoretical Mendelian ratio (i.e., null TRD) was maintained on heterozygous-by-homozygous matings. Recessive haplotypes as Holstein haplotype 0 (HH0, [26, 27], haplotype 1 (HH1, [11, 28], haplotype 3 (HH3, [11, 29] and haplotype 5 (HH5, [30], which are known to be due to embryonic lethality [21, 31], were also identified with a genotypic recessive pattern when analyzed by TRD approach presented here (Table 2). Other patterns found with the genotypic model can be related to heterosis effect. In these regions, an excess or deficiency of heterozygous offspring was observed (Table 1). A similar pattern of heterozygote excess was also observed in plants [9]. We cannot rule out that the mentioned heterozygote advantage may come from a parent-specific TRD, where different alleles have opposite preferential transmissions, resulting in an over-representation of heterozygous offspring.
In general, independently of the moment when the TRD occurred, both parameterizations showed to be complementary, allowing to capture different types of TRD, emphasizing the importance of implementing the three models. It is remarkable that the prevalence of regions with recessive TRD patterns is small in comparison to allelic TRD regions, which may potentially explain part of the reproductive inefficiency of the animals. Embryo losses during the first 42 days range from 30 to 40% in most domestic species [32] and average calving rates after single insemination are 57.5% and 37.5% for heifers and lactating Holstein cows, respectively [33], which exemplify reproductive inefficiencies that might be related to allelic TRD regions. In addition, it is worthy to highlight that the implementation of full trios allowed clear differentiation between recessive and allelic patterns, as heterozygous-by-heterozygous matings are necessary to predict the lethality of homozygous genotypes. Notice that the few or none of the trios of the heterozygous-by-heterozygous matings of the detected regions, in addition to their law chance given the low frequency, it could also be partially explained by producers’ mating decisions to avoid inbreeding.
Examining the inheritance patterns of the previously described lethal haplotypes
The inheritance patterns of already published regions with lethal alleles were examined to validate the finding of the TRD approach. Table 2 summarizes the previously describe recessive haplotype regions with TRD findings (additional results are presented in Supplementary Material 3).
Inheritance pattern of Holstein lethal haplotypes
HH0 [26], HH1 [11, 28] and HH3 [11, 29] were described previously by VanRaden et al. [11] with 22, 23 and 7 non-observed homozygous offspring from heterozygous carrier sires with heterozygous carrier maternal grandsire matings (Table 2), respectively. Haplotype-windows with recessive patterns were detected by the genotypic model using TRD approach covering HH0 haplotype or the corresponding causal mutation itself for HH1 and HH3 haplotypes. TRD corresponding results showed 43, 14 and 118 non-observed homozygous offspring from double heterozygous mating, respectively (Table 2). A haplotype displaying a genotypic TRD pattern with 156 non-observed homozygous offspring was also detected at ~ 800 Kbp from HH5 [30]. These 4 recessive Holstein haplotypes were identified displaying an additive-TRD ranging from -0.60 to -0.66 and their unfavorable effects were minimized by a favorable dominance-TRD effects (ranging from 0.29 to 0.33) in heterozygous offspring, allowing for their survival. Holstein haplotype 2 (HH2, [11, 29], haplotype 4 (HH4, [34], bovine leukocyte adhesion deficiency (HHB, [35], deficiency of uridine monophosphate synthase (HHD, [36] and Holstein cholesterol deficiency (HCD, [30] regions were also identified as exhibiting significant signals of TRD with allelic patterns. These were not only detected observing the absence (or relative absence) of homozygous offspring, but also an important reduced number of heterozygous offspring from heterozygous-by-homozygous matings (Table 2, Supplementary Material 3). For the complex vertebral malformation (HHC, [37], alleles across this region were identified with either an allelic or genotypic pattern. The genotypic pattern showed an interaction between alleles with a disadvantage for homozygous haplotypes (αg = -0.05 and δg = 0.04,Table 2, Supplementary Material 3). Moreover, a SNP (BTA18:61,231,528) and a haplotype (BTA18:58,551,307–58,696,066) of TRD regions were observed to be physically close to a QTL with a major effect on various fertility traits, such as calving ease and stillbirth. These QTL were reported by various studies during the last decades (e.g., [38, 39] and reviewed and confirmed by Müller et al. [40] and investigated at sequence level more recently by Dachs et al. [41]. In our TRD results, the haplotype (BTA18:58,551,307–58,696,066) was detected with an allelic pattern and more strongly via sire (αs = -0.44) than dam (αd = -0.21) and the SNP (BTA18:61,231,528) showed a recessive TRD pattern with 448 non-observed homozygous offspring less than expected. Furthermore, from an economic perspective, it is worth noting from that the annual loss attributed to lethal alleles in Holstein cattle has been estimated to be $7,500,265 in the US [31]. This highlights the significance of our research in identifying novel lethal alleles and improving the fertility and reproductive performance of Holstein cattle.
Concordance of TRD findings across breeds
When examining regions with known reproduction defects discovered in other cattle breeds, patterns of TRD were also identified in the Holstein data analyzed in this study. Jersey haplotype 1 (JH1; [11, 42] and Jersey haplotype 2 (JH2, [43] were identified with haplotype alleles displaying overall TRD of -0.13 and -0.15, respectively. Brown Swiss haplotype 1 (BH1, [11], Brown Swiss haplotype 2 (BH2, [11, 44], spinal muscular atrophy (BHM, [45, 46], bovine progressive degenerative myeloencephalopathy or weaver (BHW, [47, 48] and spinal dysmyelination (BHD, [49, 50] were identified with haplotype alleles displaying overall TRD of -0.46, -0.25, -0.44, -0.03 and -0.24, respectively (Table 2, Supplementary Material 3). The Ayrshire haplotype 1 (AH1, [51, 52] was also identified, with a haplotype displaying an α = -0.33. Finally, among the candidate lethal haplotypes described with absent homozygosity in Angus cattle by Hoff et al. [12], signals of TRD were also observed, overall TRD ranged from -0.17 to -0.41 on these regions (Table 2, Supplementary Material 3). All theses findings suggest, with the assumption of no recent common ancestor between these breeds and Holstein, that probably independent mutations occurred in the same genes which resulted in generated signals of TRD in the same regions, and consequently, may support the biological function of those genes on reproduction-related traits. In addition, to the best of our knowledge, there have been no investigations on these regions in Holstein cattle to date.
Novel candidate lethal allele regions
The ultimate goal of this study was to discover novel candidate lethal alleles, genes and even causal mutations directly affecting reproduction and survival. For this, potential regions were chosen to take into consideration the number of under-represented offspring and the magnitude of TRD itself. The number of under-represented offspring, considering the allelic model, is approximately equal to the number of informative offspring multiplied by twice the TRD magnitude, which also corresponds to the sum of the differences between the offspring genotypes within matings. In this context, if one assumes embryonic lethality to be the cause of a specific TRD region, the number of under-represented offspring could be viewed as the possible number of embryo losses. Thus, among the TRD findings, the number of regions with ≥ 1,000 under-represented offspring were 330 and reduced to 146, 102, 69, 35 and 11 when considering ≥ 2,500, 5,000, 10,000, 20,000 and 30,000 under-represented offspring.
On the other hand, as already introduced by Id-Lahoucine et al. [19], the magnitude of TRD describes the probability of an allele being transmitted to viable offspring suggesting its corresponding penetrance. If embryonic lethality is considered as the potential cause of observed TRD, then the probability of observing embryo losses will increase as the magnitude of TRD increases. Therefore, the usefulness of regions with strong TRD (e.g., SNP with high LD with causal mutation or haplotype harboring the causal mutation) for breeding purposes is greater compared to those with lower TRD magnitudes. Furthermore, it is worth noting that the TRD magnitudes are estimated based on population-level data. Therefore, it is possible that both low and moderate TRD signals are associated with specific causal mutations that have been generated and spread within particular families, and where further research utilizing the full pedigree is needed in order to target the origins of TRD.
In this study, after excluding previously known regions in Holsteins, 10 potential regions displayed |TRD|> 0.25 and ≥ 5,000 under-represented offspring and are summarized in Table 3. Three additional haplotypes and 2 SNPs were identified using the genotypic model as exhibiting recessive patterns (highly lethal in homozygous state), with ≥ 5,000 informative offspring and ≥ 45 non-observed homozygous offspring (Table 3). The 3 haplotype alleles with recessive patterns on BTA1 were physically close to each other (covering 10,696 Kbp) and showed similar TRD magnitudes, frequency and number of heterozygous sires, dams and non-observed homozygous offspring (Table 3), which potentially points to the same causal mutation (SNP, deletion, etc.). The LD between these 3 haplotypes ranged from 0.65 to 0.83. In addition, from 15,726 individuals carried a copy of the potential lethal allele in at least one of the 3 regions, 10,726 and 2,160 individuals carried a copy of the potential lethal allele in 3 and 2 regions simultaneously, respectively. This result gives extra evidence supporting the TRD found in this region.
Functional analyses of candidate lethal allele regions
The positional candidate genes annotated in (i) the 100 Kbp interval downstream and upstream from the potential lethal SNPs and (ii) the genes mapped within the interval of the candidate lethal TRD haplotypes are shown in Supplementary Material 4. In total, 1,400 positional candidate genes were successfully uploaded in ingenuity pathway analysis (IPA) software. The canonical pathways and diseases and functions identified by IPA are shown in Supplementary Materials 5 and 6, respectively. Two canonical pathways were significantly enriched among the genes annotated in the potentially lethal TRD regions: nucleotide excision repair (NER) pathway (p-value = 0.025; genes = HIST2H4A, POLE2) and purine nucleotides de novo biosynthesis (p-value = 0.026; genes = GMPS). Genes implicated in NER pathway are involved in the process of DNA repair, NER eliminates structural DNA lesions, such as bulky, helix-distorting adducts [53]. DNA repair, specifically NER pathway, has been shown to be crucial for fertility, as this mechanism is essential to maintain the fidelity of DNA replication during mitotic, meiotic processes in both male and female germ cells [54, 55]. On the other hand, purine de novo biosynthesis has shown to be critical during early embryo preimplantation development in mouse embryos [56].
A total of 178 enriched diseases were identified (p-value < 0.05; Supplementary Material 6). The main enriched diseases and functions, together with the canonical pathways, were represented in a network interaction between the positional candidate genes and the related terms. Two main networks were created (Figs. 2 and 3), highlighting a relevant number of genes associated with important biological processes associated with embryo development, cell survival and reproduction.

Network interaction between positional candidate genes on TRD regions and cellular development related canonical pathways and biological functions. The first layer corresponds to the canonical pathways, the second layer to the positional candidate genes and the third layer, the biological pathways. The colors of the positional candidate genes correspond to the observed TRD pattern, i.e. recessive TRD (in blue) and sire TRD (in pink)
The first network, represented in Fig. 3, is composed by the genes bta-mir-128, ARPP21, PLEKHO1, HIST2H3C, HIST2H4A, NLRP12, BCHE, and VNN1. The last three genes were associated with TRD markers displaying a recessive TRD, while the other genes were associated with markers displaying a sire TRD. This network shows an interaction between genes and biological functions associated with the development and maintaining bone marrow cells, osteoclast and bone cell lines, and differentiation of neuronal cells. Among the genes located in regions with sire TRD, it is worthy to highlight the presence of HIST2H3C and HIST2H4A genes. The HIST2H3C and HIST2H4A genes codify for Histone Cluster 2 H3 Family Member C and Histone Cluster 2 H4 Family Member A, respectively. The exchange of histone-to-protamine in sperm chromatin remodeling is a key step for fertilization, because this process determines the degree of chromatin condensation [57]. Among the genes located in regions identified showing recessive TRD, the VNN1 gene codifies a glycophosphoinositol-anchored glycoprotein highly expressed in the Sertoli cells showing dysmorphic expression between male and female gonadal cells, indicating a role in the mammalian sexual development [58]. Additionally, VNN1 plays a crucial role in the regulation of chondrogenesis [59].
The NLRP12 gene encodes for the NLRP protein 12 (nod-like receptors with a pyrin domain). Although this protein family has a major role in innate immunity, there are several studies in the last decade that highlight their importance in oocytes and early embryos [60, 61]. Lastly, the BCHE gene encodes for butyrylcholinesterase enzyme. Studies in humans suggested that impaired activity of butyrylcholinesterase in the uterus may increase uterine motility and contraction and decrease fertility [62]. Moreover, butyrylcholinesterase activity has shown to be reduced in humans with abnormal seminal parameters, such as sperm count and mobility [63].
The second interaction network (Fig. 4) is composed of the genes BCHE, TAAR1, VNN1, SLITRK3, SV2A, OPCML, and FCGR1A. The first four genes are associated with markers displaying a recessive TRD pattern, while the last three genes are associated with markers displaying a sire TRD pattern. The biological functions showed in this interaction network are associated with immune response (purple), development of the nervous system (green), and fertility (orange). Among the genes in this network, the SLITRK3 gene has been shown to be upregulated by the transcription factor gene cluster RHOX (X-linked reproductive homeobox). The RHOX gene cluster is expressed mainly in reproductive tissues and is known to have key roles in male fertility in mice and human, which suggest that SLITRK3 may be one of the genes involved in the reproductive functions promoted by RHOX [64]. The FCGR1A gene encodes Fc fragment of IgG receptor Ia, which plays an important role in the immune response. A recent study evaluating the transcriptome of corpus luteum in sheep have highlighted the importance of immune system during early pregnancy, being FCGR1A one of the genes upregulated in high prolificacy sheep [65].

Network interaction between positional candidate genes on TRD regions and cell to cell interactions. The colors of the positional candidate genes correspond to the observed TRD pattern, i.e. recessive TRD (in blue) and sire TRD (in pink). The diseases and biological functions associated with the positional candidate genes were colored based on the functional similarity. The processes associated with immune response were colored in purple, the processes associated with the development of the nervous system was colored in green, and the processes associated with fertility were colored in orange
Conclusions
Our study aimed to elucidate the prevalence of biased allele transmission in Holstein cattle. The results revealed the importance of implementing different TRD parameterizations to capture all types of distortions and to determine the corresponding inheritance patterns. The genotypic model highlighted alleles with classical recessive patterns, whereas the allelic model highlighted chromosomal regions with complete or quasi-complete absence for homozygous individuals and an under-representation (reduced viability) of the carrier heterozygous offspring as well. The full characterization of the genome allowed the identification of 604 chromosomal regions. Among them, the number of most relevant novel regions with strong allelic and recessive TRD patterns was 10 and 5, respectively. Additionally, the detection of TRD on previously published regions harboring recessive lethal alleles validated the TRD approach. Finally, novel candidate genomic regions containing lethal alleles and genes with functional and biological consequences on fertility and pre- and post-natal viability were also identified, which provides opportunities for improving breeding success in cattle.
Methods
Genotypes and trios
The dataset used in this study consisted of 441,802 Holstein genotypes from the Canadian Dairy Network (CDN) database (Lactanet, Guelph, ON). These animals were sampled from all available Holstein genotypes (> 1 million; October 2017), combining trios (sire-dam-offspring) with offspring genotyped within 90 days of birth, thus minimizing pre-selection of offspring [19, 66]. The total number of animals genotyped in trios were 340,363. The majority of trios (330,749) was structured in large paternal half-sib families with 10 to 5,725 offspring. The number of sires with at least 100 or 500 offspring was 572 and 150 with a total of 280,292 and 190,555 offspring, respectively. The animals of investigated trios, sires (n = 5,976), dams (n = 132,282) and offspring (n = 340,363) were genotyped with 22 different SNP genotyping arrays ranging from 2,900 to 777,962 SNPs (Table 4) and mapped to the UMD3.1 Bos taurus genome assembly. Given the different genotyping arrays, the number of available trios is different across SNPs. Only SNP markers with at least 10 trios were selected for TRD analyses using the original genotypes (raw-data, see Statistical Analyses section), resulting in a total of 132,991 autosomal SNPs distributed across the whole genome with different sample sizes of trios (from 10 to 340,363).
Imputation to BovineSNP50 array
A total of 43,710 animals genotyped with the BovineSNP50 BeadChip (55,647 SNPs; Illumina, Inc., San Diego, CA) were used as a reference for imputation. The total number of autosomal SNPs was 47,910, after excluding SNPs with low genotype call rate (< 90%; map file in Supplementary Material 7). In order to ensure a high accuracy for imputation, we excluded animals genotyped with the lowest density (i.e., Illumina Bovine3K BeadChip (2,900 SNP)). Thus, the number of genotyped trios reduced to 283,817. The number of parents in the imputed data were 5,224 and 117,316 for sires and dams, respectively. The total number of genotyped animals was 373,793, which were imputed and phased using FImpute [67] with the option for population and family (pedigree) imputation to provide a more accurate imputation. The 47,910 imputed SNP genotypes were then used in the TRD analyses (imputed-data, see Statistical Analyses section).
Analytical models of transmission ratio distortion
Allelic parameterization of TRD
As described by Casellas et al. [15, 16], the probability of allele transmission (P) from heterozygote parents (A/B) to offspring was parameterized either including one overall TRD effect (α) on a parent-unspecific model or differentiating between sire- (αs) and dam-specific (αd) TRD effects on a parent-specific model:
where; α, αs and αd are TRD parameters which assumed flat priors within a parametric space ranging from -0.5 to 0.5. Under a Bayesian implementation, the conditional posterior probabilities of the TRD parameters are defined as:
where y is the column vector of genotypes of the offspring generation.
Genotypic parameterization of TRD
As developed by Casellas et al. [17], genotypic parameterization can be modeled by assuming additive (αg) and dominance (or over- / under-dominance,δg) parameters, regardless of the origin of each allele. Following Casellas et al. [23], the probability of the offspring (Poff) from heterozygous-by-heterozygous mating are:
where; αg and δg are additive- and dominance-TRD parameters, respectively. For heterozygous-by-homozygous mating, correction for overall losses of individuals in terms of genotypic frequency are needed to guarantee Poff(AA) + Poff(AB) + Poff(BB) = 1. Thus, genotypic frequencies in offspring from AA × AB mating as an example become:
Under a Bayesian implementation, the conditional posterior probabilities of the TRD parameters are defined as:
where y is the column vector of genotypes of the offspring generation. Flat priors were assumed for both αg and δg within an extended parametric space. The initial range of the parametric space for αg was [-1, 1] with a p(αg) = ½ and became restricted to [-1 + δg, 1- δg] with a p(αg) = 2 / (2—2 × δg) when δg > 0. For δg, the parametric space range was [-1, |αg|] with a p(δg) = 1/(1 + αg).
TRD implementation on haplotype windows
To minimize random TRD and genotyping errors, the biallelic-haplotype procedure described by Id-Lahoucine et al. [19] was implemented to perform haplotype analyses. Assuming no specific interaction between alleles on parental generation under the same previously described parameterization for SNP markers, TRD parameters for each haplotype (Hj) are generalized to:
where; Hj is the particular j haplotype under analyses, H-j are the remaining alleles excluding j, αj, αsj and αdj are the overall, sire- and dam-specific TRD for the specific haplotype j, respectively.
The same strategy was implemented for the genotypic model, where from a heterozygous-by-heterozygous mating the probabilities of the offspring become:
where; αgj and δgj are additive- and dominance-TRD parameters for the specific haplotype j, respectively.
Statistical analyses
Transmission ratio distortion was evaluated SNP-by-SNP across 132,991 SNPs (raw-data) and 47,910 SNPs (imputed-data) and using a sliding windows haplotype approach of 2-, 4-, 7-, 10- and 20-SNP across 47,910 SNPs. The average distance in base pairs between adjacent SNPs in the imputed data was 52,248. The analyses were performed within a Bayesian framework using TRDscan v.1.0 software [19] with a unique Monte Carlo Markov chain of 110,000 iterations, where the first 10,000 iterations were discarded as burn-in. The statistical significance of TRD was evaluated using a Bayes factor [68]. Both allelic and genotypic parameterizations were compared using the deviance information criterion [24] to determine the goodness-of-fit and the inheritance pattern of each region. In order to optimize TRD analyses, TRD regions initially identified with at least one of the three models, were subsequently filtered following Id-Lahoucine et al. [19]. Firstly, a minimal number of informative parents (≥ 20 heterozygous sires and/or ≥ 100 heterozygous dams) were considered to minimize possible false TRD from genotyping errors. Secondly, regions with few heterozygous sires displaying full skewed transmission and completely explaining the observed TRD in the corresponding region, were discarded as potential genotyping errors. Third, the approximate empirical null distribution of TRD [19] at < 0.001% margin error was used in order to eliminate TRD generated by chance (i.e., gamete sampling). Subsequently, regions with a large credible interval for TRD effects (i.e., coefficient of variation > 20%) given the unstable convergence, were filtered out. Finally, in order to integrate all the results to obtain clear highlighted peaks of TRD across the whole genome, a non-parametric technique, known as kernel smoothing [69, 70] was applied. The smoothed estimate of BF for the ith base pair (bp) within the range κ1 to κn was calculated using weighted Gaussian kernel (\({\widehat{\mathrm{y}}}_{\mathrm{i}}=\sum_{\mathrm{j}=1}^{\mathrm{n}}\frac{1}{\sqrt{2{{\pi \sigma }}^{2}}}\mathrm{exp}\left(-\frac{{({\mathrm{k}}_{\mathrm{i}}-{\mathrm{k}}_{\mathrm{j}})}^{2}}{2{\upsigma }^{2}}\right)\times {\mathrm{BF}}_{\mathrm{j}}\)), where σ is the bandwidth, (κi—κj) is the distance in base pairs, and n is the total number of TRD regions included. The choice of the bandwidth for smoothing is an important component of its implementation, and different values were tested: σ = 500,000, 2,000,000 and 5,000,000 bp.
Functional analyses
Fifteen potential new candidate regions with lethal alleles were annotated using the R package: Genomic functional Annotation in Livestock for positional candidate Loci, also known as ‘GALLO’ [71]. The.gtf file corresponding to the bovine gene annotation from UMD 3.1 assembly and the.gff file with the QTL information from Animal QTL Database [72] were used for gene and QTL annotation, respectively. In order to map the genes around SNPs displaying significant TRD an interval of 100 Kilobase pairs (Kbp) upstream and downstream from the SNP coordinate was used. For those regions displaying significant TRD identified through the haplotype analyses, the positional candidate genes were annotated within the coordinates for the haplotype interval. These positional candidate genes were investigated regarding its functional profile through an enrichment analysis for Canonical pathways, diseases and functions using ingenuity pathway analysis (IPA) software (QIAGEN Inc., Fall Release 2019,http://www.ingenuity.com; [73, 74]. However, first, the R package biomaRt v. 2.40.5 [75] was used to obtain the bovine Ensembl ID, the Gene Symbol and the respective orthologous in humans and mouse for each positional candidate gene. Only the orthologous genes showing a similarity higher than 75% with the bovine annotated genes were retained as the input for the IPA software. A significance threshold of p-value < 0.05 was adopted to consider an enrichment for all the categories tested (Canonical pathways, diseases and functions).
Availability of data and materials
The data that support the findings of this study are available from Lactanet (Guelph, Canada) but restrictions apply to the availability of these data, which were used under research agreement for the current study, and so are not publicly available. Data are however available from the Angela Canovas upon reasonable request and with permission of Lactanet (Guelph, Canada).
References
Dunn LC. Evidence of evolutionary forces leading to the spread of lethal genes in wild populations of house mice. Proc Natl Acad Sci USA. 1957;43:158–63.
Eaves EA, Bennett ST, Forster P, Ferber KM, Ehrmann D, Wilson AJ, et al. Transmission ratio distortion at the INS-IGF2 VNTR. Nat Genet. 1999;22:324–5.
Fishman L, Willis JH. A novel meiotic drive locus almost completely distorts segregation in mimulus (monkeyflower) hybrids. Genetics. 2005;169:347–53.
Abdalla E, Id-Lahoucine S, Cánovas A, Casellas J, Schenkel FS, Wood BJ, et al. Discovering lethal alleles across the turkey genome using a transmission ratio distortion approach. Anim Genet. 2020;51:876–89.
Pardo-Manuel de Villena F, de la Casa-Esperon E, Briscoe TL, Sapienza C. A genetic test to determine the origin of maternal transmission ratio distortion. Meiotic drive at the mouse Om locus. Genetics. 2000;154:333–42.
Huang LO, Labbe A, Infante-Rivard C. Transmission ratio distortion: Review of concept and implications for genetic association studies. Hum Genet. 2013;132:245–63.
Zöllner S, Wen X, Hanchard NA, Herbert MA, Ober C, Pritchard JK. Evidence for extensive transmission distortion in the human genome. Am J Hum Genet. 2004;74:62.
Moore CS. Postnatal lethality and cardiac anomalies in the Ts65Dn Down Syndrome mouse model. Mamm Genome. 2006;17:1005–12.
Dai B, Guo H, Huang C, Ahmed MM, Lin Z. Identification and characterization of segregation distortion loci on cotton chromosome 18. Front Plant Sci. 2017;7:2037.
Nadeau JH. Do Gametes woo? Evidence for their nonrandom union at fertilization. Genetics. 2017;207:369–87.
VanRaden PM, Olson KM, Null DJ, Hutchison JL. Harmful recessive effects on fertility detected by absence of homozygous haplotypes. J Dairy Sci. 2011;94:6153–6.
Hoff JL, Decker JE, Schnabel RD, Taylor JF. Candidate lethal haplotypes and causal mutations in Angus cattle. BMC Genomics. 2017;18:799.
Hall MC, Willis JH. Transmission ratio distortion in intraspecific hybrids of Mimulus guttatus: Implications for Genomic Divergence. Genetics. 2005;170:375–86.
Lactanet. 10 Years of Genomic Selection: What’s Next?. 2019. https://www.cdn.ca/document.php?id=530. Accessed 20 Jan 2020.
Casellas J, Cañas-Álvarez JJ, González-Rodríguez A, Puig-Oliveras A, Fina M, Piedrafita J, et al. Bayesian analysis of parent-specific transmission ratio distortion in seven Spanish beef cattle breeds. Anim Genet. 2017;48:93–6.
Casellas J, Manunza A, Mercader A, Quintanilla R, Amills M. A flexible bayesian model for testing for transmission ratio distortion. Genetics. 2014;198:1357–67.
Casellas J, Gularte RJ, Farber CR, Varona L, Mehrabian M, Schadt EE, et al. Genome scans for transmission ratio distortion regions in mice. Genetics. 2012;191:247–59.
Jeffreys H. Theory of probability. Oxford: Clarendon Press; 1984.
Id-Lahoucine S, Cánovas A, Jaton C, Miglior F, Fonseca PAS, Miller S, et al. Implementation of bayesian methods to identify SNP and haplotypes regions with transmission ratio distortion across the whole genome: TRDscan v.1.0. J Dairy Sci. 2019;102:1–14.
Gorlov IP, Gorlova OY, Sunyaev SR, Spitz MR, Amos CI. Shifting paradigm of association studies: value of rare single-nucleotide polymorphisms. Am J Hum Genet. 2008;82:100–12.
Ghanem ME, Nishibori M. Haplotypes associated with fetal death and abortion in Holstein cows with special reference to the situation in Japan. J Anim Genet. 2018;46:25–30.
Leppälä J, Bokma F, Savolainen O. Investigating incipient speciation in Arabidopsis lyrata from patterns of transmission ratio distortion. Genetics. 2013;194:697–708.
Casellas J, Id-Lahoucine S, Cánovas A. Discriminating between allele- and genotype-specific transmission ratio distortion. Anim Genet. 2020;516:847–54.
Spiegelhalter DJ, Best NG, Carlin BP, van der Linder A. Bayesian measures of model complexity and fit. J R Stat Soc Series B Stat Methodol. 2002;64:583–639.
Khatib H, Monson RL, Schutzkus V, Kohl DM, Rosa GJM, Rutledge JJ. Mutations in the STAT5A Gene Are Associated with Embryonic Survival and Milk Composition in Cattle. J Dairy Sci. 2008;91:784–93.
Agerholm JS, McEvoy F, Arnbjerg J. Brachyspina syndrome in a Holstein calf. J Vet Diagn Invest. 2006;18:418–22.
Charlier C, Agerholm JS, Coppieters W, Karlskov-Mortensen P, Li W, de Jong G, et al. A deletion in the bovine FANCI gene compromises fertility by causing fetal death and brachyspina. PLoS ONE. 2012;7:e43085.
Adams HA, Sonstegard TS, VanRaden PM, Null DJ, Van Tassell CP, Larkin DM, et al. Identification of a nonsense mutation in APAF1 that is likely causal for a decrease in reproductive efficiency in Holstein dairy cattle. J Dairy Sci. 2016;99:6693–701.
McClure MC, Bickhart D, Null D, VanRaden P, Xu L, Wiggans G, et al. Bovine exome sequence analysis and targeted SNP genotyping of recessive fertility defects BH1, HH2, and HH3 reveal causative mutation in SMC2 for HH3. PLoS One. 2014;9:e92769.
Schütz E, Wehrhahn C, Wanjek M, Bortfeld R, Wemheuer WE, Beck J, et al. The Holstein Friesian lethal haplotype 5 (HH5) results from a complete deletion of TFB1M and cholesterol deficiency (CDH) from an ERV-(LTR) insertion into the coding region of APOB. PLoS One. 2016;11:e0154602.
Cole JB, Null DJ, VanRaden PM. Phenotypic and genetic effects of recessive haplotypes on yield, longevity, and fertility. J Dairy Sci. 2016;99:7274–88.
Zavy MT, Geisart RD. Embryonic mortality in cattle. In: Embryonic mortality in domestic species. Boca Raton: CRC Press, Inc.; 1994. p. 99–141.
Lonergan P, Fair T, Forde N, Rizos D. Embryo development in dairy cattle. Theriogenology. 2016;86:270–7.
Fritz S, Capitan A, Djari A, Rodriguez SC, Barbat A, Baur A, et al. Detection of haplotypes associated with prenatal death in dairy cattle and identification of deleterious mutations in GART, SHBG and SLC37A2. PLoS ONE. 2013;8:e65550.
Shuster DE Jr, Kehrli ME, Ackermann MR, Gilbert RO. Identification and prevalence of a genetic defect that causes leukocyte adhesion deficiency in Holstein cattle. Proc Natl Acad Sci USA. 1992;89:9225–9.
Shanks RD, Robinson JL. Embryonic mortality attributed to inherited deficiency of uridine monophosphate synthase. J Dairy Sci. 1989;72:3035–9.
Agerholm JS, Bendixen C, Andersen O, Arnbjerg J. Complex vertebral malformation in Holstein calves. J Vet Diagn Invest. 2001;13:283–9.
Thomasen JR, Guldbrandtsen B, Sorensen P, Thomsen B, Lund MS. Quantitative trait loci affecting calving traits in Danish Holstein cattle. J Dairy Sci. 2008;91:2098–105.
Cole JB, VanRaden PM, O’Connell JR, Van Tassell CP, Sonstegard TS, Schnabel RD, Taylor JF, Wiggans GR. Distribution and location of genetic effects for dairy traits. J Dairy Sci. 2009;92:2931–46.
Müller MP, Rothammer S, Seichter D, Russ I, Hinrichs D, Tetens J, Thaller G, Medugorac I. Genome-wide mapping of 10 calving and fertility traits in Holstein dairy cattle with special regard to chromosome 18. J Dairy Sci. 2017;100:1987–2006.
Dachs N, Upadhyay M, Hannemann E, Hauser A, Krebs S, Seichter D, Russ I, Gehrke LJ, Thaller G, Medugorac I. Quantitative trait locus for calving traits on Bos taurus autosome 18 in Holstein cattle is embedded in a complex genomic region. J Dairy Sci. 2023;106:1925–41.
Sonstegard TS, Cole JB, VanRaden PM, Van Tassell CP, Null DJ, Schroeder SG, et al. Identification of a nonsense mutation in CWC15 associated with decreased reproductive efficiency in Jersey cattle. PLoS ONE. 2013;8:e54872.
VanRaden P, Null D, Hutchison J, Bickhart D, Schroeder S. Jersey haplotype 2 (JH2). Changes to evaluation system (August 2014). Council on Dairy Cattle Breeding. 2014. https://aipl.arsusda.gov/reference/recessive_haplotypes_ARR-G3.html. Accessed 10 March 2019.
Schwarzenbacher H, Burgstaller J, Seefried FR, Wurmser C, Hilbe M, Jung S, et al. A missense mutation in TUBD1 is associated with high juvenile mortality in Braunvieh and Fleckvieh cattle. BMC Genomics. 2016;17:400.
El-Hamidi M, Leipold HW, Vestweber JGE, Saperstein G. Spinal muscular atrophy in Brown Swiss calves. J Vet Med A Physiol Pathol Clin Med. 1989;36:731–8.
Krebs S, Medugorac I, Röther S, Strässer K, Förster M. A missense mutation in the 3-ketodihydrosphingosine reductase FVT1 as candidate causal mutation for bovine spinal muscular atrophy. Proc Natl Acad Sci USA. 2007;104:6746–51.
McClure M, Kim E, Bickhart D, Null D, Cooper T, Cole J, et al. Fine mapping for Weaver Syndrome in Brown Swiss cattle and the identification of 41 concordant mutations across NRCAM, PNPLA8 and CTTNBP2. PLoS One. 2013;8:e59251.
Kunz E, Rothammer S, Pausch H, Schwarzenbacher H, Seefried FR, Matiasek K, et al. Confirmation of a non-synonymous SNP in PNPLA8 as a candidate causal mutation for Weaver syndrome in Brown Swiss cattle. Genet Sel Evol. 2016;48:21.
Hafner A, Dahme E, Obermaier G, Schmidt P, Dirksen G. Spinal dysmyelination in new-born Brown Swiss x Braunvieh calves. Zentralbl Veterinarmed B. 1993;40:413–22.
Thomsen B, Nissen PH, Agerholm JS, Bendixen C. Congenital bovine spinal dysmyelination is caused by a missense mutation in the SPAST gene. Neurogenetics. 2010;11:175–83.
Cooper TA, Wiggans GR, Null DJ, Hutchison JL, Cole JB. Genomic evaluation, breed identification, and discovery of a haplotype affecting fertility for Ayrshire dairy cattle. J Dairy Sci. 2014;97:3878–82.
Venhoranta H, Pausch H, Flisikowski K, Wurmser C, Taponen J, Rautala H, et al. In frame exon skipping in UBE3B is associated with developmental disorders and increased mortality in cattle. BMC Genomics. 2014;15:890.
de Boer J, Hoeijmakers JHJ. Nucleotide excision repair and human syndromes. Carcinogenesis. 2000;21:453–60.
Nudell D, Castillo M, Turek PJ, Pera RR. Increased frequency of mutations in DNA from infertile men with meiotic arrest. Hum Reprod. 2000;15:1289–94.
Gunes S, Al-Sadaan M, Agarwal A. Spermatogenesis, DNA damage and DNA repair mechanisms in male infertility. Reprod BioMed Online. 2015;31:309–19.
Alexiou M, Leese HJ. Purine utilisation, de novo synthesis and degradation in mouse preimplantation embryos. Development. 1992;114:185–92.
Ugur MR, Kutchy NA, de Menezes EB, Ul-Husna A, Haynes BP, Uzun A, et al. Retained acetylated histone four in bull sperm associated with fertility. Front Vet Sci. 2019;6:223.
Bowles J, Bullejos M, Koopman P. A subtractive gene expression screen suggests a role for vanin-1 in testis development in mice. Genesis. 2000;27:124–35.
Johnson KA, Yao W, Lane NE, Naquet P, Terkeltaub RA. Vanin-1 pantetheinase drives increased chondrogenic potential of mesenchymal precursors in ank/ank mice. Am J Pathol. 2008;172:440–53.
Van Gorp H, Kuchmiy A, Van Hauwermeiren F, Lamkanfi M. NOD-like receptors interfacing the immune and reproductive systems. FEBS J. 2014;281:4568–82.
Peng H, Liu H, Liu F, Gao Y, Chen J, Huo J, et al. NLRP2 and FAF1 deficiency blocks early embryogenesis in the mouse. Reproduction. 2017;154:245–51.
Haghnazari L, Vaisi-Raygani A, Keshvarzi F, Ferdowsi F, Goodarzi M, Rahimi Z, et al. Effect of acetylcholinesterase and butyrylcholinesterase on intrauterine insemination, contribution to inflammations, Oxidative stress and antioxidant status; a preliminary report. J Reprod Infertil. 2016;17:157–62.
Nieto-Cerón S, Vargas-López H, Pérez-Albacete M, Tovar-Zapata I, Martínez-Hernández P, Rodríguez-López JN, et al. Analysis of cholinesterases in human prostate and sperm: implications in cancer and fertility. Chem Biol Interact. 2010;187:432–5.
Borgmann J, Tüttelmann F, Dworniczak B, Röpke A, Song HW, Kliesch S, et al. The human RHOX gene cluster: target genes and functional analysis of gene variants in infertile men. Hum Mol Genet. 2016;25:4898–910.
Pokharel K, Peippo J, Weldenegodguad M, Honkatukia M, Li M, Kantanen J. Gene expression profiling of corpus luteum reveals important insights about early pregnancy in domestic sheep. Genes. 2020;11:415.
Id-Lahoucine S, Casellas J. Impact of incomplete pedigree data and independent culling level pre-selection on the genetic evaluation of livestock: A simulation study on lamb growth. Livest Sci. 2017;198:76–81.
Sargolzaei M, Chesnais JP, Schenkel FS. A new approach for efficient genotype imputation using information from relatives. BMC Genomics. 2014;15:478.
Kass RE, Raftery AE. Bayes factors. J Am Stat Assoc. 1995;90:773–95.
Parzen E. On estimation of a probability density function and mode. Ann Math Statist. 1962;33:1065–76.
Wand MP, Jones MC. Kernel smoothing. Monographs on statistics & applied probability (60). Boca Raton: Chapman and Hall/CRC; 1994.
Fonseca PAS, Suárez-Vega A, Marras G, Cánovas A. GALLO: An R package for genomic annotation and integration of multiple data sources in livestock for positional candidate loci. Gigascience. 2020;30(9):giaa149.
Hu Z, Park CA, Reecy JM. Building a livestock genetic and genomic information knowledgebase through integrative developments of Animal QTLdb and CorrDB. Nucleic Acids Res. 2019;47:701–10.
Cardoso TF, Cánovas A, Canela-Xandri O, González-Prendes R, Amills M, Quintanilla R. RNA-seq based detection of differentially expressed genes in the skeletal muscle of Duroc pigs with distinct lipid profiles. Sci Rep. 2017;7:40005.
Phillips MA, Cánovas A, Wu PW, Islas-Trejo A, Medrano JF, Rice RH. Parallel responses of human epidermal keratinocytes to inorganic SbIII and AsIII. Environ Chem. 2016;13:963–70.
Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc. 2009;4:1184.
Acknowledgements
The authors acknowledge the data provided by Lactanet (Guelph, Ontario, Canada).
Funding
Authors thank the financial support in main part by Ontario Ministry of Agriculture, Food, and Rural Affairs (OMAFRA; Ontario, Canada), the Ontario Agri-Food Innovation Alliance, Agriculture and Agri-Food Canada, and by the Sustainable Beef and Forage Science Cluster (FDE.13.17) funded by the Canadian Beef Cattle Check-Off, Beef Cattle Research Council (BCRC), Alberta Beef Producers, Alberta Cattle Feeders’ Association, Beef Farmers of Ontario, La Fédération des Productuers de bovins du Québec, and Agriculture and Agri-Food Canada’s Canadian Agricultural Partnership. This study was also supported by NSERC (Natural Sciences and Engineering Research Council).
Author information
Authors and Affiliations
Contributions
A.C., J.C. and S.I.-L. conceived the research. S.I.-L. performed TRD the analyses. A.S.-V. and P.A.S.F. performed the functional analyses. M.S. imputed genomic data. A.C. and J.C. supervised the research. A.C. secured the funding for the project. S.I.-L. wrote the first draft of the manuscript. S.I.-L., J.C., A.C., A.S.-V., P.A.S.F., M.S. and F.S.S. participated in the interpretation of the results and revision of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplementary Material 1. Threshold for the approximate empirical null distribution of additive and dominance transmissionratio distortion (TRD) for different ranges of number of informative offspring.
Additional file 2:
Supplementary material 2. Summary of the final 604 chromosomal regionsdeemed with TRD across the bovine genome.
Additional file 3:
Supplementarymaterial 3. Recessive haplotypes identified within TRD regions.
Additional file 4:
SupplementaryMaterial 4. Genes mapped within the interval of the candidate lethal TRDhaplotypes.
Additional file 5:
Supplementarymaterial 5. Canonical pathways associated with the positional candidate genesmapped within TRD regions.
Additional file 6:
Supplementarymaterial 6. Diseases and functions associated with the positional candidate genes mapped within TRDregions.
Additional file 7:
Supplementarymaterial 7. Genetic markers and the respective genomic coordinates used to identifyTRD across the bovine genome.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Id-Lahoucine, S., Casellas, J., Suárez-Vega, A. et al. Unravelling transmission ratio distortion across the bovine genome: identification of candidate regions for reproduction defects. BMC Genomics 24, 383 (2023). https://doi.org/10.1186/s12864-023-09455-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09455-6