
Abstract
Background
Genome-wide association studies (GWAS) have uncovered thousands of genetic variants that are associated with complex human traits and diseases. miRNAs are single-stranded non-coding RNAs. In particular, genetic variants located in the 3’UTR region of mRNAs may play an important role in gene regulation through their interaction with miRNAs. Existing studies have not been thoroughly conducted to elucidate 3’UTR variants discovered through GWAS. The goal of this study is to analyze patterns of GWAS functional variants located in 3’UTRs about their relevance in the network between hosting genes and targeting miRNAs, and elucidate the association between the genes harboring these variants and genetic traits.
Methods
We employed MIGWAS, ANNOVAR, MEME, and DAVID software packages to annotate the variants obtained from GWAS for 31 traits and elucidate the association between their harboring genes and their related traits. We identified variants that occurred in the motif regions that may be functionally important in affecting miRNA binding. We also conducted pathway analysis and functional annotation on miRNA targeted genes harboring 3’UTR variants for a trait with the highest percentage of 3’UTR variants occurring.
Results
The Child Obesity trait has the highest percentage of 3’UTR variants (75%). Of the 16 genes related to the Child Obesity trait, 5 genes (ETV7, GMEB1, NFIX, ZNF566, ZBTB40) had a significant association with the term DNA-Binding (p < 0.05). EQTL analysis revealed 2 relevant tissues and 10 targeted genes associated with the Child Obesity trait.
In addition, Red Blood Cells (RBC), Hemoglobin (HB), and Package Cell Volume (PCV) have overlapping variants. In particular, the PIM1 variant occurred inside the HB Motif region 37,174,641–37,174,660, and LUC7L3 variant occurred inside RBC Motif region 50,753,918–50,753,937.
Conclusion
Variants located in 3’UTR can alter the binding affinity of miRNA and impact gene regulation, thus warranting further annotation and analysis. We have developed a bioinformatics bash pipeline to automatically annotate variants, determine the number of variants in different categories for each given trait, and check common variants across different traits. This is a valuable tool to annotate a large number of GWAS result files.
Similar content being viewed by others
Background
A microRNA (miRNA) is a single-stranded, non-coding RNA that regulates gene expression. miRNAs can interact with the 3’ untranslated regions (3’UTR) of the targeting messenger RNA (mRNA) and repress protein production by inhibiting translation, a process where genetic information from DNA is transported to the ribosome for protein synthesis [1]. Because of the ability of miRNA to regulate gene expression by controlling development and differentiation, mutations affecting miRNA function could be detrimental. It is widely suggested that mutated miRNAs play a crucial role in human diseases [2].
Genetic variants, such as single nucleotide polymorphisms (SNPs), contribute to diversity among individuals by affecting gene expression. SNPs within the 3’UTR of target mRNAs can influence gene regulation by changing the binding affinity of miRNAs, thus SNPs and their target sites play a significant role in the development of complex traits and diseases [3]. Today, many functional annotation tools and sequencing methods enable convenient and deeper analysis of genetic variation. For example, genome-wide annotation of variants (GWAVA) supports prioritization of noncoding variants and helps predict the functional impact of non-coding variants [4]. Different pathway analysis tools accelerate generation of sequencing data and facilitate identification of functionally relevant variants [4, 5].
Genome-wide association studies (GWAS) uncover thousands of genetic variations associated with diverse human traits and diseases. However, it is difficult to annotate disease influence of non-coding variants (e.g., 3'UTR variants) reported from GWAS studies and explain their impact on binding miRNAs. Recent significant studies that identified GWAS SNPs alter binding sequences have been published. The SMDB database stands out by identifying both losses and gains of important somatic motifs [6]; The updated SomamiR 2.0 database includes somatic mutations for miRNA target recognition [7]. We applied MIGWAS (miRNA–target gene networks enrichment on GWAS), a pipeline to systematically estimate specific tissue enrichment over the association of miRNA and target gene networks, to analyze genetics of diverse human traits and provide a better understanding of the effects of miRNA on human diseases [8, 9].
The GTEx database contains whole-genome sequences and tissue samples from donors, helping researchers study the relationship between genetic variation and gene expression. The expression quantitative trait loci (eQTL) provided by GTEx explains gene expression variation and provides a better understanding and interpretation of GWAS results. As it is challenging to distinguish reactive and causal expression changes, incorporating eQTL analysis of GWAS data can help identify causal variants that are more prevalent in individuals with a certain trait, as well as find molecular changes within complex traits [10]. It is valuable that cis and trans-eQTLs are identified to find the position of the locus controlling expression of the target gene.
The goal of this study is to analyze patterns of GWAS functional variants located in 3’UTRs across 31 traits selected from our previous study [11] about their relevance in the context over the network between hosting genes and targeting miRNAs, and elucidate the association between the genes harboring these variants and genetic traits. Due to the large amounts of GWAS variants, it is often difficult to process and analyze the data efficiently by hand. In our study, we have created a bioinformatics pipeline that annotates GWAS files automatically by quantifying the number of variants in different categories for any given trait and identifying and characterizing common variants across multiple different traits.
Materials and methods
ANNOVAR, a software tool, is used to efficiently annotate genetic variants by examining their functional importance on genes and different variant regions [12]. By using gene-miRNA target pairs for 31 traits from MIGWAS, in which miRNA-gene target pairs were identified in our other study [11], we annotated variation for every trait using ANNOVAR to report functional importance of the targeted genes. We categorized the variants and checked for the prevalence of the 3'UTR variant category for each trait. Specifically, genetic variants located in the 3’UTR region could alter the interactions of the UTRs and the miRNAs. To normalize the proportion, we divided the occurrences of 3’UTR by the unique genes as a measurement matrix for comparison.
By utilizing the annotation tool DAVID, we conducted a Gene Ontology analysis on 15 targeted genes harboring 3’UTR variants for Child Obesity traits, which we found had the highest 3’UTR prevalence among the 31 traits. DAVID is a web-accessible program that utilizes a multitude of tools that enables users to translate genome-scale datasets by converting the data into biological meaning [13, 14].
In addition, in order to have deeper insights about GWAS data, we applied eQTL analysis to GWAS Child Obesity results. All tested gene-SNP pairs and significant eQTL pairs in the GTEx cohort (version 8) were downloaded from the GTEx portal. The GTEx dataset includes 581 Adipose Subcutaneous samples and 469 Adipose Visceral Omentum samples with available genotype and gene expression data. The GTEx eQTL mapping results were filtered Storey Q-value < 0.05 and SNPs with minor allele frequency (MAF) > 0.05 in each tissue respectively. A total of 12,795 eGenes in the GTEx Adipose Subcutaneous samples and 10,410 eGenes in the GTEx Adipose Visceral Omentum samples were retained with this filtering.
We also performed GO and KEGG pathway enrichment analyses to investigate the biological function among the 16 miRNA target genes (ZNF566, RC3H1, KCTD15, FTO, ARSJ, GMEB1, CEP120, PIGN, ETV7, ZBTB40, ETV7, NFIX, WDR55, C1orf173, FAM114A1, ARSJ, CD59, CLVS1) identified by MIGWAS for the Child Obesity trait [11]. We employed the g:GOSt tool (https://biit.cs.ut.ee/gprofiler/gost) available on the web-based software g:Profiler [15] and considered all annotated genes as background. We used the default option g:SCS method in g:Profiler for multiple testing correction in our analysis.
We then extracted 3’UTR sequences and locations for all genes in the traits from Ensembl Biomar and searched for motifs of each gene using MEME (Multiple EM for Motif Elicitation), a popular tool used to identify signals, or motifs, in genetic DNA/proteins [16]. We compared the motif region to the variant positions identified and reported from ANNOVAR, which elucidates the functional significance of miRNA binding. Variants located in the motif regions of genes likely play a functionally important role in miRNA binding. The matched motifs regions are reported and visualized through various formats like position.
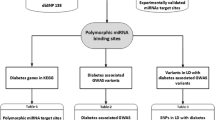
The detailed workflow of identifying variants in 3’UTR regions of miRNA-targeted genes for 31 traits is shown in Fig. 1.

Workflow of elucidating variants in 3’UTR region
Results
GWAS variants annotation pipeline (TACG-Var)
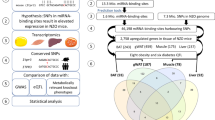
Our developed bioinformatics pipeline named as TACG-Var: Tool for Annotation and Classification of Genes and Variants efficiently annotates large numbers of variants files and is available at https://github.com/JuDong1214/Pipeline/blob/main/README.md. The pipeline includes 4 python scripts and 1 bash script, which calls 3 of the python scripts: 3'UTR Variant Prevalence.py, Common Genes.py, and Common Variants.py. The detailed pipeline calling process is shown in Fig. 2.

Pipeline calling process of annotating variants for the large number of GWAS traits
The ANNOVAR python calling script annotates traits and reports functional importance in ANNOVAR format. The other 3 python scripts, 3'UTR Variant Prevalence.py, Common Genes.py, and Common Variants.py. can be run by the bash script under the same directory. Under a Unix system, one can run the python calling bash script to efficiently run all python scripts on all files under the same directory.
The 3'UTR Variant Prevalence.py script finds the prevalence of 3’UTR regions in each trait. This script takes in ANNOVAR formatted.csv files and counts the number of unique regions (intergenic, intronic, ncRNA_intronic, UTR3, exonic, downstream, upstream, ncRNA_exonic, UTR5, upstream;downstream, splicing, ncRNA_splicing, and UTR5;UTR3) in each ANNOVAR formatted trait file. By utilizing the bash script to automate this process for all traits, the outputs are stored in a created output file.
The Common Variants.py script is used to find the common variants between two individual traits. The bash script runs this script multiple times and automatically compares all the variant files under the same directory. This python script takes in ANNOVAR formatted.csv or.xlsx files, and outputs in standard output format when run alone.
The Common Genes.py script is used to find the common genes between two individual traits. Similar to the process of the common variants script, the bash script runs this script multiple times and automatically compares all the variant files under the same directory. This python script takes in.txt files with a gene name in each line, and outputs in standard output format when running this script alone.
To top off the pipeline, the bash script is needed to automate the 3'UTR Variant Prevalence.py, Common Genes.py, and Common Variants.py script for all the traits. By utilizing this bash script, all outputs from the 4 python scripts can be stored in created output files.
Overlap analysis of MIGWAS traits
Since some variants can be shared across multiple traits, we used the miRNA targeted gene list from multiple traits to check overlap across traits. We found that Red Blood Cells (RBC), Hemoglobin (HB), and Package Cell Volume (PCV) all have high overlaps in variants for miRNA targeted genes among the three traits (shown in Fig. 3). In addition, we found that the Height trait had overlaps with 22 out of the 30 traits. The common variants between 31 MIGWAS traits based on ANNOVAR annotation results are reported in Additional file 1.

Number of common variants shared between 31 studied traits
Our study found that these three traits have four common genes: TFPI, HBS1L, MED1, PIK3R3. TFPI encodes a Kunitz-type serine protease inhibitor that initiates the extrinsic pathway of blood coagulation [17]. The intergenic region of HBS1L and the MYB gene control fetal hemoglobin level. Additionally, this region influences erythrocyte, platelet, erythrocyte volume and hemoglobin content [18].
Prevalence of 3’UTR variants and functional enrichment and eQTL analysis on genes associated with child obesity
Comparing the prevalence of 3’UTR variants in each trait, we identified that Child Obesity had the highest percentage of variants (75%) out of total unique genes occurring in the 3’UTR region (shown in Fig. 4). The detailed number of gene and variants in 3’UTR for 31 MIGWAS traits based on the ANNOVAR results are reported in Additional file 2.

Percentage of genes and variants occurring in the 3'UTR region for 31 traits
The DAVID analysis discovered that genes ETV7, GMEB1, NFIX, ZNF566, ZBTB40 with term DNA-binding have significant association with Child Obesity (p-value < 0.05).
The result of the list of miRNA targeted genes for Child Obesity trait with significant associated GO term identified as the top cluster by DAVID is shown in Table 1. The detailed DAVID results are reported in Additional file 3.
We also used eQTL to find the overlap between target genes with eGenes in GTEx v8 eQTL for Child Obesity trait. We discovered that there are 10 target genes in eGenes of Adipose_Subcutaneous and 9 target genes in eGenes of Adipose_Visceral_Omentum. Adipose is a widely studied tissue in child obesity [19, 20]. The overlapping eGenes for two tissues are shown in Table 2. The detailed eQTL results are reported in Additional file 4.
We also conducted the functional enrichment analyses using the g:GOSt tool to identify GO and KEGG pathways among the 16 miRNA targeted genes reported for Child Obesity trait. We identified a significantly enriched GO Term (GO:0,008,484 sulfuric ester hydrolaseactivity) with an adjusted p-value < 0.05. This GO:0,008,484 sulfuric ester hydrolase activity term is enriched in brown adipose tissue (BAT) [21], and two genes (ARSJ and PIGN) associated with this term are found to be in the set of intersections. Significant terms and associated genes related to human phenotype ontology (HP) with adjusted p-values were identified and reported in Additional file 5. Interestingly, PIGN, FTO, and CEP120 associated with HP were also identified through the eQTL analysis as eGenes for Adipose Subcutaneous and Adipose Visceral Omentum tissues.
Variants in motif region for traits HB and RBC
After matching the motifs regions from MEME with the variants positions from ANNOVAR, we discovered that the PIM1 variant (C > T) occurred at location 37,174,646 inside the HB Motif region 37,174,641–37,174,660 (Fig. 5), and LUC7L3 variant (T > C) occurred at location 50,753,923 inside RBC Motif region 50,753,918–50,753,937 (Fig. 6). PIM1 is a threonine kinase often overexpressed in gastric cancers [22], potentially causing HB to be lower than usual. LUC7L3 is a gene associated with Type 1 Diabetes and codes for a protein that localizes with a speckled pattern in the nucleus [23]. The variant locations inside the Motif region are reported in Additional files 6 and 7.

A variant of PIM1 annotated by ANNOVAR falls within the motif region identified by MEME for Hemoglobin levels (HB) trait

A variant of LUC7L3 annotated by ANNOVAR falls within the motif region identified by MEME for Red Blood Cell count (RBC) trait
Discussions
In our study, we use several bioinformatics tools (MIGWAS, ANNOVAR, MEME, DAVID, eQTL) to elucidate and pinpoint functional variants located in 3’UTRs across 31 traits from GWAS. MIGWAS reported miRNA enrichment over the target gene network for the 31 traits. ANNOVAR annotated the variants harbored by genes associated with the traits and reported the number of variants in the 3’UTR category. MEME provided the motifs regions of the targeted genes, which we used to compare to the variants locations. DAVID was used to conduct pathway analysis on the Child Obesity Trait. eQTL was applied to find tissues relevant to Child Obesity Trait.
In our study, we discovered that Child Obesity had the highest prevalence of variants inside the 3'UTR region. Obesity is strongly tied to distribution of adipose tissue, which mainly accumulates in the intra-abdominal site, which is visceral fat surrounding the omentum, and subcutaneous site [19]. Obesity can also be interpreted as a low-grade inflammatory state with adipose tissue generating large quantities of pro-inflammatory molecules, thus underlying a relationship between the immune system and adipose tissue. For children, obesity is the most common cause of abnormal growth. In addition, children with obesity have higher mortality rates. People who experienced Child Obesity were more susceptible to coronary disease and atherosclerosis.
MiRNAs can regulate gene expression through targeting 3’UTR regions of mRNAs. There were 14 miRNAs (hsa-mir-320c-2, hsa-mir-10b, hsa-mir-4694, hsa-mir-574, hsa-mir-4458, hsa-mir-653, hsa-mir-3682, hsa-mir-4284, hsa-mir-4297, hsa-mir-4652, hsa-mir-4517, hsa-mir-4421, hsa-mir-4721, hsa-mir-576) targeting into 3’UTR regions of candidate genes containing variants identified by MIGWAS for Child Obesity trait. A complete list of targeting miRNAs for all traits reported from MIGWAS can be found in our previously published study [11]. Indeed, it has been reported that miRNA seed region could be a key factor to drive the regulation change [6]. The mutation that occurs at miRNA seed and/or binding region could have a serious cascading effect on miRNA's regulation function on its targeting mRNAs/genes.
The five genes identified by DAVID (ETV7, GMEB1, NFIX, ZNF566, ZBTB40) that have significant associations with Child Obesity belong to the term DNA-Binding, which refers to proteins that bind to DNA sequences in order to modify the DNA or regulate gene expression [24]. Among the 10 genes associated with Child Obesity and the two adipose tissues identified by eQTL, we noticed that ETV7 and ZNF566 were also reported by DAVID. ETV7 is a member of the ETS family of transcription factors that is involved in oncogenesis and plays an important role in a variety of cellular processes throughout development and differentiation [25]. The protein encoded by this gene is predominantly expressed in hematopoietic tissues [26]. GMEB1 is a transcriptional factor that is essential for parvovirus DNA replication and modulates the transactivation of the glucocorticoid receptor [27]. NFIX is a member of a family of CCAAT-binding transcription factors that can initiate transcription of both vertebrate and viral genes [28]. Malan et al. (2010) reported that there is a nearly ubiquitous expression of NFIX in the central nervous system and the peripheral nervous system. Their study of distal femoral growth plates of 1- to 5-week-old mice and human fetus revealed strong NFIX expression in bone and in pre-hypertrophic chondrocytes [29].
Our study has several limitations. Firstly, since our method is based on GWAS results, if GWAS misreports, such as only reporting a single variant of the trait instead of the whole genetic region, our results could be affected. Secondly, our pipeline also has many limitations, especially with regards to the format of input files. The files that take ANNOVAR format input require the headings of the graphs to be exactly written as the original ANNOVAR file. Moreover, the output files produce results in text format, so conversion into excel file will have to be done manually.
Conclusions
Variants located in 3’UTR can often impact gene regulation by altering the binding affinity of miRNA. Therefore, it is important to annotate the functional important variants and see how they are related to their harboring genes and traits. Our study aimed to pinpoint GWAS functional variants located in 3’UTRs across 31 traits selected from our previous study in order to elucidate the association between the genes harboring these variants and genetic traits. The bioinformatics bash pipeline we developed is a valuable tool to annotate a large number of GWAS result files. Lastly, our analysis expands our knowledge on the disease causativeness annotation and provides clarification of non-coding variants’ effect on genetic traits and paves the way for future human disease studies.
Availability of data and materials
The input and output datasets associated with the pipeline are available at: https://github.com/JuDong1214/TACG-Var
Abbreviations
- miRNA:
-
Micro ribonucleic acid
- RNA:
-
Ribonucleic acid
- mRNA:
-
Messenger ribonucleic acid
- DNA:
-
Deoxyribonucleic acid
- SNP:
-
Single-nucleotide polymorphism
- 3′UTR:
-
Three prime untranslated region
- GWAS:
-
Genome-wide association study
- MIGWAS:
-
MiRNA–target gene networks enrichment on GWAS
- ANNOVAR:
-
Annotate Variation
- DAVID:
-
Database for annotation, visualization, and integrated discovery
- eQTL:
-
Expression Quantitative trait loci
References
O’Brien J, Hayder H, Zayed Y, Peng C. Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol (Lausanne). 2018;9:402.
Tufekci KU, Oner MG, Meuwissen RL, Genc S. The role of microRNAs in human diseases. Methods Mol Biol. 2014;1107:33–50.
Xie L, Li S, Jin J, He L, Xu K, Zhu L, Du M, Liu Y, Chu H, Zhang Z, et al. Genetic variant in miR-21 binding sites is associated with colorectal cancer risk. J Cell Mol Med. 2019;23(3):2012–9.
Ritchie GR, Dunham I, Zeggini E, Flicek P. Functional annotation of noncoding sequence variants. Nat Methods. 2014;11(3):294–6.
Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc. 2015;10(10):1556–66.
Jiang L, Duan M, Guo F, Tang J, Oybamiji O, Yu H, Ness S, Zhao YY, Mao P, Guo Y. SMDB: pivotal somatic sequence alterations reprogramming regulatory cascades. NAR Cancer. 2020;2(4):zcaa030.
Bhattacharya A, Cui Y. SomamiR 2.0: a database of cancer somatic mutations altering microRNA-ceRNA interactions. Nucleic Acids Res. 2016;44(D1):D1005-1010.
Sakaue S, Hirata J, Maeda Y, Kawakami E, Nii T, Kishikawa T, Ishigaki K, Terao C, Suzuki K, Akiyama M, et al. Integration of genetics and miRNA-target gene network identified disease biology implicated in tissue specificity. Nucleic Acids Res. 2018;46(22):11898–909.
Okada Y, Muramatsu T, Suita N, Kanai M, Kawakami E, Iotchkova V, Soranzo N, Inazawa J, Tanaka T. Significant impact of miRNA-target gene networks on genetics of human complex traits. Sci Rep. 2016;6:22223.
Nica AC, Dermitzakis ET. Expression quantitative trait loci: present and future. Philos Trans R Soc Lond B Biol Sci. 2013;368(1620):20120362.
Li B, Dong J, Yu J, Fan Y, Shang L, Zhou X, Bai Y. Pinpointing miRNA and genes enrichment over trait-relevant tissue network in genome-wide association studies. BMC Med Genomics. 2020;13(Suppl 11):191.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164.
Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4(5):P3.
Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC, Lempicki RA. The DAVID gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8(9):R183.
Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, Vilo J. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019;47(W1):W191–8.
Bailey TL, Williams N, Misleh C, Li WW. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006;34(Web Server issue):W369-373.
Gene [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; 2004 – [cited 2021 02 06]. Available from: https://www.ncbi.nlm.nih.gov/gene/7035
Gene [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; 2004 – [cited 2021 02 06]. Available from: https://www.ncbi.nlm.nih.gov/gene/10767.
Slyper AH. Childhood obesity, adipose tissue distribution, and the pediatric practitioner. Pediatrics. 1998;102(1):e4.
Leoni MC, Valsecchi C, Mantelli M, Marastoni L, Tinelli C, Marchi A, Castellazzi A. Impact of child obesity on adipose tissue physiology: assessment of adipocytokines and inflammatory cytokines as biomarkers of obesity. Pediatr Rep. 2010;2(2):e19.
Aldiss P, Lewis JE, Lupini I, et al. Cold Exposure Drives Weight Gain and Adiposity following Chronic Suppression of Brown Adipose Tissue. Int J Mol Sci. 2022;23(3):1869. https://doi.org/10.3390/ijms23031869.
Magnuson NS, Wang Z, Ding G, Reeves R. Why target PIM1 for cancer diagnosis and treatment? Future Oncol. 2010;6(9):1461–78.
Gene [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; 2004 – [cited 2021 02 06]. Available from: https://www.ncbi.nlm.nih.gov/gene/51747.
Morgat ALT, Coudert E, Axelsen K, Neto TB, Gehant S, Bansal P, Bolleman J, Gasteiger E, de Castro E, Baratin D, Pozzato M, Xenarios I, Poux S, Redaschi N, Bridge A, UniProt Consortium. Enzyme annotation in UniProtKB using Rhea. Bioinformatics. 2019;36(6):1896–901.
Potter MD, Buijs A, Kreider B, van Rompaey L, Grosveld GC. Identification and characterization of a new human ETS-family transcription factor, TEL2, that is expressed in hematopoietic tissues and can associate with TEL1/ETV6. Blood. 2000;95(11):3341–8.
Gene [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; 2004 – [cited 2021 02 06]. Available from: https://www.ncbi.nlm.nih.gov/gene/51513.
Kotsaris G, Kerselidou D, Koutsoubaris D, Constantinou E, Malamas G, Garyfallos DA, Etaatzivassiliou EG. TRAF3 can interact with GMEB1 and modulate its anti-apoptotic function. J Biol Res (Thessalon). 2020;27:7.
Santoro C, Mermod N, Andrews PC, Tjian R. A family of human CCAAT-box-binding proteins active in transcription and DNA replication: cloning and expression of multiple cDNAs. Nature. 1988;334(6179):218–24.
Malan V, Rajan D, Thomas S, Shaw AC, Louis Dit Picard H, Layet V, Till M, van Haeringen A, Mortier G, Nampoothiri S, et al. Distinct effects of allelic NFIX mutations on nonsense-mediated mRNA decay engender either a Sotos-like or a Marshall-Smith syndrome. Am J Hum Genet. 2010;7(2):189–98.
Acknowledgements
The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS.
We thank Judy Bai for helping with paper review. We also thank Li Fang and Kai Wang for offering valuable ANNOVAR annotation advice.
About this supplement
This article has been published as part of BMC Genomics Volume 23 Supplement 4, 2022: Selected articles from the International Conference on Intelligent Biology and Medicine (ICIBM 2021): genomics. The full contents of the supplement are available online at https://bmcgenomics.biomedcentral.com/articles/supplements/volume-23-supplement-4.
Funding
This research was partially supported from the University of Michigan to XZ. The publication costs for this article are funded by Next-Gen Intelligent Science Training. The funders had no role in the study design, data collection and analysis, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
MS participated in study design, data curation, and manuscript drafting. JY generated figures, created python scripts, performed MEME and ANNOVAR analysis and participated to draft the manuscript. BL generated graphs, performed DAVID analysis, and participated to draft the manuscript. JD performed ANNOVAR analysis, created the bash pipeline, and participated to draft the manuscript. JG drafted the eQTL and GTEx introduction. LS ran the eQTL and functional enrichment analysis and wrote the eQTL analysis methods and results. XZ provided the data and biological interpretation and offered study advice. YB designed and supervised the project, provided biological interpretation including the functional enrichment analysis results, and offered study advice. All authors read, revised, and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Common variants between 31 MIGWAS traits.
Additional file 2.
Number of gene and variants in 3’UTR for 31 MIGWAS traits.
Additional file 3.
Genes and GO terms associated with trait child obesity identified by DAVID.
Additional file 4.
GTEx eQTL mapping results for GWAS genes.
Additional file 5.
Functional enrichment results from gProfiler for 16 miRNA targeted genes for Child Obesity trait.
Additional file 6.
Variants in the Motif Region of HB.
Additional file 7.
Variants in the Motif Region of RBC.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Song, M., Yu, J., Li, B. et al. Identification of functionally important miRNA targeted genes associated with child obesity trait in genome-wide association studies. BMC Genomics 23 (Suppl 4), 360 (2022). https://doi.org/10.1186/s12864-022-08576-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-022-08576-8