
Abstract
Background
Bean pyralid is one of the major leaf-feeding insects that affect soybean crops. DNA methylation can control the networks of gene expressions, and it plays an important role in responses to biotic stress. However, at present the genome-wide DNA methylation profile of the soybean resistance to bean pyralid has not been reported so far.
Results
Using whole-genome bisulfite sequencing (WGBS) and RNA-sequencing (RNA-seq), we analyzed the highly resistant material (Gantai-2-2, HRK) and highly susceptible material (Wan82–178, HSK), under bean pyralid larvae feeding 0 h and 48 h, to clarify the molecular mechanism of the soybean resistance and explore its insect-resistant genes. We identified 2194, 6872, 39,704 and 40,018 differentially methylated regions (DMRs), as well as 497, 1594, 9596 and 9554 differentially methylated genes (DMGs) in the HRK0/HRK48, HSK0/HSK48, HSK0/HRK0 and HSK48/HRK48 comparisons, respectively. Through the analysis of global methylation and transcription, 265 differentially expressed genes (DEGs) were negatively correlated with DMGs, there were 34, 49, 141 and 116 negatively correlated genes in the HRK0/HRK48, HSK0/HSK48, HSK0/HRK0 and HSK48/HRK48, respectively. The MapMan cluster analysis showed that 114 negatively correlated genes were clustered in 24 pathways, such as protein biosynthesis and modification; primary metabolism; secondary metabolism; cell cycle, cell structure and component; RNA biosynthesis and processing, and so on. Moreover, CRK40; CRK62; STK; MAPK9; L-type lectin-domain containing receptor kinase VIII.2; CesA; CSI1; fimbrin-1; KIN-14B; KIN-14 N; KIN-4A; cytochrome P450 81E8; BEE1; ERF; bHLH25; bHLH79; GATA26, were likely regulatory genes involved in the soybean responses to bean pyralid larvae. Finally, 5 DMRs were further validated that the genome-wide DNA data were reliable through PS-PCR and 5 DEGs were confirmed the relationship between DNA methylation and gene expression by qRT-PCR. The results showed an excellent agreement with deep sequencing.
Conclusions
Genome-wide DNA methylation profile of soybean response to bean pyralid was obtained for the first time. Several specific DMGs which participated in protein kinase, cell and organelle, flavonoid biosynthesis and transcription factor were further identified to be likely associated with soybean response to bean pyralid. Our data will provide better understanding of DNA methylation alteration and their potential role in soybean insect resistance.
Similar content being viewed by others
Background
Bean pyralid (Lamprosema indicata (Fabricius)) is an important leaf-feeding insect, is widely distributed throughout the world and is found in China, Korea, Japan, India, the Americas and Africa [1]. During the years when serious damage has occurred, it can produce 4 to 5 generations a year, with overlapping of generations in the soybean producing areas. Bean pyralid larva spins or wraps leaves to form wrapped leaves and hide in them. After feeding on leaves, only veins and petioles remain, which leads to the difficulty in the normal photosynthesis of soybean and affects plant growth, in turn leading to serious yield losses [2, 3]. In view of the serious damages caused by bean pyralid, the highly resistant and highly susceptible soybeans were identified [3]. The resistance of soybean to bean pyralid is inherited by two pairs of major genes plus polygenes, and the resistance loci are mainly located in linkage groups A2, C2, D1a, D1b, H, K and O [4,5,6]. The contents of soluble sugar, JA, CAT and PPO are related to the induction of bean pyralid larvae. Meanwhile, the contents of SOD, ET and ABA are related to the pest induction and genotypes [7]. Trypsin inhibitor A-like; chalcone isomerase 4-like; lipoxygenase-9; alpha-dioxygenase 1-like; lectin precursor; peroxidase 12-like; stress-induced protein SAM22; and so on, may be the potential target proteins (genes) for soybean to resist bean pyralid larvae [8,9,10]. In addition, such miRNAs as gma-miR156q; gma-miR166u; gma-miR166b; gma-miR319d; gma-miR394a-3p; and gma-miR396e, may also participate in the regulation of soybean resistance to bean pyralid larvae [11]. However, very little is known regarding the mechanism of epigenetic regulation related to soybean resistance to bean pyralid.
DNA methylation can turn off the activities of some genes, while demethylation can induce gene reactivity and expression. In addition, it can control the networks of gene expressions, thereby playing an important role in plant growth, development, and responses to biotic stress, and is an important means of regulating genome function [12]. Plants are often attacked by pathogens and pests during their growth and development processes. Such attacks can induce the plants to produce physiological or even gene level variations and changes in gene expressions in order to avoid or endure adversity. However, the main research involves the epigenetic effects of biotic stress on plants undergoing disease stress, such as xanthomonas oryzae pv.oryzae [13, 14]; tobacco mosaic virus [15, 16]; soybean cyst nematode [17] and arabidopsis cyst nematode [18]. There have been few studies conducted regarding epigenetic inheritance caused by insect. Therefore, DNA methylation can be used as an entry point to explore soybean resistance to bean pyralid.
In this study, we performed methylome and transcriptome analyses to different insect resistant material in soybean. We used the leaves of Gantai-2-2 (highly resistant material) and Wan82–178 (highly susceptible material) [3] before and after exposure to bean pyralid larvae as the experimental materials. This is the first time to deepen the understanding of the regulatory relationship between DNA methylation and gene expression in soybean undergoing insect stress, to explore the role of related genes in soybean resistance to bean pyralid larvae, and how gene expression is regulated in the whole genome of the soybean resistance to bean pyralid larvae, so as to lay a foundation for further research regarding the molecular mechanism of soybean response to insect stress at the epigenetic level.
Results
Bisulfite sequencing of genomes of different soybean cultivars
To study the characteristics and patterns of DNA methylation in the leaves of different insect resistant materials, we used the Illumian HiSeq4000 platform to construct DNA libraries of the highly resistant material (Gantai-2-2, HRK) and highly susceptible material (Wan82–178, HSK), respectively. The leaves were subjected to bean pyralid larvae feeding for 0 h and 48 h. The results showed that each sample produced 40 G filtered clean bases on average, the Q20 were 98.00, 97.99, 98.14 and 98.30%, respectively (Table 1). Meanwhile, more than 99.50% cytosines were converted, which indicated that the adopted high-throughput sequencing technology had a high recognition rate (Table 1). There were 89.57, 89.92, 92.09 and 90.50% clean reads were mapped to the reference soybean genome of Glycine_max_v2.0 (https://www.ncbi.nlm.nih.gov/assembly/GCF_000004515.4) (Table 1). The comparison results indicated that DNA methylation sequencing conversion rate was high and sequencing quality was qualified.
In addition, to estimate whether or not the sequencing depth could satisfy the coverage of the sequencing data, the sequencing coverages of four DNA libraries were counted. Then, when the sequencing depth was 25×, it was considered that more than 93.40% reading segments had been successfully covered (Table 1). Therefore, it was inferred that the overall sequencing quality of the four DNA libraries was relatively good and the vast majority of base sites had been covered.
To enhance the current understanding of the epigenetic regulation and DNA methylation levels of soybean leaves with different resistance to bean pyralid larvae, we further compared the genome-wide methylation levels of the four samples. It was determined that methylated cytosine ranged from 18.37 to 21.30%. Meanwhile, the average level of methylated cytosine in each context was also calculated, in the CG context ranged from 68.27 to 74.71%; in the CHG context ranged from 42.15 to 47.64%; and in the CHH context ranged from 4.90 to 5.81% (Table 2). It was observed that DNA methylation level was highest in the CG context, and lower in the CHG and CHH contexts. These findings indicated that the CG context was the most important methylation context for soybean. There were significant differences in DNA methylation levels among the different resistance materials. For example, the DNA methylation levels of the resistant material were lower than those of the susceptible material. Also, the DNA methylation levels of the resistant material increased while those of the susceptible material decreased after insect feeding stress. The increasing and decreasing results obtained in our study were similar to the inconsistent variations of methylation observed in different types of rice undergoing saline-alkali stress [19].
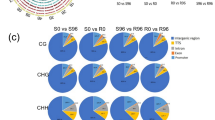
To investigate the DNA methylation patterns in different soybean genomic regions, we analyzed the methylation profiles in gene regions (Table 2). It was observed that DNA methylation level was highest in the CG context followed by CHG and CHH contexts in each gene region of the four samples. Repeat and CpG-island regions had the highest methylation levels of the different gene regions, which indicated that these two regions may be epigenetic regulatory regions which alter gene expressions. Meanwhile, the methylation levels were much higher in mRNA than in exon. After bean pyraild larvae feeding, the average methylation levels of the CG, CHG and CHH contexts in the resistant material were increased among the different transcription regions. However, the opposite effects were observed in the susceptible material. In the CG, CHG and CHH contexts, the methylation levels were more abundant in upstream and downstream regions than in exon regions (Fig.1). In addition, methylation was more frequent in the CHG context than in the CG and CHH contexts in upstream, first intron, and downstream regions, which indicated that there were some tendencies and differences among the different transcription regions.

Canonical DNA methylation profiles of the entire transcriptional units
Distribution ratios of the mCG, mCHG, and mCHH in the methylated C-base
To further analyze the distributions of the different methylated sites, all of the methylated C sites were selected and recombined (Table 3). After bean pyralid larvae feeding for 48 h, in the resistant material, mCG decreased from 36.16 to 34.47%, mCHG decreased from 34.42 to 32.80% and mCHH increased from 29.42 to 32.73%. In the susceptible material, mCG decreased from 36.20 to 34.78%, mCHG decreased from 34.22 to 32.86% and mCHH increased from 29.58 to 32.39%. The methylation levels of mCG, mCHG and mCHH in the resistant and susceptible materials were similar before and after bean pyralid larvae feeding. Therefore, the results indicated that the genotype had little effect on the methylation levels of the mCG, mCHG and mCHH, and the methylation levels were mainly affected by insect stress.
The methylation levels of each type of methylated C were statistically analyzed (Fig.2). The analysis results revealed that when the methylation levels ranged from 0 to 60%, the distribution proportions of the methylated C followed the order of mCHH > mCHG > mCG. However, when the methylation levels were between 60 and 80%, the distribution proportions followed the order of mCHG > mCHH > mCG. Furthermore, when the methylation levels were higher than 80%, the mCG site was significantly increased.

Distribution of methylation level of mC in each sequence context
Identification of the differentially methylated genes
To study the differential methylation among different resistant soybean varieties, we successfully identified the differentially methylated regions (DMRs). The number of DMRs identified in HRK0/HRK48 in the CG, CHG and CHH contexts were 664, 1550 and 0, respectively; in HSK0/HSK48 were 2200, 4670 and 2, respectively; in HSK0/HRK0 were 19,200, 20,272 and 31, respectively; and in HSK48/HRK48 were 19,178, 20,807 and 33, respectively. Also, DMRs detected at the CG and CHG contexts were significantly higher than that at the CHH context.
We divided the DMRs into the following two groups: DMR-associated genes (DMGs) and DMR-associated promoters (DMPs), which DMRs overlapped with the genes and promoters [20]. In HRK0/HRK48, 497 no-repeated DMGs (207 hyper-DMGs and 290 hypo-DMGs) and 223 no-repeated DMPs (99 hyper-DMPs and 124 hypo-DMPs) were identified, of which 48 DMGs appeared in both promoter regions and gene bodies, simultaneously. In HSK0/HSK48, 1594 no-repeated DMGs (687 hyper-DMGs, 882 hypo-DMGs and 25 shared-DMGs) and 612 no-repeated DMPs (235 hyper-DMPs and 377 hypo-DMPs) were identified, of which 186 DMGs appeared in both promoter regions and gene bodies, simultaneously. In HSK0/HRK0, 9596 no-repeated DMGs (2717 hyper-DMGs, 6577 hypo-DMGs and 302 shared-DMGs) and 3173 no-repeated DMPs (1357 hyper-DMPs, 1786 hypo-DMPs and 30 shared-DMPs) were identified, of which 1479 DMGs appeared in both promoter regions and gene bodies, simultaneously. In HSK48/HRK48, 9554 no-repeated DMGs (2944 hyper-DMGs, 6302 hypo-DMGs and 308 shared-DMGs) and 3217 no-repeated DMPs (1542 hyper-DMPs, 1636 hypo-DMPs and 39 shared-DMPs) were identified, of which 1379 DMGs appeared in both promoter regions and gene bodies, simultaneously. In HRK0/HRK48, HSK0/HSK48, HSK0/HRK0 and HSK48/HRK48, there were more hypo-DMGs and hypo-DMPs than hyper-DMGs and hyper-DMPs. Therefore, it was confirmed that the decreases of genome-wide DNA methylation levels may be one of the causes of the responses of plants to insect stress.
Gene ontology (GO) annotation and Kyoto encyclopedia of genes and genomes (KEGG) pathway enrichment analyses of DMGs
To better understand the functions of DMGs, GO annotation analysis was conducted. DMGs in the four comparisons were involved in the biological process, cellular component and molecular function. During the biological process, DMGs were mainly enriched in cellular process, metabolic process, biological regulation, regulation of biological process and response to stimulus. In the cellular component, DMGs were mainly concentrated in the cell, cell part, membrane, membrane part and organelle. In the molecular function, DMGs were mainly involved in binding and catalytic activity. It was speculated that the methylation patterns of the different contexts among the four comparisons were consistent in subcellular localization, molecular function, and biological process.
To further identify the metabolic pathways and functions of DMGs, the obtained DMGs were compared in the KEGG database (Table 4). In HRK0/HRK48, 151 DMGs at the CG context had participated in 71 pathways, with one pathway being significantly enriched; 259 DMGs at the CHG context had participated in 80 pathways, with one pathway being significantly enriched. In HSK0/HSK48, 671 DMGs at the CG context had participated in 108 pathways, with 8 pathways being significantly enriched; 667 DMGs at the CHG context had participated in 103 pathways, with 3 pathways being significantly enriched. In HSK0/HRK0, 6923 DMGs at the CG context had participated in 132 pathways, with 5 pathways being significantly enriched; 1907 DMGs at the CHG context had participated in 134 pathways, with 11 pathways being significantly enriched. In HSK48/HRK48, 6851 DMGs at the CG context had participated in 134 pathways, with 11 pathways being significantly enriched; 1943 DMGs at the CHG context had participated in 125 pathways, with 2 pathways being significantly enriched. Therefore, it was speculated that the methylation of the different contexts may have had a tendency to participate in the regulation of the biological functions. These pathways provided a useful reference for studying the biological processes and functions of the genes.
Interconnection of DMGs and DEGs
To further the current understanding of the relationships between transcriptome and methylation of soybean resistance to bean pyralid larvae, the data from WGBS and RNA-Seq [10] were jointly analyzed. The correlation analysis results showed that 512 DEGs were identified as DMGs in the four comparisons, of which 265 genes showed negative regulation (Table S1), the up-regulated genes correlated with hypo-DMGs and down-regulated genes correlated with hyper-DMGs, were screened as the negatively correlated genes. In addition, 247 genes showed positive correlations, the up-regulated genes correlated with hyper-DMGs and down-regulated genes correlated with hypo-DMGs, were screened as the positively correlated genes. About 64, 93, 236 and 194 DEGs in HRK0/HRK48, HSK0/HSK48, HSK0/HRK0 and HSK48/HRK48, respectively, were associated with DMGs. There were 34, 49, 141 and 116 negatively correlated genes were identified in the four comparisons, respectively. And 11, 10, 98 and 84 negatively correlated genes in the four comparisons, respectively, were occurred in the promoter regions. Therefore, it was speculated that the changes in DNA methylation levels of 265 negatively correlated genes may be one of the reasons for the significant differences in the gene transcription levels induced by bean pyralid larvae feeding. Meanwhile, the changes in DNA methylation levels of 247 positive correlated genes may not have been the reason for the direct regulation of the gene transcription levels. Subsequently, we will focus on negatively correlated genes, which are considered to be of significance of the biological processes in plant responses to insect stimulus, whether for in-depth explorations of gene functions or pattern analyses of DNA methylation.
KEGG enrichment analysis of negatively correlated genes
KEGG enrichment analysis of negatively correlated genes located in the gene bodies showed that (Table S2), in HRK0/HRK48, 10 negatively correlated genes (29.41%) were enriched in 11 pathways, with 4 pathways being significantly enriched, namely RNA transport, ascorbate and aldarate metabolism, fatty acid biosynthesis, and porphyrin and chlorophyll metabolism. In HSK0/HSK48, 13 negatively correlated genes (26.53%) were enriched in 23 pathways, with 5 pathways being significantly enriched, namely ubiquinone and other terpenoid-quinone biosynthesis, sulfur relay system, thiamine metabolism, selenocompound metabolism, and ether lipid metabolism. In HSK0/HRK0, 56 negatively correlated genes (39.72%) were enriched in 65 pathways, with 6 pathways being significantly enriched, namely pyruvate metabolism, propanoate metabolism, glyoxylate and dicarboxylate metabolism, carbon metabolism, lipoic acid metabolism, carotenoid biosynthesis. In HSK48/HRK48, 72 negatively correlated genes (62.07%) were enriched in 58 pathways, with 5 pathways being significantly enriched, namely carbon metabolism, propanoate metabolism, lipoic acid metabolism, pyruvate metabolism, biosynthesis of amino acids. The results suggested that various defense responses would be activated when soybean were subjected to bean pyralid larvae stress.
In addition, KEGG enrichment analysis of negatively correlated genes located in the promoter regions showed that (Table S3), in HRK0/HRK48, two negatively correlated genes (18.18%) were enriched in 4 pathways. In HSK0/HSK48, 5 negatively correlated genes (50.00%) were enriched in 10 pathways, with 2 pathways being significantly enriched, including monoterpenoid biosynthesis, and SNARE interactions in vesicular transport. In HSK0/HRK0, 30 negatively correlated genes (30.61%) were enriched in 42 pathways, with 5 pathways being significantly enriched, including endocytosis, glycolysis/gluconeogenesis, pyruvate metabolism, RNA degradation, sesquiterpenoid and triterpenoid biosynthesis. In HSK48/HRK48, 31 negatively correlated genes (36.90%) were enriched in 40 pathways, with 3 pathways being significantly enriched, including endocytosis, monoterpenoid biosynthesis, sesquiterpenoid and triterpenoid biosynthesis.
Functional analysis of the negatively correlated genes
For further understanding the resistance mechanism of the soybean to bean pyralid larvae, Mercator software was used to obtain the classification statistics of 265 negatively correlated genes, of which 114 were annotated into 24 categories (Table S4). These genes were determined to be mainly related to such pathways as protein biosynthesis and modifications; primary metabolism; secondary metabolism; cell cycle, cell structure and component; phytohormone action; external stimuli responses, and so on.
A total of 31 DEGs related to protein metabolism and modification (Table S4), among them, two genes were up-regulated in HRK0/HRK48; five genes were up-regulated and five genes were down-regulated in HSK0/HSK48; three genes were up-regulated and one gene was down-regulated in HSK0/HRK0; one gene was up-regulated and three genes were down-regulated in HSK48/HRK48; six genes were up-regulated and three genes were down-regulated in HSK0/HRK0 and HSK48/HRK48; one gene was up-regulated in HRK0/HRK48 and HSK0/HSK48. 27 DEGs related to cell cycle, cell structure, and cell component, of which five genes were up-regulated in HRK0/HRK48; seven genes were up-regulated in HSK0/HSK48; nine genes were up-regulated and two genes were down-regulated in HSK0/HRK0; two genes were up-regulated and one gene was down-regulated in HSK48/HRK48; one gene was up-regulated in HSK0/HRK0 and HSK48/HRK48. It was found that some genes related to primary metabolism, a total of six DEGs related to lipid metabolism; two DEGs related to amino acid metabolism; three DEGs related to carbon metabolism; and five DEGs related to coenzyme metabolism were identified. Among them, one gene was up-regulated in HRK0/HRK48; three genes were up-regulated and two genes were down-regulated in HSK0/HSK48; three genes were up-regulated and one gene was down-regulated in HSK0/HRK0; two genes were up-regulated and two genes were down-regulated in HSK48/HRK48; one gene was up-regulated and one gene was down-regulated in HSK0/HRK0 and HSK48/HRK48. Two DEGs related to flavonoid biosynthesis were identified, of which one gene was down-regulated in HSK0/HRK0; one gene was up-regulated in HSK48/HRK48. Six DEGs related to transcription factor were identified, of which one gene was down-regulated in HRK0/HRK48; one gene was up-regulated in HSK0/HSK48; three genes were up-regulated in HSK0/HRK0; and one gene was up-regulated in HSK48/HRK48. The results indicated that DEGs regulated by the methylation were involved in many biological pathways after bean pyralid larvae feeding.
Validation analysis of negatively correlated genes
To further verify that the negative correlations between DNA methylation and the transcriptome were not random, five negatively correlated genes were randomly selected. PS-PCR and qRT-PCR technologies were used to analyze their DNA methylation patterns and gene expression patterns. The results revealed that the PS-PCR expressed patterns and WGBS sequencing expressed patterns of five DMRs were all the same. Moreover, the qRT-PCR expression patterns of five DEGs were consistent with the RNA-Seq expression patterns (Fig. 3). These findings indicated that the sequencing results of WGBS and RNA-Seq were reliable, and that the DNA methylation may regulate the responses of soybean to pest stress by regulating the expression levels of genes related to insect resistance.

Expression levels of five DMGs validated by PS-PCR and qRT-PCR. A Differently methylated levels in HSK0/ HRK0 and HSK48/ HRK48. B qRT-PCR analysis of the genes in HSK0/ HRK0 and HSK48/ HRK48
Discussion
Genome-wide DNA methylation characteristics of soybean resistance to bean pyralid larvae
Since DNA methylation may potentially participate in the regulations of gene expressions, as well as the maintenance of genome stability, gene silencing in plants, it thereby plays important regulatory roles in plant growth, development, and stress resistance [19, 21, 22]. We found that the genome-wide DNA methylation levels of the four samples ranged from 18.37 to 21.30%, which is consistent with the DNA methylation levels reported in other plants [23,24,25]. DNA methylation was found in the CG, CHG, and CHH contexts, and different contexts have different modification patterns, the methylation levels in the CG context were significantly highest than that in the CHG and CHH contexts, which suggested that the CG context was the most important methylation context in soybean. These results were consistent with the type and level of DNA methylation detected in soybean root, steam, cotyledon and leaf [26, 27]. These findings indicated that the differences of DNA methylation in all patterns may have played important roles in the soybean responses to insect stress.
DMGs involved in regulation of soybean resistance to bean pyralid larvae
When plants are attacked by herbivorous insects, they activate a series of molecular signals in order to start their biotic defense responses [28]. By combining KEGG pathway and MapMan enrichment analysis, we found that some defense-related candidate genes had different methylation and transcription reactions after bean pyralid larvae feeding.
Protein kinases are involved in plant signaling pathways [29]. Cysteine-rich receptor-like protein kinase (CRK) and LRR-receptor-like kinase play important regulatory roles in plant growth, biotic and abiotic stresses [30, 31]. For example, 76 CRK promoter regions in soybean which contained biotic stress response elements [32]. When plants are exposed to stress, such as insect feeding, salt damage, and so on, serine/threonine protein kinase (STK) is rapidly activated by phosphorylation as serine and threonine residue. This further activates downstream signaling molecules through cascade phosphorylation for the purpose of activating specific signal transduction pathways, and finally transmits external signals to the nuclei in order to activate or inhibit the expressions of specific genes [32, 33]. The expression of STK in arabidopsis [34] and soybean [35] could be induced by insect stress. Mitogen-activated protein kinase (MAPK or MAP) is widely involved in the signal transmission of plant to stress, then activates the expression of anti-stress genes, so that plants have certain adaptability to stress [36,37,38,39,40]. It was also related to the defense response induced by insect stress. For example, MAPK was activated in tobacco after insect stress [41]. NaMEK1, NaMEK2, NaMKK1, NaSIPKK and NaNPK2 in Nicotiana attenuata play important roles in the defense of Manduca sexta. They could resist plant attack by herbivorous insects [42, 43]. The growth of Spodoptera littoralis was inhibited by AtMKK3 [44]. Lectin-like receptor kinases (LecRLKs) play important roles in plant pest resistance, they can combine with exogenous glycosyl to protect plants from insect stress [45, 46]. For example, LecRK1 in Nicotiana attenuata plays an important role in plant defense responses triggered by Manduca sexta [47, 48]. The resistance of rice to brown planthopper (BPH) was enhanced after over-expression of OsLecRK [49]. LecRK-1.8 plays an important role in the recognition of inducers derived from insect eggs in arabidopsis [50]. Our results obtained that 11 DEGs related to protein kinase, such as CRK40; CRK62; probable LRR receptor-like serine/threonine-protein kinase; STK; MAPK9; L-type lectin-domain containing receptor kinase VIII.2, and so on, were identified. It was speculated that protein kinases, as important signaling molecules, play very important regulatory roles in soybean resistance to bean pyralid larvae.
The activation and production of insect-resistant substance metabolism in plants usually consume a certain amount of growth energy, when the anti-insect defense mechanism of plants is activated, it will also lead to some changes and recombination in the morphology of the affected plants [51]. Plant cell walls are the primary cell structures sensing external stress signals, and are involved in maintaining cell morphology and related to such physiological activities as extracellular signal recognition. They are essentially the first line of defense against pathogens or pests [52,53,54]. Cellulose synthase complex (CSC) is assembled by cellulose synthase monomer (CesA) on the Golgi complex, and is transported by secretory vesicles and bound to cell membranes [55,56,57]. Plant cells can regulate cell wall formation through CSC assembly and transportation, thereby participating in plant morphogenesis and stress responses [57, 58]. It was observed that following IAA treatments of cotton, GhCesA1 and GhCesA2 were significantly up-regulated [59]. CSI1 is known to be involved in the formation of SmaCC/MASC and participates in the rapid recovery of CSC in plasma membrane after the stress conditions have subsided [60, 61]. Moreover, CSI1 directly mediates the interactions between CSCs and microtubules. In the absence of CSI1, the arrangements of CSCs and microtubules will be disrupted [62]. As a microfilament binding protein, fimbrin is one of the important regulatory factors of microfilament skeletons [63]. Kinesin (KIN) uses the energy produced by its hydrolysis of ATP to move along microtubules and provide power for intracellular material transport. For example, FRA1 of the arabidopsis KIN-4 family is a driver protein which moves to the positive ends of microtubules, and its function deficient mutant FRA1 showed irregular depositions of cellulose microfibrils on cell walls, making the stem brittle [64,65,66]. CLASP can be used as a regulatory protein of microtubule binding proteins [67, 68]. We found that a significant number of genes induced by bean pyralid larvae related to cell wall and cell cycle tissue metabolic pathways, such as CesA, CSI1, fimbrin-1, KIN-14B, KIN-14 N, KIN-4A, CLASP, and so on. The expression levels of those genes were all up-regulated after bean pyralid larvae feeding. This up-regulation may assist in the plant cell wall structuring processes in order to create a stronger physical protective layer against insects and reduce the damages to soybean undergoing insect stress, and maintain the stability of the cells and organelles. It was speculated that when soybean is subjected to pest stress, the anti-insect signaling pathways are activated after sensing cell wall damage, which activates a series of self-cell defense responses in soybean and greatly enhances the resistance of soybean. Moreover, genes related to cell cycle tissue can also effectively regulate plant tolerance to insects [69].
Cytochrome P450 (CYP) is a class of plant antioxidant inducers and detoxification genes, which can catalyze a number of substances which have defense functions in organisms, and plays an important role in the defense of organisms from diseases and insects stresses [70,71,72]. For example, cyanogen glycosides synthesized by CYP79A and CYP71E1 in sorghum were toxic to pests [73]. The expressions of CYP71A1 in rice [74] and CYP51 in tobacco [75] were induced by insect stresses, thus improving plant resistance to pests. CYP71A26 and CYP71B34 were involved in the response to pest stress in tea plants [76]. We observed that the expression of cytochrome P450 81E8 in the resistant material was higher than that in the susceptible material after bean pyralid larvae feeding. The results indicated that the release of terpenoids from the resistant material could be induced by pest stress. It was speculated that soybean can utilize cytochrome P450 family to reduce the threats caused by pests.
Transcription factors can regulate the expressions of multiple genes related to biotic stress, and improve the resistance of plant to disease and insects [77, 78]. ERF transcription factor plays an important regulatory role in plant resistance to insect infestations [79]. For example, the combination of SSaERF1 and GCC-box can enhance the resistance of arabidopsis to Prodenia litura [80], and BrERF11b can enhance the resistance of tobacco to Myzus persicae and Prodenia litura [81], thus indicating that plant resistance to insect stress can be improved through the over-expression of ERF. BEE1, bHLH and GATA transcription factors play important role in plant resistance to biotic and abiotic stresses [82, 83]. Before bean pyralid larvae feeding, the expression levels of ERF and BEE1 in the resistant material were higher than those in the susceptible material, which indicated that these two transcription factors were related to the genotypes of the resistant material. After insect stress, bHLH25 and GATA 26 were induced in the resistant material, and bHLH79 was induced in the susceptible material. Therefore, it was speculated that the differential expressions of these transcription factors may be an important reason for the differences of induced resistance levels and the persistence of resistant and susceptible soybean varieties.
Conclusions
In order to further understand the molecular mechanism of soybean responses to bean pyralid larvae, we used WGBS to analyze the genome-wide methylation of highly resistant and highly susceptible soybean leaves before and after bean pyralid larvae feeding. It was found that DNA methylation levels of specific genes changed in response to insect stress. At the same time, according to the DNA methylation and transcriptome association analysis, we concluded that there was a mainly negative correlation between DNA methylation and gene expression to a certain extent. In addition, the response to bean pyralid may be related to the pathways, such as protein biosynthesis and modification; primary and secondary metabolisms; cell cycle, cell structure and component; phytohormone action; RNA biosynthesis and processing, and so on. Meanwhile, by analyzing the expression levels and DNA methylation levels of those genes, the relationships between their methylation status and expression levels in different materials were revealed, and the roles of these related genes in the induction processes could be explored. This research investigation comprehensively analyzed the molecular mechanism of soybean undergoing insect stress from the transcription levels and methylation levels, which was of great significance to the research of soybean insect resistance.
Materials and method
Experimental materials
Both the highly resistant material (Gantai-2-2) and highly susceptible material (Wan 82–178)(Fig. S1) were planted in gray insect-proof net room on a test field at the Guangxi Academy of Agricultural Sciences in Nanning, Guangxi, China. When the plant growth reached 10 compound leaves, the fourth instar larvae of bean pyralid were grafted to each seedling according to a density of five larvae. Samples were taken at 0 h and 48 h after grafting. The samples were quickly frozen in liquid nitrogen and stored at − 80 °C for further use.
Total DNA extraction and detection
Total genomic DNA was extracted from soybean leaves using a DNeasy Plant Mini Kit (Qiagen, Valencia, CA, USA). The degradation of DNA in the samples was detected by agarose gel electrophoresis. The OD260/280 values of DNA were detected using a Nano Drop 2000 spectrophotometer (Thermo Fisher Scientific, MA, USA), and the concentration levels of DNA were accurately quantified by Qubit Fluorometer (Invitrogen, CA, USA). The qualified DNA samples were used for the library construction.
Sequencing analysis of the bisulfite
For WGBS librarys constructing, the DNAs were broken into fragments with a mean size of 250 bp using Bioruptor (Diagenode, Belgium). Following end repair and adenylation, the sonicated DNA fragments were ligated to cytosine-methylated barcodes according to manufacturer’s instruction. The DNA fragments were treated with bisulfite using the ZYMO EZ DNA Methylation-Gold kit (Zymo research, Orange County, CA, USA). Different Insert size fragments were excised from the same lane of a 2% TAE agarose gel. The products were purified by using a QIAquick Gel Extraction kit (Qiagen, Valencia, CA, USA) and then amplified by PCR. Finally, the qualified DNA libraries were sequenced on the Illumina Hiseq4000 platform (BGI-Shenzhen, Shenzhen, China).
Data filtering and sequence alignment
The raw data were filtered by removing adapter sequences, contamination and low-quality reads. After the filtering process was completed, BSMAP software [84] was used to map the clean reads with the soybean reference genome (https://www.ncbi.nlm.nih.gov/assembly/GCF_000004515.4), and the comparison rates and bisulfite conversion rates were calculated. In order to calculate the methylation levels of each site, we calculated the proportion of the number of reads supporting methylation to the total number of reads covering the site [85]. The formula was as follows: 
Where Nm and Nn represent the reads number of mC and nonmethylation-C, respectively.
DMR detection
A window containing at least five CG (CHG or CHH) was found at the same position in two of the sample genomes, and the differences in the CG methylation levels between the two samples of that window were compared. The region with significant differences (Fisher’s Exact, 2-fold change, and p-value ≤0.05) in the methylation between the two samples was referred to as DMR. If the contiguous region formed by the two adjacent DMRs differed significantly in methylation levels in the two samples, the two DMRs were combined into a single contiguous DMR. Otherwise, they were considered to be two independent DMRs.
CIRCOS software was used to compare the methylation levels of DMR in the different samples [86]. The degree of difference of a methyl-cytosine (mCG, mCHG, mCHH) was also calculated using the following formula:  .
.
Where Rm1 and Rm2 represent the methylation levels of mC for sample 1 and sample 2, respectively. If the value of Rm1 or Rm2 is 0, it shall be replaced by 0.001.
GO and KEGG analysis
Gene Ontology (GO) enrichment analysis method was used to provide all the GO terms which were significantly enriched in the DMGs, and to filter the DMGs with specific biological functions. Based on the GO TermFinder (http://www.yeastgenome.org/help/analyze/go-term-finder) [87], the number of genes in each term was calculated. Then, a hypergeometric test method was used to find the GO terms which were significantly enriched in the DMGs when compared with the entire genome background. The GO terms with a p-value≤0.05 were regarded as significantly enriched.
KEGG is the main public database for those pathways [88]. Through significant enrichment analyses of the pathways, it can be determined which pathways are significantly enriched in the DMGs when compared with the whole genome background, taking the KEGG pathway as a unit. Pathways with a p-value≤0.05 were regarded as significantly enriched.
Conjoint analysis of genome-wide DNA methylation and transcriptome
The original data obtained from WGBS and RNA-Seq [10] were analyzed and compared. The intersections of DNA methylation levels and gene expression levels were taken for conjoint analysis, and the DEGs in DMGs were screened out. The correlation between the methylation level of DMR and the expression level of DEG was detected by pearson correlation analysis. There were five overlapping situations involved: DMR_genes_VS_DEG_genes; DMR_Hypergenes_VS_DEG_upgenes; DMR_Hypergenes_VS_DEG_downgenes; DMR_Hypogenes_VS_DEG_upgenes; DMR_Hypogenes_VS_DEG_downgenes. The criterion for selecting the intersection genes were p-value < 0.05 [20].
MapMan biological function annotation
The amino acid sequence of the unigene coding protein obtained by CDS analysis was submitted to the MapMan website application online software mercator (http://mapman.gabipd.org/web/guest/mercator) for annotation of the biological functions of the encoding protein. The mapping information of the biological processes of the species was obtained.
Pyrosequencing PCR (PS-PCR) validation
Five genes with negative correlations between DNA methylation and gene expression were randomly selected. DNA methylation validation was conducted using the PS-PCR method. The DMR regions corresponding to those five genes were identified. All primers were designed using PyroMark Assay Design 2.0 (Table S5) and commercially synthesized (BGI, Shenzhen, China). The PCR was conducted in the following conditions: a total volume of 50 μL, containing 10.0 μL 5× buffer GC (KAPA), 1.0 μL dNTP, 1.0 μL of each primer, 2.0 μL template, 0.2 μL Taq Master Mix and 34.8 μL ddH2O. The thermal cycling conditions were as follows: 95 °C for 3 m; followed by 40 cycles of heating at 94 °C for 30 s, 50 °C for 30 s, 72 °C for 1 m and annealing at 72 °C for 7 m.
Quantitative real-time PCR (qRT-PCR) validation
The primer sequence was designed with Primer Premier 5.0 software (Premier Biosoft International, Palo Alto, CA) (Table S5). Next, 1.0 μg of total RNA was reverse-transcribed by reverse transcriptase according to the protocol of iScript cDNA Synthesis Kit (Bio-Rad, CA, USA), and used as the template for the following qRT-PCR amplification. The qRT-PCR reaction mixture (25.0 μL) contained 10.0 μL SybrGreen qPCR Master Mix (2× concentration, Ruian Biotechnologies, Shanghai, China), 0.6 μL upstream primer, 0.6 μL downstream primer (10.0 μM), 1.0 μL cDNA, and 7.8 μL ddH2O. The thermal cycling conditions were as follows: pre-denaturation at 95 °C for 2 m; followed by 40 cycles of heating at 95 °C for 10 s and annealing at 60 °C for 40 s. β-actin gene was used as the internal control gene. The relative level of genes expression was evaluated by the 2-ΔΔct method.
Availability of data and materials
All data were submitted to the National Center for Biotechnology Information (NCBI) under SRA number SRA549176.
Change history
22 December 2021
A Correction to this paper has been published: https://doi.org/10.1186/s12864-021-08252-3
Abbreviations
- WGBS:
-
Whole-genome bisulfite sequencing
- RNA-Seq:
-
RNA-sequencing
- DMG:
-
Differentially methylated gene
- DMR:
-
Differentially methylated region
- DMP:
-
Differentially methylated promoter
- DEG:
-
Differentially expressed gene
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- PS-PCR:
-
Pyrosequencing PCR
- qRT-PCR:
-
Quantitative real-time PCR
- CRK:
-
Cysteine-rich receptor-like protein kinase
- STK:
-
Serine/threonine protein kinase
- CSC:
-
Cellulose synthase complex
- CesA:
-
Cellulose synthase monomer
- KIN:
-
Kinesin
- SBT:
-
Subtilisin-like protease
- CYP:
-
Cytochrome P450
References
Choi KH, Hong YK, Chang YJ, Moon JS, Kim CS, Choi DC, et al. Development under constant temperatures and seasonal prevalence in soybean field of the bean pyralid, Omiodes indicatus (Lepidoptera: Crambidae). Korean J Appl Entomol. 2008;47(4):353–8. https://doi.org/10.5656/KSAE.2008.47.4.353.
Li GJ. Detection of QTL for photooxidation- related traits, dynamic chlorophyll content and resistance to bean pyralid (Lamprosema indicata Fabricius) in soybean (Glycine max Merrill). Nanjing: Nanjing Agricultural University. 2010; p. 38.
Sun ZD, Yang SZ, Chen HZ, Li CY, Long LP. Identification of soybean resistance to bean pyralid (Lamprosema indicata Fabricicus) and obiposition preference of bean pyralid on soybean varieties. Chin J Oil Crop Sci. 2005;27(4):69–71.
Xing GN, Zhou B, Wang YF, Zhao TJ, Yu DY, Chen SY, et al. Genetic components and major QTL confer resistance to bean pyralid (Lamprosema indicata Fabricius) under multiple environments in four RIL populations of soybean. Theor Appl Genet. 2012;125(5):859–75. https://doi.org/10.1007/s00122-012-1878-7.
Li GJ, Cheng LG, Zhang GZ, He XH, Zhi HJ, Zhang YM. Mixed major-gene plus polyegens inheritance analysis for resistance in soybean to bean pyralid (Lamprosema indicata Fabricius). Soybean Sci. 2008;27(1):33–6, 41.
Li GJ, Li HN, Cheng LG, Zhang YM. Mapping quantitative traitloci for resistance in soybean to bean pyralid (Lamprosema indicata Fabricius). Chin J Oil Crop Sci. 2009;31(3):365–9.
Zeng WY, Cai ZY, Zhang ZP, Chen HZ, Yang SZ, Tang XM, et al. Physiological and biochemical characteristics of Lamprosema indicata (Fabricius)-resistant soybean. J Southern Agric. 2016;46(12):2112–6.
Zeng WY, Sun ZD, Lai ZG, Cai ZY, Chen HZ, Yang SZ, et al. Correlation analysis on transcriptomic and proteomic of soybean resistance to bean pyralid (Lamprosema indicata). Sci Agric Sin. 2018;51(7):1244–60.
Zeng WY, Sun ZD, Cai ZY, Chen HZ, Lai ZG, Yang SZ, et al. Proteomic analysis by iTRAQ-MRM of soybean resistance to Lamprosema indicate. BMC Genomics. 2017;18(1):444. https://doi.org/10.1186/s12864-017-3825-0.
Zeng WY, Sun ZD, Cai ZY, Chen HZ, Lai ZG, Yang SZ, et al. Comparative transcriptome analysis of soybean response to bean pyralid larvae. BMC Genomics. 2017;18(1):871. https://doi.org/10.1186/s12864-017-4256-7.
Zeng WY, Sun ZD, Lai ZG, Yang SZ, Chen HZ, Yang XH, et al. Determination of the MiRNAs related to bean pyralid larvae resistance in soybean using small RNA and transcriptome sequencing. Int J Mol Sci. 2019;20(12):2966. https://doi.org/10.3390/ijms20122966.
Dowen RH, Pelizzola M, Schmitz RJ, Lister R, Dowen JM, Nery JR, et al. Widespread dynamic DNA methylation in response to biotic stress. Proc Natl Acad Sci U S A. 2012;109(32):E2183–91. https://doi.org/10.1073/pnas.1209329109.
Sha AH, Lin XH, Huang JB, Zhang DP. Analysis of DNA methylation related to rice adult plant resistance to bacterial blight based on methylation - sensitive AFLP (MSAP) analysis. Mol Gen Genomics. 2005;273(6):484–90. https://doi.org/10.1007/s00438-005-1148-3.
Akimoto K, Katakami H, Kim HJ, Ogawa E, Sano H. Epigenetic inheritance in rice plants. Ann Bot (Lond). 2007;100(2):205–17. https://doi.org/10.1093/aob/mcm110.
Kovalchuk I, Kovalchuk O, Kalck V, Boyko V, Filkowski J, Heinlein M, et al. Pathogen-induced systemic plant signal triggers DNA rearrangements. Nature. 2003;423(6941):760–2. https://doi.org/10.1038/nature01683.
Boyko A, Kathiria P, Zemp FJ, Yao YL. Transgenerational changes in the genome stability and methylation in pathogen-infected plants. Nucl Acids Res. 2007;35(5):1714–25. https://doi.org/10.1093/nar/gkm029.
Rambani A, Rice JH, Liu JY, Lane T, Mazarei M, Pantalone V, et al. The methylome of soybean roots during the compatible interaction with the soybean cyst nematode. Plant Physiol. 2015;168(4):1364–77. https://doi.org/10.1104/pp.15.00826.
Hewezi T, Lane T, Piya S, Rambani A, Rice JH, Staton M. Cyst nematode parasitism induces dynamic changes in the root epigenome. Plant Physiol. 2017;174(1):405–20. https://doi.org/10.1104/pp.16.01948.
Wang WS, Pan YJ, Zhao XQ, Dwivedi D, Zhu LH, Fu BY, et al. Drought-induced site-specific DNA methylation and its association with drought tolerance in rice (Oryza sativa L.). J Exp Bot. 2011;62(6):1951–60. https://doi.org/10.1093/jxb/erq391.
Jiang SH, Sun QG, Chen M, Wang N, Xu HF, Fang HC, et al. Methylome and transcriptome analyses of apple fruit smatic mutations reveal the difference of red phenotype. BMC Genomics. 2019;20(1):117. https://doi.org/10.1186/s12864-019-5499-2.
Chinnusamy V, Zhu JK. Epigenetic regulation of stress responses in plants. Curr Opin Plant Biol. 2009;12(2):133–9. https://doi.org/10.1016/j.pbi.2008.12.006.
Gao DH, Gao X, Liu J, Kimatu JN. Methylation sensitive amplified polymorphism (MSAP) reveals that alkali stress triggers more DNA hypomethylation levels in cotton (Gossypium hirsutum L) roots than salt stress. Afr J Biotechnol. 2011;10(82):18971–80.
Hsieh PH, He SB, Buttress T, Gao HB, Couchman M, Fischer RL, et al. Arabidopsis male sexual lineage exhibits more robust maintenance of CG methylation than somatic tissues. Proc Natl Acad Sci U S A. 2016;113(52):15132–7. https://doi.org/10.1073/pnas.1619074114.
Bewick AJ, Schmitz RJ. Gene body DNA methylation in plants. Curr Opin Plant Biol. 2017;36:103–10.
Kong XJ, Khan A, Li B, Zheng J, Dawar FU, Li M, et al. Deviat DNA methylation play a key role in the pollen abortion of Glssypium barbadense L cytoplasmic male sterility. Ind Crop Prod. 2020;154:112622. https://doi.org/10.1016/j.indcrop.2020.112622.
Song QX, Lu X, Li QT, Chen H, Hu XY, Ma B, et al. Genome-wide analysis of DNA methylation in soybean. Mol Plant. 2013;6(6):1961–74. https://doi.org/10.1093/mp/sst123.
Li YW, Ding XL, Wang X, He TT, Zhang H, Yang LS, et al. Genome-wide comparative analysis of DNA methylation between soybean cytoplasmic male- sterile line NJCMS5A and its maintainer NJCMS5B. BMC Genomics. 2017;18(1):596. https://doi.org/10.1186/s12864-017-3962-5.
Howe GA, Jander G. Plant immunity to insect herbivores. Annu Rev Plant Biol. 2008;59(1):41–66. https://doi.org/10.1146/annurev.arplant.59.032607.092825.
León J, Rojo E, Sánchez-Serrano J. Wound signaling in Plants. J Epx Bot. 200l;52(354):1–9.
Bourdais G, Burdiak P, Gauthier A, Nitsch L, Salojärvi J, Rayapuram C, et al. Large-scale phenomics identifies primary and fine-tuning roles for CRKs in responses related to oxidative stress. PLoS Genet. 2015;11(7):e1005373. https://doi.org/10.1371/journal.pgen.1005373.
Afzal AJ, Wood AJ, Lightfoot DA. Plant receptor-like serine threonine kinases: roles in signaling and plant defense. Mol Plant-Microbe In. 2008;21(5):507–17. https://doi.org/10.1094/MPMI-21-5-0507.
Delgado-Cerrone L, Alvarez A, Mena E, León IPD, Montesano M. Genome wide analysis of the soybean CRK-family and transcriptional regulation by biotic stress signals triggering plant immunity. PLoS One. 2018;13(11):e0207438. https://doi.org/10.1371/journal.pone.0207438.
Zipfel C. Pattern-recognition receptors in plant innate immunity. Cum Opin Immunol. 2008;20(1):10–6. https://doi.org/10.1016/j.coi.2007.11.003.
Zhang J. Differential proteomic analysis of the Arabidopsis thaliana in response to insect feeding and mechanical wounding. Master's thesis of Jilin Agriculture University. 2011.
Fan R, Wang H, Wang YL, Yu DY. Proteomic analysis of soybean defense response induced by cotton worm (prodenia litura Fabricius) feeding. Proteome Sci. 2012;10(5):16–21. https://doi.org/10.1186/1477-5956-10-16.
Link VL, Hofmann MG, Sinha AK, Ehness R, Strnad M, Roitsch T. Biochemical evidence for the activation of distinct subsets of mitogen-activated protein kinases by voltage and defence-related stimuli. Plant Physiol. 2002;128(1):271–81. https://doi.org/10.1104/pp.010569.
Hamel LP, Nicole MC, Sritubtim S, Morency MJ, Ellis M, Ehlti Ng J, et al. Ancient signals: comparative genomics of plant MAPK and MAPKK gene families. Trends Plant Sci. 2006;11(4):192–8. https://doi.org/10.1016/j.tplants.2006.02.007.
Kong XP, Pan JW, Zhang D, Jiang SS, Cai GH, Wang L, et al. Identification of mitogen-activated protein kinase kinase gene family and MKK-MAPK interaction network in maize. Biochem Bioph Res Co. 2013;441(4):964–9. https://doi.org/10.1016/j.bbrc.2013.11.008.
Pan JW, Zhang MY, Kong XP, Xing X, Liu Y, Zhou YK, et al. ZmMPK17, a novel maize group D MAP kinase gene, is involved in multiple stress responses. Planta. 2012;235(4):661–76. https://doi.org/10.1007/s00425-011-1510-0.
Sun HW, Zhang HB, Xu ZS, Wang Y, Li YY, Tian B, et al. TMT-based quantitative proteomic analysis of the effects of pseudomonas syringae pv. Tabaci (Pst) infection on photosynthetic function and the response of the MAPK signaling pathway in tobacco leaves. Plant Physol Bioch. 2021;166:657–67. https://doi.org/10.1016/j.plaphy.2021.06.049.
Wu JQ, Hettenhausen C, Meldau S, Baldwin IT. Herbivory rapidly activates MAPK signaling in attaeked and unattaeked leaf regions but not between leaves of Nicoliana attenuate. Plant Cell. 2007;19(3):1096–122. https://doi.org/10.1105/tpc.106.049353.
Heinrich M, Baldwin IT, Wu J. Two mitogen-activated protein kinase kinases, MKK1 and MEK2, are involvedin wounding and specialist lepidopteran herbivore Manduca sexta-induced responses in Nicotiana attenuata. J Exp Bot. 2011;62(12):4355–65. https://doi.org/10.1093/jxb/err162.
Heinrich M, Baldwin IT, Wu J. Three MAPK kinases, MEK1, SIPKK, and NPK2, are not involved in activation of SIPK after wounding and herbivore feeding but important for accumulation of trypsin proteinase inhibitors. Plant Mol Biol Rep. 2012;30(3):731–40. https://doi.org/10.1007/s11105-011-0388-0.
Sözen C, Schenk ST, Boudsocq M, Chardin C. Wounding and insect feeding trigger two independent MAPK pathways with distinct regulation and kinetics. Plant Cell. 2020;32(6):1988–2003. https://doi.org/10.1105/tpc.19.00917.
Brill LM, Evans CJ, Hirsch AM. Expression of MsLEC1- and MsLEC2-antisense genes in alfalfa plant lines causes severe embryogenic, developmental and reproductive abnormalities. Plant J. 2001;25(4):453–61. https://doi.org/10.1046/j.1365-313x.2001.00979.x.
Rüdiger H, Gabius HJ. Plant lectins: occurrence, biochemistry, functions and applications. Glycoconj J. 2001;18(8):589–613. https://doi.org/10.1023/A:1020687518999.
Gilardoni PA, Hettenhausen C, Baldwin IT, Bonaventure G. Nicotiana attenuata LECTIN RECEPTOR KINASE 1 suppresses the insect-mediated inhibition of induced defense responses during Manduca sexta herbivory. Plant Cell. 2011;23(9):3512–32. https://doi.org/10.1105/tpc.111.088229.
Bonaventure G. The Nicotiana attenuata lectin receptor kinase 1 is involved in the perception of insect feeding. Plant Signal Behav. 2011;6(12):2060–3. https://doi.org/10.4161/psb.6.12.18324.
Liu YQ, Wu H, Chen H, Liu YL, He J, Kang HY, et al. A gene cluster encoding lectin receptor kinases confers broad-spectrum and durable insect resistance in rice. Nat Biotechnol. 2015;33(3):301–5. https://doi.org/10.1038/nbt.3069.
Gouhier-Darimont C, Schmiesing A, Bonnet C, Lassueur S, Reymond P. Signaling of Arabidopsis thaliana response to Pieris brassicae eggs shares similarities with PAMP-triggered immunity. J Exp Bot. 2013;64(2):665–74. https://doi.org/10.1093/jxb/ers362.
Schwachtje J, Baldwin IT. Why does herbivore attack reconfigure primary metabolism? Plant Physiol. 2008;146(3):845–51. https://doi.org/10.1104/pp.107.112490.
Keegstra K. Plant cell walls. Plant Physiol. 2010;154(2):483–6. https://doi.org/10.1104/pp.110.161240.
Houston K, Tucker MR, Chowdhury J, Shirley N, Little A. The plant cell wall: a complex and dynamic structure as revealed by the responses of genes under stress conditions. Front Plant Sci. 2016;7:228. https://doi.org/10.3389/fpls.2016.00984.
Tenhaken R. Cell wall remodeling under abiotic stress. Front Plant Sci. 2015;5:771. https://doi.org/10.3389/fpls.2014.00771.
McFarlane HE, Dring A, Persson S. The cell biology of cellulose synthesis. Annu Rev Plant Biol. 2014;65(1):69–94. https://doi.org/10.1146/annurev-arplant-050213-040240.
Carroll A, Somerville C. Cellulosic biofuels. Annu Rev Plant Biol. 2009;60(1):165–82. https://doi.org/10.1146/annurev.arplant.043008.092125.
Crowell EF, Bischoff V, Desprez T, Desprez T, Rolland A, Stierhof YD, et al. Pausing of Golgi bodies on microtubules regulates secretion of cellulose synthase complexes in Arabidopsis. Plant Cell. 2009;21(4):1141–54. https://doi.org/10.1105/tpc.108.065334.
Gutierrez R, Lindeboom JJ, Paredez AR, Emons AMC. Arabidopsis cortical microtubules position cellulose synthase delivery to the plasma membrane and interact with cellulose synthase trafficking compartments. Nat Cell Biol. 2009;11(7):797–806. https://doi.org/10.1038/ncb1886.
Liu TM, Zhu SY, Tang QM. De Novo assembly and characterization of transcriptome using illumian paired-end sequencing and identification of CesA gene in ramie (Boehmeria nivea L. gaud). BMC Genomics. 2013;14:125. https://doi.org/10.1186/1471-2164-14-125.
Lei L, Singh A, Bashline L, Li S, Yingling YG, Gu Y. CELLULOSE SYNTHASE INTERACTIVE1 is required for fast recycling of cellulose synthase complexes to the plasma membrane in Arabidopsis. Plant Cell. 2015;27(10):2926–40. https://doi.org/10.1105/tpc.15.00442.
Zhu XY, Li SD, Pan SQ, Xin XR, Gu Y. CSI1, PATROL1, and exocyst complex cooperate in delivery of cellulose synthase complexes to the plasma membrane. Proc Natl Acad Sci U S A. 2018;115(15):E3578–87. https://doi.org/10.1073/pnas.1800182115.
Li SD, Lei L, Somerville CR, Gu Y. Cellulose synthase interactive protein1 (CSI1) links microtubules and cellulose synthase complexes. Proc Natl Acad Sci U S A. 2012;109(1):185–90. https://doi.org/10.1073/pnas.1118560109.
Karpova TS, Tatchell K, Cooper JA. Actin filaments in yeast are unstable in the absence of capping protein or fimbrin. J Cell Biol. 1995;131(6):1483–93. https://doi.org/10.1083/jcb.131.6.1483.
Zhong R, Burk DH, Morrison WH, Ye ZH. A kinesin-like protein is essential for oriented deposition of cellulose microfibrils and cell wall strength. Plant Cell. 2002;14(12):3101–17. https://doi.org/10.1105/tpc.005801.
Zhu C, Dixit R. Single molecule analysis of the Arabidopsis FRA1 kinesin shows that it is a functional motor protein with unusually high processivity. Mol Plant. 2011;4(5):879–85. https://doi.org/10.1093/mp/ssr077.
Kong ZS, Ioki M, Braybrook S, Li SD, Ye ZH, Julie Lee YRJ, et al. Kinesin-4 functions in vesicular transport on cortical microtubules and regulates cell wall mechanics during cell elongation in plants. Mol Plant. 2015;8(7):1011–23. https://doi.org/10.1016/j.molp.2015.01.004.
Ambrose C, Allard JF, Cytrynbaum EN, Wasteneys GO. A CLASP-modulated cell edge barrier mechanism drives cell-wide cortical microtubule organization in Arabidopsis. Nat Commun. 2011;2(1):430. https://doi.org/10.1038/ncomms1444.
Lindeboom JJ, Nakamura M, Saltini M, Hibbel A, Walia A, Ketelaar T, et al. CLASP stabilizes microtubule plus ends created by serving to drive cortical array reorientation. J Cell Biol. 2018;218(1):190–205. https://doi.org/10.1083/jcb.201805047.
Shen ZJ. Mechanism of herbivorous insect resistance and salt tolerance in mangrove plant Avicennia marina. Doctoral Dissertation of Xiamen University. 2018.
Harvey PJ, Campanella BF, Castro PML, Harms H, Lichtfouse E, Schoffner AR, et al. Phytoremedition of polyaromatic hydrocarbons, anilines and phenols. Envi Sci Pollut Res. 2002;9(1):29–47. https://doi.org/10.1007/BF02987315.
Schuler MA, Daniele WR. Functional genomics of P450s. Annu Rev Plant Biol. 2003;54(1):627–9.
Morant M, Bak S, Moller BL, Werck-Reichhart D. Plant cytochromes P450: tools for pharmacology, plant protection and phytoremediation. Curr Opin Biotech. 2003;14(2):151–62. https://doi.org/10.1016/S0958-1669(03)00024-7.
Kasai S. Role of cytochrome P450 in mechanism of pyrethroid resistance. J Pestic Sci. 2004;29(3):220–1. https://doi.org/10.1584/jpestics.29.220.
Lu HP, Luo T, Fu HW, Wang L, Tan YY, Huang JZ, et al. Resistance of rice to insect pests mediated by suppression of serotonin biosynthesis. Nat Plants. 2018;4(6):338–44. https://doi.org/10.1038/s41477-018-0152-7.
Wang EM, Wang R, DeParasis J, Loughrin JH, Gan S, Wagner GJ. Suppression of a P450 hydroxylase gene in plant trichome glands enhances natural-product-based aphid resistance. Nat Biotechnol. 2001;19(4):371–4. https://doi.org/10.1038/86770.
Shan RY, Lin ZH, Chen ZH, Zhong QS, You XM, Chen CS. Molecular cloning and expression analysis of cytochrome P450 CYP71A26 and CYP71B34 genes in tea plants (Camellia sinensis). J Tea Sci. 2018;38(5):450–60.
Verma V, Ravindran P, Kumar PP. Plant hormone-mediated regulation of stress responses. BMC Plant Biol. 2016;16(1):86. https://doi.org/10.1186/s12870-016-0771-y.
Gibbs DJ, Conde JV, Berckhan S, Prasad G, Mendiondo GM, Holdsworth MJ. Group VII ethylene response factors coordinate oxygen and nitric oxide signal transduction and stress responses in plants. Plant Physiol. 2015;69(1):23–31. https://doi.org/10.1104/pp.15.00338.
Li JJ, Guo X, Zhang MH, Wang X, Zhao Y, Yin ZG, et al. OsERF71 confers drought tolerance via modulating ABA signaling and proline biosynthesis. Plant Sci. 2018;270:131–9. https://doi.org/10.1016/j.plantsci.2018.01.017.
Takafuji K, Rim H, Kawauchi K, Mujiono K, Shimokawa S, Ando Y, et al. Evidence that ERF transcriptional regulators serve as possible key molecules for natural variation in defense against herbivores in tall goldenrod. Sci Rep. 2020;10(1):5352. https://doi.org/10.1038/s41598-020-62142-4.
Wu J, Gao H, Zhu XW, Li DF. An ERF transcription factor enhances plant resistance to Myzus persicae and Spodoptera litura. Biotechnol Biotec Eq. 2020;34(1):946–54. https://doi.org/10.1080/13102818.2020.1813051.
Friedrichsen DM, Nemhauser J, Muramitsu T, Maloof NJ, Alonso J. Three redundant brassinosteroid early response genes encode putative bHLH transcription factors required for normal growth. Genetics. 2002;162(3):1445–56. https://doi.org/10.1093/genetics/162.3.1445.
Zhang CJ, Hou YQ, Hao QN, Chen HF, Chen LM, Yuan SL, et al. Genome-wide survey of the soybean GATA transcription factor gene family and expression analysis under low nitrogen stress. PLoS One. 2015;10(4):e0125174. https://doi.org/10.1371/journal.pone.0125174.
Xi Y, Li W. BSMAP: Whole genome bisulfite sequence MAPping program. BMC Bioinformatics. 2009;10(1):232.
Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base reolution show widespread epigeomic differences. Nature. 2009;462(7271):315–22. https://doi.org/10.1038/nature08514.
Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A. Distinct DNA methylomes of newborns and centenarians. Proc Natl Acad Sci U S A. 2012;109(26):10522–7. https://doi.org/10.1073/pnas.1120658109.
Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, et al. GO: TermFinder-open source software for accessing gene ontology information and finding significantly enriched gene ontology terms associated with a list of genes. Bioinformatics. 2004;20(18):3710–5. https://doi.org/10.1093/bioinformatics/bth456.
Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40(D1):109–14. https://doi.org/10.1093/nar/gkr988.
Funding
This work was supported by the grants from the Natural Science Fundation of Guangxi (2017GXNSFDA198037, 2016GXNSFAA380238), and the Development Foundation of Guangxi Academy of Agricultural Sciences (2020YM116, 2021YT054).
Author information
Authors and Affiliations
Contributions
ZS and WZ conceived and designed the experiments. WZ, RT, SL, ZS, ZL, SY, HC and XQ performed the experiments. WZ, RT and SL analyzed the data. ZS and WZ contributed reagents/materials/analysis tools. ZS and WZ conceived the experiments and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable for this research. Soybean seeds for this study were obtatined from the Guangxi Academy of Agricultual Scinence.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised: the original publication mistakenly listed five corresponding authors.
Supplementary Information
Additional file 1: Table S1
The negatively correlated genes between transcription and methylation of soybean resistance to bean pyralid larvae.
Additional file 2: Table S2
Pathway analysis of the negatively correlated genes in the gene bodies.
Additional file 3: Table S3
Pathway analysis of the negatively correlated genes in the promoter regions.
Additional file 4: Table S4
MapMan cluster analysis of the negatively correlated genes.
Additional file 5: Table S5
Primers for the qRT-PCR and PS-PCR.
Additional file 6: Fig. S1
The resistant material and susceptible material under bean pyralid larvae feeding for 48 h. A: Gantai-2-2; B: Wan82–178.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zeng, WY., Tan, YR., Long, SF. et al. Methylome and transcriptome analyses of soybean response to bean pyralid larvae. BMC Genomics 22, 836 (2021). https://doi.org/10.1186/s12864-021-08140-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-08140-w