
Abstract
Background
Improving feed efficiency (FE) is an important goal due to its economic and environmental significance for farm animal production. The FE phenotype is complex and based on the measurements of the individual feed consumption and average daily gain during a test period, which is costly and time-consuming. The identification of reliable predictors of FE is a strategy to reduce phenotyping efforts.
Results
Gene expression data of the whole blood from three independent experiments were combined and analyzed by machine learning algorithms to propose molecular biomarkers of FE traits in growing pigs. These datasets included Large White pigs from two lines divergently selected for residual feed intake (RFI), a measure of net FE, and in which individual feed conversion ratio (FCR) and blood microarray data were available. Merging the three datasets allowed considering FCR values (Mean = 2.85; Min = 1.92; Max = 5.00) for a total of n = 148 pigs, with a large range of body weight (15 to 115 kg) and different test period duration (2 to 9 weeks). Random forest (RF) and gradient tree boosting (GTB) were applied on the whole blood transcripts (26,687 annotated molecular probes) to identify the most important variables for binary classification on RFI groups and a quantitative prediction of FCR, respectively. The dataset was split into learning (n = 74) and validation sets (n = 74). With iterative steps for variable selection, about three hundred’s (328 to 391) molecular probes participating in various biological pathways, were identified as important predictors of RFI or FCR. With the GTB algorithm, simpler models were proposed combining 34 expressed unique genes to classify pigs into RFI groups (100% of success), and 25 expressed unique genes to predict FCR values (R2 = 0.80, RMSE = 8%). The accuracy performance of RF models was slightly lower in classification and markedly lower in regression.
Conclusion
From small subsets of genes expressed in the whole blood, it is possible to predict the binary class and the individual value of feed efficiency. These predictive models offer good perspectives to identify animals with higher feed efficiency in precision farming applications.
Similar content being viewed by others

Background
Peripheral blood is widely used in human medicine and veterinary fields as a relevant and easy sampling source of biological information, since it transports a large variety of molecules including DNA, coding and non-coding regulatory RNA, proteins and metabolites from all over the body. Their dynamics reflects homeostatic regulation [1,2,3], physiological changes [4, 5] and variations in immune capacity [6, 7]. Circulating molecules also provide valuable insights into complex phenotypes such as obesity and diabetes [8, 9], health status [10], sensitivity to heat stress [11] and nutrient efficiency for productive outputs [3, 7, 12]. Therefore, they hold much promise for the identification of biomarkers for particular phenotype prediction [13]. Both hypothesis-based and discovery-based procedures are used for the search of biomarkers. For a discovery-based procedure, high-throughput expression studies analyzed by linear model statistics and functional annotation bioinformatics are often used to enlighten how expressed genes and related biological pathways are discriminants between treatments. However, a plethora of machine learning (ML) approaches applied on data gathered in a learning base from characterized samples have the potential to surpass these traditional approaches in predicting class membership and individual values of unknown samples gathered in a test base [14]. In conditions where small variations in the data may cause significant changes in the prediction, these methods generally overcome complex, noisy and hidden relationships when ranking the most important genes for prediction and avoid the pitfalls of overfitting.
Feed efficiency (FE) has become a research priority in growing pigs to support competitive and sustainable meat production. Improving FE is a strategy to reduce the amount of feed needed to produce meat and to reduce environmental wastes and emissions. Feed efficiency is measured on a farm as feed conversion ratio (FCR), calculated as the ratio of an amount of feed intake to body weight (BW) gain. Residual feed intake (RFI) has also been proposed as a refined measure of net FE in selection experiments [15]. It is defined as the difference between the observed feed intake and the feed intake predicted from growth and maintenance requirements. For RFI, BW gain and indicators of body composition such as backfat thickness must be recorded during a test period for each animal. This is time-consuming and costly, especially when animals are housed in group. Moreover, FE is underlined by variations in the transcripts of several genes participating in many functional pathways in different tissues [16], which adds to its complexity. Therefore, there is a need to find molecular biomarkers that accurately differentiate high and low FE animals and that can be further used for improving FE of growing animals in breeding programs or nutritional decision tools. So far, various studies have revealed differences in the whole blood transcriptome between low RFI (most feed efficient) and high RFI pigs (less feed efficient) at post-weaning [12] and during the growing period [3, 7]. Moreover, the concentration of IGF-1 in blood plasma of juvenile post-weaned pigs was correlated with RFI measured during the growing period [17], suggesting that circulating molecules may even serve as early indicators for FE. However, among genes identified as differentially expressed between steers with low or high BW gain and feed intake, only few of them were similarly found across different cohorts [18]. This highlights the importance of incorporating different datasets to cover various experimental conditions and to avoid the limits of each design (number of samples/number of treatments) for biomarker discovery.
This study aimed to identify reliable sets of expressed genes in the whole blood to predict the RFI group or individual FCR value. For that, ML algorithms were applied on a merged transcriptomic dataset from three independent experiments where meta-data for RFI and FCR were also available in growing pigs.
Results
Animals and FE traits
Three independent experiments [19,20,21] were merged to reanalyze gene expression levels in whole blood from a total of 148 females and barrows. These experiments all included purebred French Large White pigs of two lines divergently selected for RFI during 7th to 9th generations, and were based on different dietary treatments. The distribution of FCR values for the 148 pigs considered in the merged dataset was illustrated in Fig. 1, according to the RFI group and their experiment of origin. The FCR averaged 2.85 kg feed/kg BW, and covered a large range of values (Min = 1.92; Max = 5.00). It was generally lower for pigs of the low RFI line than for pigs of the high RFI line, but there was an interpenetration between the two lines within each experiment and between experiments.

Distribution of feed conversion ratio across the dataset. Pigs of low and high residual feed intake (RFI) lines were considered in three different experiments unraveling different periods for blood sampling. Feed conversion ratio (FCR) was measured for each pig during specific test periods. The first dataset included 21 pigs, the second dataset included 48 pigs and the third dataset included 79 pigs. In the merged dataset, 148 pigs were thus analyzed. Feeding conditions, test periods and age and body weight of pigs when blood sampling was performed, are detailed in Material and Methods
Model performance in RFI classification
Merging the transcriptomic data of the three independent experiments resulted in a new dataset of 26,687 annotated expressed probes across the 148 blood samples. The random forest (RF) and gradient tree boosting (GTB) procedures were applied to this merged dataset to find the most important transcripts that allow the classification of pigs (low RFI/high RFI). These algorithms were considered to produce an excellent fit of predicted to observed values even when the specific nature of the relationships between the predictor variables and the dependent variable was very complex [22]. In the two procedures, a randomly selected bootstrap sample set was used as a learning dataset (n = 74 pigs), whereas the remaining samples (n = 74 pigs) were used in a test dataset for validation. Learning and validation datasets including transcriptomic data and meta-data (RFI group, FCR) are freely available at https://doi:10.15454/J4XOPD.
From the RF procedure, a total of 778 probes (out of the 26,687 annotated probes) were first selected to provide an accurate classification of pigs into low and high RFI groups during the training step. In the validation step (Supp. Table S1), the RF model further selected 328 probes (out of the 778 probes) as very important variables (VIP) for RFI classification. The accuracy of the model was estimated by the proportion (%) of good classification, and the optimal model was selected according to the receiver operating characteristic curve (ROC) as a diagnostic ability of the binary classifier system. Iterative steps allowed to obtain the best model (96% of success on average) with a subset of 50 molecular probes (out of the 328 VIP). It provided a good prediction for 94.74% of the high RFI pigs and 97.22% of the low RFI pigs, respectively (Table 1), so that the prediction accuracy was similar for the two RFI lines (chi2 = 0.59). The 50 VIP corresponded to 25 unique identified expressed genes since 17 probes had no consolidated annotation and some genes were represented by two up to four probes (GPX3, CD1A, and SERPINF1). The list of these 50 probes, the encoded genes, and the score attributed to each probe in the predictive RF model is given in Supp. Table S2.
From the GTB procedure, a total of 728 probes (out of the 26,687 annotated probes) were similarly retained as providing an accurate classification on low/high RFI during the training step. In the validation step, the GTB model further identified 391 probes (out of 728 probes) as the best VIP to classify pigs on low or high RFI (Supp. Table S1). Iterative steps led to select a subset of 50 molecular probes (out of the 391 VIP) allowing 100% of good classification (Table 1). These 50 probes corresponded to 34 unique annotated expressed genes (Table 2); these genes were all represented by a single probe in the model but 16 probes had no consolidated annotation. The top five transcripts for classifying pigs into low or high RFI were the following: PSEN1, SERPINF1, TMEM63B, EPAS1 and MX1 genes (Table 2).
Overall, 12 annotated expressed genes (25% of the genes retained by each method) were commonly proposed by the RF and GTB models as top VIPs to classify pigs on low or high RFI. They were PSEN1, SERPINF1, EPAS1, GPX3, CLU, WDHD1, HTRA1, PARVG, HMG20A, RPS18, SLC46A3 and DCT.
Model performance in FCR prediction
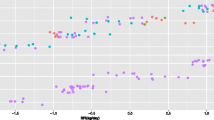
When addressing continuous variables such as FCR, regression trees rather than classification trees must be built. The transcriptomic dataset was similarly split into training (n = 74) and validation (n = 74) datasets. About 1393 probes (out of the 26,687 annotated probes) were selected during the training procedure. The performance of the models was then evaluated by using the validation set, and models with the best R2 and the lowest Root Mean Squared Error (RMSE) were retained (Table 3). The accuracy of prediction by the GTB algorithm (R2 ~ 0.80; RMSE ~ 0.23) exceeded that obtained by the RF procedure (R2 ~ 0.65 and RMSE ~ 0.29). Due to this large difference in model performance between the two algorithms in regression, also mentioned by others [23], only the results of the GTB model for FCR prediction were described in this study. The GTB procedure first identified 428 probes as a top VIP to predict FCR values. Iterative steps led to a good compromise between a lower number of VIP and increased accuracy of the prediction, which was obtained with 50 molecular expressed probes. These 50 probes corresponded to 27 unique annotated genes (Table 4). Finally, the predicted (X) values were compared with the observed (Y) values for the pigs included in the validation set (n = 74). The quality of the relationships was evaluated based on the RMSE of prediction (RMSEP) obtained by a leave-one-out cross-validation from the value of the predicted residual sum of squares. Observed and predicted values for FCR were very close (R2 = 0.80, RMSEP = 0.15; Fig. 2). The mean of predicted FCR values was 2.83 and the mean for observed FCR values was 2.85, respectively, and the error made by the model was evaluated at 7% on the average. The samples (n = 5) having the highest residual (> 0.15) all corresponded to pigs of the high RFI line but from different experiments (1 pig from experiment 1, 1 pig from experiment 2, and 3 pigs from experiment 3, Supp. Fig. S1), suggesting no bias due to the independent datasets. Without these few samples (5 out of 74), the prediction accuracy was improved (R2 = 0.94).

Regression analysis of the relationship between observed and predicted FCR. A predictive model to identify the most important annotated expressing probes able to predict feed-conversion-ratio (FCR) was built from the whole blood transcriptome merged from three independent experiments, and using a Gradient TreeNet Boosting (GTB) algorithm. Randomly selected bootstrap pig samples (n = 74) were used for learning, whereas the remaining samples (n = 74) were used for validation. Iterative steps led to retain a set of 50 very important variables. The graph was then computed between observed and predicted FCR values. Accuracy of the prediction was estimated by using R squared (R2) and root mean square error of prediction (RMSEP). Pigs considered in the study were from two divergent selection lines for residual feed intake (RFI), a measure of net feed efficiency. The red square represents pigs of the high RFI line, and the blue dot represents pigs of the low RFI group. No specific bias in prediction was observed due to RFI line
Overview of the biological pathways shared by the molecular predictors of RFI and FCR traits
To progress in the knowledge of the most important biological pathways underlying the variation of FE among pigs, the expressed genes selected by the GTB models as top VIP allowing binary diagnostic for RFI (low/high) or predicting FCR (individual values) were submitted to a functional analysis using n bioinformatics tool. The 391 molecular probes initially selected to split pigs into low and high RFI groups, corresponded to 253 annotated unique genes that were clustered into 14 biological pathways (Table 5). The lipid metabolic process and transport, response to oxidative stress, phosphorylation, and positive regulation of defense response were among the top functional pathways identified across these genes. The 728 molecular probes selected to predict FCR values corresponded to 477 unique annotated genes that were clustered in 10 biological pathways (Table 5). Significant pathways were related to immune and defense response (regulation of leukocyte activation, regulation of cytokine production, regulation of acute inflammatory response and positive regulation of immune response), glycoprotein metabolic process, regulations of protein transport and of peptidase activity, and protein amino acid auto-phosphorylation.
The subset of the 50 best VIP to predict FCR values are participating in a variety of pathways, such as the regulation of immune system response (CD84, IGF2, PSMB9, TRIM38, PIKFYVE, KLF1, IRF2BP2), protein metabolism and especially ubiquitination process (SHPRH, PSMB9, FEM1C, TRIM38), response to peptides and organic substances (GNG12, KLF1, IGF2, PSMB9, TRIM38), lipid and cholesterol metabolic process (MORC2 CYP51A1, DHCR24), oxido-reduction (SDR39U1) or intracellular transport (CD84, GEMIN5, PIKFVE, AAGAB, SLC36A4). This suggested equal importance of many biological routes in the variation of FCR, and underlined genes as pivots in inter-related pathways.
Overall, 63 unique genes (i.e., 8% of all VIPs) expressed in the whole blood were identified as common VIP for the two FE traits (Table 6). Among them, BCO2, CREBRF, GPX3, HMG20A, JPH4, PAG1 and SPTLC2 were notably included in the list of top 50 VIP for RFI, while IRF2BP2, MACF1, MORC2, SDR39U1, TRIM46 and ZNF644 were included in the list of top 50 VIP for FCR.
Discussion
Due to the integrative nature of FE and the difficulties to record it accurately for each pig, there is a strong need to identify relevant biomarkers of FE traits. Also, because transcriptomic differences in muscle and liver segregated pigs on RFI better than their genotype and farm of origin [24], we hypothesized that the landscape of gene expression levels in the whole blood, a compartment that summarizes the variations in tissue metabolism, may be used to find biomarkers of FE in growing pigs. The data presented herein confirmed that the gene expression profiling in the whole blood represented a relevant source to identify small sets of candidate biomarkers for two FE traits. Previous studies have identified about 1000 genes [1] and even more [2] that were differentially expressed in the whole blood between low and high RFI pig lines. But none have tried to identify molecular predictors for low/high RFI (binary classification) and quantitative values of FCR (prediction of individual values). For that, ML procedures have proven their capability to develop highly precise prediction models including FE [25]. In the current study, it was possible to discriminate pigs according to RFI (low/high) by using a subset of few blood transcripts (< 50) with 96 to 100% of success when using RF and GTB procedures, respectively. Moreover, it was possible to predict individual FCR, and not only the assignment of animals to divergent groups, by using another subset of 50 transcripts corresponding to 25 unique annotated genes with a good (~ 0.65; RF model) and very good (~ 0.80; GTB model) accuracy. Similarly, recent studies using ML algorithms [26, 27] in pigs showed that it was possible to predict the binary class of RFI by using the expression of 200 genes in the liver (accuracy: 0.78), 100 genes in duodenum (accuracy: 0.69) and 50 genes in skeletal muscle (accuracy: 0.61–0.70). In the current study, only 8% of all VIPs were identified as common predictors for RFI and FCR. These two traits are not equivalent, with only a moderate (0.39) genetic correlation between RFI and FCR [15], and in some studies, higher correlations between FCR and production traits than between FCR and RFI [28]. The common predictors in our study may correspond to the RFI part of FCR variability. Irrespective of the FE trait, the GTB procedures had better performance than the RF algorithms. This confirms that, despite a significant amount of overlap between the two methods and although RF performs well for class object detection, the gradient boosting methods result in better performance on other assessments like regression [22, 29]. Indeed, the GTB algorithm combines the gradient descent error minimization approach with boosting, and encapsulates an ensemble of weak prediction models added sequentially to improve the robustness of predictors [14]. In cattle, different ML methods have been tested to identify candidate genes for growth prediction, and the authors concluded that the better performance was obtained with the gradient boosting machine algorithm followed by the RF [30]. As compared with the individual ML method alone, combining RF and GTB together may further produce the highest value of prediction accuracy with the smallest subsets of genes that are biologically relevant to FE, as suggested in beef cattle [25]. This deserves further studies.
The subsets of genes combined in predictive models of FE in growing pigs were involved in several functional pathways that might be of equivalent importance in the definition of RFI and FCR. For some of them, they shared common transcriptional regulators. Finding relevant biological categories across the VIP attested to the reliability of the proposed candidates. Indeed, many genes in the immune/inflammatory system were identified as top predictors for RFI (PSNE1, SERPINF1, MX1), for FCR (CD84, PIKFYVE, IRF2BP2) and for both traits (JPH4, PAG1). This is consistent with findings that low RFI pigs had specificities in their immune tissue profile and capacity to respond to infectious or inflammatory challenges as compared with high RFI pigs [31, 32]. Especially, IRF2BP2 (Interferon Regulatory Factor 2 Binding Protein 2) has emerged as an important transcriptional co-regulator in the immune system [33]. Moreover, JPH4, a gene that was also identified as differentially expressed in the liver of pigs that differed in FE [24], stimulates the expression of activation markers and cytokines [34]. However, PAG1 negatively regulates T-cell activation [35]. Finding these two genes having opposite actions on T cell activation in the prediction model suggests subtle modifications in the regulation of immune signaling in pigs ranked for FE. Interestingly, SERPINF1, HTRA1 and NPR3 proposed for binary classification of pigs on RFI, have been previously identified as having the biggest changes in expression level in the whole blood between low and high RFI pig lines [3]. In accordance, Chen and colleagues [25] indicated that the GTB method picked up the top-ranked differentially expressed genes identified by t-test for FE in beef cattle. In the current study, some candidate biomarkers for FE were also associated with roles in the ubiquitination and protein modification process. The importance of the ubiquitin pathway may be over-estimated here, since we considered the whole blood where this process is specifically enriched [36]. This could be also related to the higher protein turnover identified in the liver of the most efficient pigs compared with less efficient pigs [31]. Because immunity, inflammation and ubiquitin-related protein modification are inter-related pathways, it is not surprising to find common genes in these pathways among the proposed biomarkers. For instance, TRIM38 encoding the E3 ubiquitin ligase has multifaceted roles in innate immunity and inflammation [37]. Different genes related to antioxidant response and oxido-reduction activity, such as GPX3 (glutathione peroxidase-3) and BCO2 (beta-carotene oxygenase-2), were among the top-ranked VIPs for RFI classification. This is consistent with previous studies showing a difference in susceptibility to oxidative stress between low and high feed efficient pigs [38, 39]. Finally, lipid transport and catabolism, including SPTLC2 and MORC2 identified as top predictors, were underlined as biological pathways able to classify pigs according to RFI. Similarly, molecular alterations in lipid metabolism have been observed in the liver of low/high RFI pigs, having consequences on triglycerides, phospholipids or cholesterol concentrations in the blood of pigs from the same [3] or different [40] RFI lines. Among others, PLA2G4A (phospholipase A2 group IVA) identified as a common VIP for RFI and FCR traits, was recently suggested as a key regulator of fat deposition in chicken [41]. Increased circulating levels of IL-15 correlated with variations in adipose tissue mass and FCR in male mice fed a high-fat diet [42]. In the current study, it is unlikely that differences in diets between experimental pigs biased the results. Indeed, we did not observe any marked changes in the accuracy of prediction model for FCR reasoned on (net) energy intake or feed intake [26].
Altogether, this study reduced the complexity of FE into small subsets (< 50) of predictive transcripts. Among these candidate biomarkers for accurate predictions of RFI groups and FCR values, several genes have been already proposed as top molecular contributors to differences between low and high RFI lines, and even, in predictive models of FE. For instance, the expression level of GPX3 was identified as affected by RFI selection in the whole blood [3], muscle, adipose tissues and liver of the same lines of pigs [16]. Similarly, HTRA1 was listed as differentially expressed in both muscle and liver between low and high RFI pigs of two different populations [24]; this serine protease mediates multiple biological processes by antagonizing IGF-binding proteins and proteins of the TGF-beta family [43]. In line, the expression level of IGF2 in muscle was identified as a reliable predictor for RFI breeding values [26] and increasing muscle growth through the IGF-1/2 signaling pathway was proposed as a potential strategy for the improvement of FE in Yorkshire pigs from 30 to 90 kg BW [44]. Expression level of PSNE1 in muscle was also included in a predictive model for RFI breeding value in pigs of Large White breed [26]. The expression level of SLC46A3 in liver, a gene involved in macromolecule degradation process [45], was identified as important to categorize pigs into RFI groups in the Hermitage Maxgro genotype [27]. Finding the same molecular candidates in different tissues is not surprising, since only < 10% of protein coding genes are tissue specific [46]. This reinforces the interest in using readily accessible samples in living animals such as blood to predict complex phenotypes. The proposed biomarkers can be also confronted to genomic regions identified by genome-wide association study (GWAS) as affecting FE traits. For instance, TEX2 (Testis Expressed 2 protein) identified here as a predictive biomarker for RFI classification, was identified as a positional candidate in SNP detected for average daily feed intake (a FE related trait) in Landrace pigs [47].
Conclusion
This study identified small sets of transcripts in the whole blood as candidate biomarkers for FE traits, namely RFI group (low/high) and FCR values measured in growing pigs. Since nutritional requirements for the most efficient pigs might be slightly greater than usual feed recommendations [48], these circulating biomarkers could be further used as a decision support tool for feeding animals with an appropriate diet. Therefore, this study offers encouraging perspectives for assigning animals to phenotypic groups and to be used as proxy of FE in large numbers of animals. Additional studies are required to confirm the generality of the predictions in other pig breeds and crossbreds before these circulating biomarkers could be readily used for precision farming strategies.
Methods
General design
This study reused phenotypic data obtained in pigs from the three independent experiments that were previously published [19,20,21], to avoid the needs of new sampling in living animals while obtaining a high number of animals allowing robust predictions. The application of ML procedures on the merged dataset (n = 148 pigs) avoided the overfitting often observed when simple classification or regression procedures are used for a limited number of animals and a high number of dependent variables, and the leave-one-out method was an additional way to resampling the datasets. Thus, this study fits with the 3R (Replacement, Reduction and Refinement) principles.
Pigs and blood samples
The three independent datasets referred to purebred French Large White pigs produced in a divergent selection experiment for RFI. The selection program was described in full details elsewhere [49], including the equation to calculate RFI from a regression between observed feed intake and that expected based on requirements for maintenance (based on the metabolic BW) and performance (average daily gain, backfat thickness). From birth to weaning, all pigs were reared in the selection farm of INRAE (UE Genesi, Le Magneraud & Rouillé, France; https://doi.org/10.15454/1.5572415481185847E12). All pigs were weaned at 28 days (d), and were first fed ad libitum with standard starter and weaner diets. During subsequent test periods in dedicated buildings, pigs have undergone different feeding conditions depending on the experiments as described below. As indicated in the referenced publications [19,20,21], the three experiments were conducted in accordance with the French legislation on animal experimentation, and the protocols were approved by regional ethical committees evaluating the research question, design, plan analysis, animal care and monitoring, and ways to minimize pain and consider limit points (especially regarding jugular blood sampling). At the end of each experiment, pigs were slaughtered using approved procedures, including electronarcosis followed by jugular exsanguination.
The first dataset [19] included 21 castrated males from the 7th generation of selection (n = 10 low RFI pigs and n = 11 high RFI pigs) housed at thermo-neutrality (24 °C) and reared at the INRAE experimental pig facility at Saint-Gilles, France (UE3P, https://doi.org/10.15454/1.5573932732039927E12). At 80 d of age, pigs were transferred in individual cages, and were fed a standard diet that met nutritional requirements for growth. At 87 d of age (59.2 kg BW on average), blood was collected from the jugular vein and prepared for RNA extraction. The feed conversion ratio (FCR) was calculated from individually measured daily feed intake and average daily gain for the 14 d of the trial (i.e., from 87 d to 100 d of age).
The second dataset [20] included 48 castrated males from the 8th generation of selection (n = 24 low RFI pigs and n = 24 high RFI pigs). Pigs were reared at the INRAE experimental pig facility at Saint-Gilles, France (UE3P, https://doi.org/10.15454/1.5573932732039927E12). At 74 d of age, pigs were transferred in individual cages and after 2 d of transition, the first half was fed a standard diet and the second half was fed a high-fiber high-fat diet during the growing and finishing phases. At 132 d of age (average BW of 75.6 kg), blood was sampled from the jugular vein and prepared for RNA extraction. The FCR was then calculated from 76 d to 132 d of age.
The third dataset [21] included 79 castrated males and females from the 9th generation of selection (n = 37 low RFI pigs and n = 42 high RFI pigs). Pigs were reared at the experimental INRAE pig facility at Le Magneraud, France (UE Genesi; https://doi.org/10.15454/1.5572415481185847E12). Blood was sampled at 40 d of age from the jugular vein. At 70 d of age, pigs were transferred to group-housing facilities equipped with single-place electronic feeders. The first half of the pigs were fed standard diets, whereas the second half was fed a high-fiber diet during the growing-finishing phases [21]. The FCR was then calculated from 90 d to 161 d of age.
In the three datasets, the reference to low or high RFI line was indicated for each pig, and the FCR value was individually attributed. Other factors (sex, season, generation, diet) were not taken into account.
Microarrays data
Microarray data considered in the current study were obtained from the referenced publications in the first [19] and second [3] experiments, and were newly acquired from RNA extracted from the stored blood samples in the third experiment. All experiments followed the same procedures for RNA extraction and expression data generation. The porcine commercial Agilent-026440 microarray (V2, 44 K, GPL15007, Agilent Technologies, Massy, France) had been used in the first experiment (representing about 12,332 unique annotated genes). The custom porcine microarray (8x60K, GPL16524 Agilent Technologies) that contained the same probes as the Agilent-026444 and an additional set of probes enriched with immune system, muscle and adipose tissue genes, has been used in the second and third experiments (representing about 14,466 unique annotated genes). In the three transcriptomic datasets, raw spot intensities have been submitted to quality filtration based on four criteria: background intensity value, diameter, saturation and uniformity of the spot, and intensities of filtered spots were log2 transformed and median-centered to correct for microarray effect.
For the current study, the three microarray datasets were then merged into a single new dataset. There was no exclusion of any animals in this merged dataset. To obtain consolidated expression values across the three independent datasets, the molecular data have been normalized by mean centering, i.e. subtracting the mean value across all probes from all raw values for each pig sample in the merged dataset. The merged dataset also included meta-data such as the experiment of origin (1, 2, and 3), RFI group (n = 71 pigs of low RFI line, n = 77 pigs of high RFI line) and FCR value (n = 148 pigs). All data were deposited in a publicly available repository at https://doi.org/10.15454/J4XOPD.
Supervised methods to identify important variables for the prediction of FE traits
The merged dataset was used to search the most important molecular predictors for the RFI group and FCR value, by using ML methods. The experimental unit was the pig. Among the panel of ML methods for dimensionality reduction, classification and regression used in livestock breeding [14], the RF and GTB procedures were chosen in the current study and were compared for performance in classification (RFI group) and regression (FCR value) procedures. These two ML methods use decision trees, but RF uses a large number of trees combined by averaging or “majority rules” at the end of the process [50], whereas GTB starts the combining process of decision trees at the beginning [27, 51, 52]. Other differences include how trees are built: RF builds each tree independently, while GTB builds one tree at a time but in an additive model proceeding in a forward stage-wise sequential error–correcting process to combine results along the way and converge to an accurate model [29]. Sequential steps for learning, validation, and finally, selection of the best models were performed according to standards described by Fernandez-Lozano and colleagues [53]. Models were generated from RF and GTB algorithms with Salford Predictive Modeler 8.0 (SPM 8.0®).
The RF models were generated with about 1500 trees for classification of RFI and regression for FCR. For that, a randomly selected bootstrap sample set was created by using 50% of the original dataset for learning (n = 74 pigs). Consequently, each bootstrap sample called “out-of-bag” data (OOB) excluded 50% of the data that were further used for validation (n = 74 pigs), and the leave-one-out method assessed the performance by resampling the training set. The test dataset allowed cross-validation ensuring that the training of the model was not biased. To split branches of a tree, a random sample of m variables was chosen from the full set of p variables. The partition of probes between learning and validation datasets was shown in Supp. Fig. S2. We checked that the three experiments of origin were included in both the training and validation datasets.
The GTB prediction models were also generated using 1500 small decision trees for classification or regression, and using a randomly selected bootstrap sample set for learning (n = 74 pigs) and the remaining data (n = 74 pigs) for validation. As recommended, each tree typically contained about six terminal nodes. The model was like to Fourier or Taylor series, which is a sum of factors that becomes progressively more accurate as the expansion continues. After each step of boosting, the algorithm scaled the newly added weights, which balanced the influence of each tree. The accuracy of the algorithm was improved by introducing randomization through training the base learner on different randomly selected samples at each iteration.
In both procedures, significant variables were selected using the Gini index to evaluate the discriminant ability of the potential selected feature, defined as:
Where p2 (j | t) is the estimated class probability for feature t or node t in a decision tree and j is an output data or class. Only the variables that improved Gini index and minimized the OOB error rate were retained as very important variables in prediction (VIP).
Multiple runs for each ML methods were performed (ten times) to consider variations in the observations used for the training step (using permutations and leave-one-out procedures) and the stability of the techniques. The iteration steps were also applied to reduce the number of VIPs in the selected models. At each run, the accuracy of classification models was estimated with the proportion (%) of good classification and the optimal models were selected according to the ROC curve. In regression, RMSE was calculated as the square root of the difference between the realized and the predicted observation within the OOB data after permuting each predictor variable in the training dataset divided by the number of trees for the regression procedure. The adjusted coefficient of determination (R2) was also computed. The predicted (X) values for FCR obtained by the best GTB model and the observed (Y) values measured on the pigs were compared (X-Y) using the GLM procedure. The model was considered unbiased when the intercept obtained by the GLM model was not different from 0 and the slope was not significantly different from 1. The quality of the relationships was evaluated based on RMSE of prediction (RMSEP) obtained by a leave-one-out cross-validation from the value of the predicted residual sum of squares.
Pathway enrichment analysis
Gene-annotation enrichment analyses among the VIP identified for binary classification of pigs on RFI and prediction of FCR were performed on encoded genes by using DAVID bioinformatics tool on default settings [54].
Availability of data and materials
The datasets generated and analyzed in the current study are deposited in a publicly available repository at https://doi.org/10.15454/J4XOPD
References
Konieczna J, Sanchez J, van Schothorst EM, Torrens JM, Bunschoten A, Palou M, et al. Identification of early transcriptome-based biomarkers related to lipid metabolism in peripheral blood mononuclear cells of rats nutritionally programmed for improved metabolic health. Genes Nutr. 2014;9:1–15.
Díaz-Rúa R, Keijer J, Caimari A, van Schothorst EM, Palou A, Oliver P. Peripheral blood mononuclear cells as a source to detect markers of homeostatic alterations caused by the intake of diets with an unbalanced macronutrient composition. J Nutr Biochem. 2015;26:398–407.
Jégou M, Gondret F, Vincent A, Tréfeu C, Gilbert H, Louveau I. Whole blood transcriptomics is relevant to identify molecular changes in response to genetic selection for feed efficiency and nutritional status in the pig. PLoS One. 2016;11:e0146550.
Shen J, Zhou C, Zhu S, Shi W, Hu M, Fu X, et al. Comparative transcriptome analysis reveals early pregnancy-specific genes expressed in peripheral blood of pregnant sows. PLoS One. 2014;9:e114036.
Wojciechowicz B, Kołakowska J, Zglejc-Waszak K, Martyniak M, Kotwica G, Franczak A. The whole blood transcriptome at the time of maternal recognition of pregnancy in pigs reflects certain alterations in gene expression within the endometrium and the myometrium. Theriogenology. 2018;126:159–65.
Mach N, Gao Y, Lemonnier G, Lecardonnel J, Oswald I, Estellé J, et al. The peripheral blood transcriptome reflects variations in immunity traits in swine: towards the identification of biomarkers. BMC Genomics. 2013;14:894.
Liu H, Feye K, Nguyen Y, Rakhshandeh A, Loving C, Dekkers J, et al. Acute systemic inflammatory response to lipopolysaccharide stimulation in pigs divergently selected for residual feed intake. BMC Genomics. 2019;20:728.
Ghosh S, Dent R, Harper ME, Gorman S, Stuart J, McPherson R. Gene expression profiling in whole blood identifies distinct biological pathways associated with obesity. BMC Med Genet. 2010;3:56.
Te Pas M, Koopmans S, Kruijt L, Calus M, Smits M. Plasma proteome profiles associated with diet-induced metabolic syndrome and the early onset of metabolic syndrome in a pig model. PLoS One. 2013;8:e73087.
Ye MH, Bao H, Meng Y, Guan L, Stothard P, Plastow G. Comparative transcriptomic analysis of porcine peripheral blood reveals differentially expressed genes from the cytokine-cytokine receptor interaction pathway related to health status. Genome. 2017;60(12):1021–8.
Dou S, Villa-Vialaneix N, Liaubet L, Billon Y, Giorgi M, Gilbert H, et al. 1HNMR-based metabolomic profiling method to develop plasma biomarkers for sensitivity to chronic heat stress in growing pigs. PLoS One. 2017;12:e0188469.
Liu H, Nguyen YT, Nettleton D, Dekkers JC, Tuggle CK. Post-weaning blood transcriptomic differences between Yorkshire pigs divergently selected for residual feed intake. BMC Genomics. 2016;17:73.
Liew CC, Ma J, Tang HC, Zheng R, Dempsey A. The peripheral blood transcriptome dynamically reflects system wide biology: a potential diagnostic tool. J Lab Clin Med. 2006;147:126–32.
Nayeri S, Sargolzaei M, Tulpan D. A review of traditional and machine learning methods applied to animal breeding. Anim Health Res Rev. 2019;20:31–46.
Gilbert H, Billon Y, Brossard L, Justine F, Gatellier P, Gondret F, et al. Review. Divergent selection for residual feed intake in the growing pig. Animal. 2017;11:1–13.
Gondret F, Vincent A, Houée-Bigot M, Siegel A, Lagarrigue S, Causeur D, et al. A transcriptome multi-tissue analysis identifies biological pathways and genes associated with variations in feed efficiency of growing pigs. BMC Genomics. 2017;18:244.
Bunter K, Lewis C, Hermesch S, Smits R, Luxford B. Maternal Capacity, Feed Intake and Body Development in Sows. In: Proc. 9th World Cong. Genet. Appl. Livest. Prod: Leipzig, Germany; 2010.
Lindholm-Perry AK, Freetly HC, Oliver WT, Rempel LA, Keel BN. Genes associated with body weight gain and feed intake identified by meta-analysis of the mesenteric fat from crossbred beef steers. PLoS One. 2020;15:e022.
Campos P, Noblet J, Jaguelin-Peyraud Y, Gilbert H, Mormède P, Donzele RF, et al. Thermoregulatory responses during thermal acclimation in pigs divergently selected for residual feed intake. Int J Biometeorol. 2014;58:1545–57.
Gondret F, Louveau I, Mourot J, Duclos M, Lagarrigue S, Gilbert H, et al. Dietary energy sources affect the partition of body lipids and the hierarchy of energy metabolic pathways in growing pigs differing in feed efficiency. J Anim Sci. 2014;92:4865–77.
Gilbert H, Terenina E, Ruesche J, Gress L, Billon Y, Mormede P, et al. Responses of pigs divergently selected for cortisol level or feed efficiency to a challenge diet during growth. In: Proc. World Congress on Genetics Applied to Livestock Production; 2018. p. 11–9.
Lee HC, Yoon SB, Yang SM, Kim WH, Ryu HG, Jung CW, et al. Prediction of acute kidney injury after liver transplantation: machine learning approaches vs logistic regression model. J Clin Med. 2018;7:428.
Truong VH, Vu QV, Thai HT, Ha MH. A robust method for safety evaluation of steel trusses using gradient tree boosting algorithm. Adv Eng Softw. 2020;147:102825.
Vigors S, O'Doherty J, Bryan K, Sweeney T. A comparative analysis of the transcriptome profiles of liver and muscle tissue in pigs divergent for feed efficiency. BMC Genomics. 2019;20:461.
Chen W, Alexandre PA, Ribeiro G, Fukumasu H, Sun W, Reverter A, et al. Identification of predictor genes for feed efficiency in beef cattle by applying machine learning methods to multi-tissue transcriptome data. Front Genet. 2021;12:619857.
Messad F, Louveau I, Koffi B, Gilbert H, Gondret F. Investigation of muscle transcriptomes using gradient boosting machine learning identifies molecular predictors of feed efficiency in growing pigs. BMC Genomics. 2019;20:659.
Piles M, Fernandez-Lozano C, Velasco-Galilea M, González-Rodríguez O, Sanchez JP, Torrallardona D, et al. Machine learning applied to transcriptomic data to identify genes associated with feed efficiency in pigs. Genet Sel Evol. 2019;51:10.
Wen C, Yan W, Zheng J, Ji C, Zhang D, Sun C, et al. Feed efficiency measures and their relationships with production and meat quality traits in slower growing broilers. Poult Sci. 2018;97:2356–64.
Friedman JH. Greedy function approximation: a gradient boosting machine. Ann Stat. 2001;29:1189–232.
Li B, Zhang N, Wang Y-G, George AW, Reverter A, Li Y. Genomic prediction of breeding values using a subset of SNPs identified by three machine learning methods. Front Genet. 2018;9:237.
Horodyska J, Hamill R, Reyer H, Trakooljul N, Lawlor P, McCormack U, et al. RNA-seq of liver from pigs divergent in feed efficiency highlights shifts in macronutrient metabolism, hepatic growth and immune response. Front Genet. 2019;10:117.
Vigors S, O'Doherty J, Ryan MT, Sweeney T. An analysis of the basal colonic innate immune response of pigs divergent in feed efficiency and following an ex-vivo lipopolysaccharide challenge. Physiol Genomics. 2019;51:443–8.
Chen HH, Keyhanian K, Zhou X, Vilmundarson RO, Almontashiri NA, Cruz SA, et al. IRF2BP2 reduces macrophage inflammation and susceptibility to atherosclerosis. Circ Res. 2015;117:671–83.
Woo JS, Srikanth S, Nishi N, Ping P, Takeshima H, Gwack Y. Junctophilin-4, a component of the endoplasmic reticulum–plasma membrane junctions, regulates Ca2+ dynamics in T cells. Proc Natl Acad Sci U S A. 2016;113:2762–7.
Davidson D, Bakinowski M, Thomas ML, Horejsi V, Veillette A. Phosphorylation-dependent regulation of T-cell activation by PAG/Cbp, a lipid raft-associated transmembrane adaptor. Mol Cell Biol. 2003;23:2017–28.
Désert C, Merlot E, Zerjal T, Bed'Hom B, Härtle S, Cam A, et al. Transcriptomes of whole blood and PBMC in chickens. Comp Biochem Physiol Part D Genomics Proteomics. 2016;20:1–9.
Hu MM, Shu HB. Multifaceted roles of TRIM38 in innate immune and inflammatory responses. Cell Mol Immunol. 2017;14:331–8.
Patterson B, Outhouse A, Helm E, Dekkers J, Schwartz K, Gabler N, et al. Novel observations of peroxiredoxin-2 profile and protein oxidation in skeletal muscle from pigs that differ in residual feed intake and health status. Meat Muscle Biol. 2018;2:171.
Sierżant K, Perruchot MH, Merlot E, Le Floc’h N, Gondret F. Tissue-specific responses of antioxidant pathways to poor hygiene conditions in growing pigs divergently selected for feed efficiency. BMC Vet Res. 2019;15:341.
Jégou M, Gondret F, Lalande J, Tea I, Baeza E, Louveau I. NMR-based metabolomics highlights differences in plasma metabolites in pigs exhibiting diet-induced differences in adiposity. Eur J Nutr. 2015;55:1189–99.
Xiao C, Sun T, Yang Z, Xu W, Wang J, Zeng L, et al. Transcriptome landscapes of differentially expressed genes related to fat deposits in Nandan-Yao chicken. Funct Int Genomics. 2021;21:113–24.
Quinn LS, Anderson BG, Strait-Bodey L, Stroud AM, Argilés JM. Oversecretion of interleukin-15 from skeletal muscle reduces adiposity. Am J Physiol Endocrinol Metab. 2009;296:191–202.
Oka C, Tsujimoto R, Kajikawa M, Koshiba-Takeuchi K, Ina J, Yano M, et al. HtrA1 serine protease inhibits signaling mediated by Tgfbeta family proteins. Development. 2004;131:1041–53.
Jing L, Hou Y, Wu H, Yuanxin M, Li X, Cao J, et al. Transcriptome analysis of mRNA and miRNA in skeletal muscle indicates an important network for differential residual feed intake in pigs. Sci Report. 2015;5:11953.
Bissa B, Beedle AM, Govindarajan R. Lysosomal solute carrier transporters gain momentum in research. Clin Pharmacol Ther. 2016;100:431–6.
Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, et al. Landscape of transcription in human cells. Nature. 2012;489:101–8.
Fu L, Jiang Y, Wang C, Mei M, Zhou Z, Song H, et al. A genome-wide association study on feed efficiency related traits in landrace pigs. Front Genet. 2020;11:692.
Saintilan R, Brossard L, Vautier B, Sellier P, Bidanel J, van Milgen J, et al. Phenotypic and genetic relationships between growth and feed intake curves and feed efficiency and amino acid requirements in the growing pig. Animal. 2015;9:18–27.
Gilbert H, Bidanel JP, Gruand J, Caritez JC, Billon Y, Guillouet P, et al. Genetic parameters for residual feed intake in growing pigs, with emphasis on genetic relationships with carcass and meat quality traits. J Anim Sci. 2007;85:3182–8.
Breiman L. Machine Learning Springer Link, vol. 45; 2001. p. 5–32.
Friedman JH. Stochastic gradient boosting. Comput Stat Data Analysis. 2002;38:367–78.
Hastie T, Tibshirani R, Friedman JH. The elements of statistical learning data mining, inference, and prediction. 2nd ed. New York: Springer; 2009. p. 337–84.
Fernandez-Lozano C, Gestal M, Munteanu C, Dorado J, Pazos A. A methodology for the design of experiments in computational intelligence with multiple regression models. Peer J. 2016;4:e2721.
Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, et al. DAVID bioinformatics resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35(Web Server issue):W169–75.
Acknowledgements
The authors are grateful to Annie Vincent (PEGASE, INRAE) and to Yannick Lippi and Claire Naylies (Get-TRiX facility, Genotoul, Toulouse, France) who performed RNA extraction from blood and/or produced the original transcriptomic datasets. Thanks are also due to staff of UE3P (https://doi.org/10.15454/1.5573932732039927E12) and Genesi (https://doi.org/10.15454/1.5572415481185847E12) Experimental Units (France) for animal care and line selection procedure.
Funding
The Feed-a-Gene project has received funding from the European Union’s H2020 Programme under grant agreement no 633531. Farouk MESSAD was supported by a Regional grant (SAD, Brittany region) from France. Funders approved the aim of the study but had no roles in its design, data analysis, data interpretation, or in the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
FG: conceived the study; FM: implemented the analysis, performed the machine learning analyses and functional analysis, and wrote the initial draft; FG, IL, DR, HG: provided datasets; FG, FM: drafted the manuscript; FG, IL, DR, HG: discussed the data; All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was based on previous published studies. The original publications have included a statement on ethics approval to use animals into genetics and feeding experiments.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supp. Table S1
Iterative steps for model reduction to predict RFI class using different machine learning algorithms
Additional file 2: Supp. Table S2
List of probes identified as important to classify pigs in low or high RFI using random forest algorithm on transcripts levels of genes in the whole blood
Additional file 3: Supp. Fig. S1
Regression analysis of the relationship between observed and predicted FCR according to the dataset of origin Partition of pigs used in the test dataset for validating
Additional file 4: Supp. Fig. S2
Partition of molecular probes expressed in the whole blood between trained and validation datasets to analyze traits related to feed efficiency in pigs
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Messad, F., Louveau, I., Renaudeau, D. et al. Analysis of merged whole blood transcriptomic datasets to identify circulating molecular biomarkers of feed efficiency in growing pigs. BMC Genomics 22, 501 (2021). https://doi.org/10.1186/s12864-021-07843-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-07843-4