
Abstract
Background
Cryptocaryon irritans is an obligate parasitic ciliate protozoan that can infect various commercially important mariculture fish species and cause high lethality and economic loss. Current methods of controlling this parasite with chemicals or antibiotics are widely considered to be environmentally harmful. Piscidins with broad spectrum antibacterial, antifungal and antiviral activities were found to have potent activity against C. irritans. Little, however, has been understood about the killing mechanisms of piscidins in parasites.
Results
In total, 57.12, 50.44, 55.86 and 47.87 million raw reads were generated from untreated theront and trophont, and piscidin (Lc-pis) treated theront and trophont libraries, respectively. After de novo assembly, 966,609 unigenes were generated with an average length of 420 bp: among these, 618,629 unigenes showed identity with sequences in one or more databases, with some showing to be significantly manipulated by Lc-pis treatment. The species classification showed that more than 25.8% unigenes from trophonts were homologous to the large yellow croaker (Larimichthys crocea) and less than 3.8% unigenes from theronts were matched. The homologous unigenes demonstrated that the tissue from host could exist in trophonts and might be transported to parasite via vesicular transports. Our analysis showed that regulatory transcripts were involved in vesicular trafficking. Among transcripts induced by Lc-pis, most genes up-regulated in treated and untreated theronts were involved in cell migration and apoptosis related pathways. Few transcripts were found to be down-regulated in treated and untreated trophonts related to cell structure and migration after treatment.
Conclusions
This is the first transcriptome analysis of C. irritans exposed to Lc-pis, which enhanced the genomic resources and provided novel insights into molecular mechanisms of ciliates treated by cationic antimicrobial peptide. Our comprehensive transcriptome analysis can facilitate the identification of potential drug targets and vaccines candidates for controlling this devastating fish pathogen.
Similar content being viewed by others


Background
Cryptocaryon irritans, the pathogen of the marine white spot disease (cryptocaryonosis), is an obligate parasitic ciliate protozoan that undergoes four developmental stages, including the infective stage (theront), the feeding stage (trophont), the pre-reproductive stage (protomont) and the reproductive stage (tomont) [1, 2]. Cryptocaryonosis has had a significant negative impact on the mariculture industry worldwide, resulting in huge economic losses [2]. In southern China, C. irritans has afflicted mariculture fish species including groupers (e.g. Epinephelus spp.), sea breams (Sparidae) and large yellow croaker (Larimichthys crocea), which can result in up to 75% mortality [3]. The large yellow croaker has been widely cultured since the 1990s, mainly in floating cages; in terms of the estimated national marine fish culture production (volume) at species level the croaker is currently at the first rank [4]. However, this industry has suffered significant economic losses due to cryptocaryonosis [5]. Current methods of controlling this parasite with chemicals or antibiotics are widely considered to be environmentally harmful.
Antimicrobial polypeptides (AMPs) have been receiving considerable attention as effective and environmentally friendly commercial therapeutics against white spot disease. Piscidins, one of the most common AMPs families, exhibit potent broad spectrum antibacterial, antifungal and antiviral activities [6]. The biological activity of different piscidins isolated from fishes have been tested against gram-positive and gram-negative bacteria (prokaryotes) as well as protozoans (eukaryotes) [7, 8]. Previous studies showed that piscidins were also active against several fish ectoparasites in addition to C. irritans, including Amyloodinium ocellatum dinospore, Ichthyophthirius multifiliis and Trichodina spp. [9]. The killing mechanism of AMPs are known to bind lipopolysaccharide, cell wall components and DNA [10, 11]. Many studies focused on the bacterial phospholipid membrane as the main target of these peptides. The barrel-stave model as a possible mechanism of action of piscidins in bacteria has shown to be similar with that in fungi [12,13,14]. Only in cancer cells, peptides were reported to induce membrane destruction, intracellular calcium mechanism and apoptosis triggered by binding to the mitochondrial membrane [15, 16]. Few studies, however, attempted to undertake any molecular analysis about killing mechanism of piscidins in parasites.
According to our previous study, a gill-expressed piscidin identified in the large yellow croaker (Lc-pis) was lethal to C. irritans [17]. Killing the detached trophont or the infective theront can stop the reproductive cycle and prevent spread of this disease. RNA sequencing (RNA-Seq) is a powerful approach for whole-transcriptome analysis and can provide an extremely precise measurement in model and non-model organisms [18, 19]. It has been broadly applied in studies of the molecular mechanisms including stress resistance, models of development and the immune defenses [20, 21]. Transcriptomic analyses of C. irritans have been conducted on different developmental stages and transformations after low temperature treatments [22,23,24,25]. We analyzed transcriptome variances among different developmental stages and compared the changes in transcriptomes after treated by Lc-pis. Furthermore, we documented the evidence for the modulation of the C. irritans transcriptome when exposed to the antiparasitic Lc-pis and revealed different responses in different developmental stages of C. irritans.
Methods
Test peptide
Lc-pis (IWGLIAHGVGHVGRLIHGLIRG) with a purity of > 98% was synthesized (China Peptides Co. Ltd., Shanghai, China). After synthesis, reversed phase high-performance liquid chromatography (RP-HPLC) was used to purify Lc-pis. The chromatographic condition was: 0.1% trifluoroacetic acid (TFA) in water as Buffer 1 while 0.1% TFA in acetonitrile as Buffer 2, and the chromatographic condition was 15%–60% Buffer 2 on a Kromasil 100–5 C18 (4.6 mm × 250 mm, 5 μm) column at the flow rate of 0.2 mL/min for 15 min at 35 °C. Lc-pis was detected by its absorbance at 220 nm. After purification, an API-150EX mass spectrometer with the ion source of electrospray ionization was used to detect and identify the peptide. Sterile deionized water was used to dilute Lc-pis prior to C. irritans treatment.
Sample preparation and treatment
Forty juveniles of the large yellow croaker (weight 78.5 ± 9.1 g, total length 19.4 ± 0.9 cm) were obtained from a commercial fish farm at Gulf of Sandu, Fujian Province, China in October, 2015. Then the fish were infected with a non-lethal concentration of C. irritans (about 8000 theronts/fish) in 800 L of seawater. The collection method for C. irritans was described previously [26]. The water was oxygenated continuously and replaced every three days, and the salinity, water temperature and photoperiod were maintained at 23–25‰, 23 ± 1 °C and 12 L: 12D, respectively. The trophont were gently scraped from skin and gills 3 days after infection and washed with sterilized seawater more than three times to clear fish tissues. Within four days, mature trophonts were obtained using standard procedures [27]. Theronts incubated within 2 h in sterilized seawater were filtered (pore size 0.22 μm; GSWP04700, Millipore Corp.) and collected. In order to understand the genetic basis for differences after treated by Lc-pis, four cDNA libraries isolated from trophonts (control: ZYT-c and treatment: ZYT-t) and theronts (control: YC-c and treatment: YC-t) were established. The four treatment groups were divided. Theronts and trophonts were treated with Lc-pis at a final concentration of 1.5 μM for 90 min in order to maintain the RNA quality of C. irritans population. Theronts and trophonts without Lc-pis treatment were used as controls. Each sample was performed in triplicate. Cryptocaryon irritans were immediately cryopreserved in liquid nitrogen until RNA isolation. Animal treatment in this study was performed in strict accordance with the recommendations of Animal Care Quality Assurance in China.
Library construction and Illumina sequencing
Total RNA was extracted using MiniBEST Universal RNA Extraction Kit (TaKaRa 9767) according to the manufacturer’s instructions. Triplicate RNA were merged into one sample. RNA degradation and contamination was monitored on 1% agarose gels. RNA concentration and integrity were measured using a Qubit RNA Assay Kit in Qubit2.0 Flurometer (Life Technologies, CA, USA) and RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). The four mRNA-seq libraries were performed at Novogene Co., Ltd. (Beijing, China) with a NEBNext Ultra RNA Library Prep Kit for Illumina (New England Biolabs, USA) following the manufacturer’s recommendations. mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. First strand cDNA was synthesized using random hexamer primers and M-MLV Reverse Transcriptase (RNaseH-). Second strand cDNA synthesis was subsequently performed using DNA Polymerase I and RNase H. DNA fragments were treated for end-repairing, adenylation of 3′ ends and ligation of adaptors. The library fragments were purified with AMPure XP system (Beckman Coulter, CA, USA) to preferentially select cDNA fragments of 150~ 200 bp in length, and suitable fragments were enriched by PCR amplification.
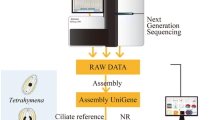
Assembly of sequencing and gene annotation
The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumia) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina Hiseq platform and 2 × 125 bp paired-end reads were generated. Raw sequences were deposited to NCBI Short Read Archive (SRA) database (http://www.ncbi.nlm.nih.gov/Traces/sra/). After removing adaptor sequences, ambiguous ‘N’ nucleotides (with the ratio of ‘N’ to be more than 10%) and low quality sequences (with quality score to be less than 5), the remaining clean reads were assembled using Trinity for transcriptome assembly without reference genome [19]. The longest transcript of each single gene was selected as a unigene. For annotation analysis, unigenes were BLASTX-searched against seven databases, including the National Center for Biotechnology Information (NCBI) non-redundant protein sequence (Nr) database, non-redundant nucleotide sequence (Nt) database, Protein family (Pfam), Clusters of Orthologous Groups (KOG/COG), Gene ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology (KO) database, and the Swiss-Prot, using a cut-off E-value of 10–5. Differentially expressed genes (DEGs) between untreated therents and trophonts were identified with DEGseq analysis on adjusted read count data. To identify unigenes involved in C. irritans responsing to antiparasitic peptide treatment, pairwise comparisons for differential expression analysis were conducted among theront treatment and its control group, and trophont treatment and its control group. Unigenes were annotated based on BLASTX results, and the best alignments were used for downstream analyses. KEGG database were used to predict the functions of unigenes [28, 29].
Experimental validation of transcription levels
RNAs from four treatment groups (theronts and trophonts treated by Lc-pis; theronts and trophonts without Lc-pis treatment) were extracted with method mentioned above. Replicate samples were run for real time quantitative-PCR (RT-qPCR) validation of Illumina sequencing. First-strand cDNA was synthesized using the PrimeScript RT reagent Kit with gDNA Eraser (TaKaRa DRR034A) according to the manufacturer’s protocol. Specific primer for five genes (heat shock protein 90, GTP-binding protein, serine/threonine kinase, serum/glucocorticoid regulated kinase, Rab 5) were designed (Additional file 1). PCR products were purified and cloned using pMD™19-T Vector (TaKaRa, Japan). The plasmids carrying target genes were extracted and purified using AxyPrep Plasmid DNA Miniprep Kit (Axygen, America). Calibration curve was generated for qPCR using tenfold serial dilution of the plasmid. Quantitative PCR was performed using ABI Prism 7500 RT PCR machine with SYBR Premix DimerEraser (TaKaRa RR091A). Thermo cycling condition was 95 °C for 30 s, followed by 40 cycles at 95 °C for 3 s, 60 °C for 30 s and 72 °C for 30 s. For direct comparison with the results in transcriptome, qPCR results were converted to log2-fold changes. A Pearson’s correlation test was calculated using GraphPad Prism 5.0.
Scanning electron microscopy (SEM)
Sample preparation for SEM was conducted according to previous methods [30]. Collected samples were added to 2.5% glutaraldehyde at 4 °C for 24 h. Theronts were fixed in a 6:1 mixture of saturated HgCl2 and 1% OSO4 at 4 °C for 10 min. All solutions were diluted in sodium cacodylate buffer (pH 7.2). After washing, samples were dehydrated in a graded ethanol series, critical-point dried, placed on aluminium stubs, and sputter coated with platinum. Prepared samples were observed with a Hitachi S-4800 SEM.
Data availability
The sequence data of the present study have been deposited into SRA (the accession numbers: SRX2417025, SRX2417144, SRX2417145, and SRX2417163).
Results
Effect of Lc-pis on C. irritans
Given our goal of investigating the ways C. irritans are effected, it is important to explore both structures and genes regulated by Lc-pis. The light microscopy results showed that Lc-pis quickly induced the membrane and cytoplasmic leakage rupture of theront and trophont ruptur in cells (data not shown). Under SEM, theronts lost part of cilium and changed their shapes from pyriform to round. The uneven surface of treated trophonts could be ascribed to specific or non-specific interaction of Lc-pis with the membrane (Fig. 1). Only few parts of trophonts post stimulus had cytoplasmic leakage.

Images of microscopic structure of the theront and trophont under scanning electron microscopy (SEM). Surface of the theronts for control group (a) and for 1.5 μM Lc-pis treated (b), arrow indicates the region without cilium; the trophonts for control group (c) and for Lc-pis treated (d), arrows mark the uneven surface
Transcriptome sequencing and assembly
In total, 57.12, 50.44, 55.86 and 47.87 million raw reads were generated from untreated theront and trophont, and Lc-pis treated theront and trophont libraries, respectively (Table 1). A reference transcriptome was obtained from all raw reads from the four cDNA libraries. Clean reads were acquired with a Q20 percentage of over 96.48%, which were assembled into 966,609 transcripts with a mean length of 420 bp and an N50 length of 463 bp (Table 2). An overview of the sequencing results was summarized in Additional file 2.
Homology analysis and gene functional annotation
In total, 884,341 non-redundant unigenes were found, among these, 214,488 matched to known proteins in the Nr database, 416,413 matched to putative homologues in the Nt database and only 33,287 unigenes matched to KO database. In total, 618,629 unigenes (69.95%) showed identity with sequences in one or more databases (Additional file 3). The e-value distribution showed that 24.4% of the annotated unigenes had strong homology (e-value <1e-45), whereas 33.8% of the unigenes had low homology (1e-15 < e-value<1e-5). The species classification showed that more than 25.8% unigenes of trophonts were homologous to the large yellow croaker but less than 3.8% unigenes of theronts were matched. Sequence homologues for over 68% unigenes of the predicted peptides of theronts and about 42% of trophonts were found to be associated with others including bacteria. Unigenes associated with many kinds of protozoa showed that the unigenes reveal high conservation between protozoa. Homology analysis is shown in pie charts in Fig. 2.

Species distribution of gene annotations in transcriptional profile. BLASTx top-hit species distribution of gene annotations in transcriptional profile of theronts (a), treated theronts (b), trophonts (c) and treated trophonts (d) of Cryptocaryon irritans
In total, 201,906 DEGs were classified into three major functional categories (biological process, cellular component and molecular function) and 55 subcategories (Fig. 3). Additional file 4 represents GO enrichment analysis of DEGs from above three groups [31]. With the KOG classification, 128,322 matched unigenes were clustered into 26 categories. Among these categories, the largest group was group O-posttranslational modification, protein turnover and chaperones (15,942; 12.42%), followed by group T-signal transduction mechanisms (15,763; 12.28%), while only 8 unigenes were classified as unnamed protein (Fig. 3).

GO annotation and histogram presenting clusters of orthologous groups (KOG) classifications. Of 884,341 unigenes, 114,478 sequences were assigned to 26 KOG classifications. The GO results were summarized in three main GO categories: biological process, cellular component and molecular function. The x-axis indicates the subcategories, and the y-axis indicates the numbers related to the total number of GO terms present
Hierarchical clustering of DEGs among transcriptomes
Expression levels of treatment and control groups were divided into 18 categories based on K-means clustering. Detailed expression profile clusters were shown in Additional file 5. The largest subcluster 17 contained 3589 DEGs with decreasing expression levels from the theront to trophont; the same trend was also observed in subclusters 2 (2664 DEGs), 8 (1516 DEGs), 9 (2884 DEGs), 11 (759 DEGs), 12 (1022 DEGs) and 18 (559 DEGs). The expression patterns in most subclusters (e.g. 1 (1506 DEGs), 3 (508 DEGs), 4 (900 DEGs) and 5 (467 DEGs)) showed that DEGs regulated positively in theronts after Lc-pis treatment while DEGs were down-regulated in treated trophonts (Fig. 4).

The top six clusters determined by K-means clustering, including 15,274 DEGs (68.8% of all genes). The x-axis indicates different group. The y-axis indicates the log2 (ratio) of gene expression. Each grey row represents the relative expression of DEGs in a cluster. The blue line represents the average value of all members. The red line denotes reference, the line above the red line represents up-regulation, and the line below the red line represents down-regulation. The number of DEGs within a cluster is shown after that subcluster
Stage-specific gene expression of theronts and trophonts
Functions of the 8751 transcripts were assigned to GO classes with 4626 functional terms. The profile between YC-c and ZYT-c is shown in Additional file 6. Of 17,373 unigenes that were differentially expressed, 8575 were up-regulated and 8798 down-regulated. The pathways with most genes were the ribosome (ko 03010), vibrio cholerae infection (ko 05110), prion diseases (ko 05020), pathogenic Escherichia coli infection (ko 05130), cocaine addiction (ko 05030) and phagosome (ko 04145) (Table 3). Among them, the phagosome pathway contained 83 DEGs, including actin-associated protein coronin, dynein, Rab5 and major histocompatibility complex class I (MHC I). Genes such as Rab11b, Rab7a, dynamin mapping to endocytosis pathway (ko 04144) and extracellular signal-regulated kinase1/2 (ERK 1/2), cofilin, gelsolin and actin related proteins 2/3(Arps 2/3) mapping to Fc gamma receptor-mediated phagocytosis pathway (ko 04666) were also related to vesicular trafficking.
Comparative gene expression of response to Lc-pis treatment
The GO profiles showed that 8751 transcripts with significantly differing transcript levels occurred in the theront group but not found in the trophont group. The unigenes were mapped to the reference pathways recorded in the KEGG database. In the theront group, the ribosome related pathways were also the most enriched when compared to the stage of trophonts (Additional file 7). Transcripts such as tenascin and histones were highly regulated in group treated with piscidins. Beyond that, a number of DEGs were involved in different pathways causing cell death (i.e. apoptosis, antigen processing and presentation, lysosome, mammalian target of rapamycin and phosphatidylinositide 3-kinase signaling pathway) after being treated with Lc-pis.
Only 283 transcripts with significantly differing transcript levels were found in the trophont group; among these, 215 DEGs were down-regulated. DEGs in the trophont group were classified into protein polymerization (GO:0051258), generation of precursor metabolites and energy (GO:0006091), chemokine activity (GO:0008009), chemokine receptor binding (GO:0042379), receptor binding (GO:0005102), actin monomer binding (GO:0003785). All function terms above were involved in the theront group (Additional file 7); however, only few DEGs, such as alpha-tubulin, beta tubulin and cytochrome c oxidase subunit were involved in the trophont group and transcripts with significant alteration were of low amplitude. Other terms including leukocyte transendothelial migration, tight junction, phagosome, regulation of actin cytoskeleton, focal adhesion, adherens junction and gap junction were associated with membrane or cytoskeleton structure, while others were associated with stimulus induced or apoptosis related pathways (Fig. 5).

KEGG pathway enrichment analysis of the differently expressed genes between the theront group (a) and trophont group (b)
Corroboration of illumina sequencing approach by RT qPCR
Expression values determined by the RT-qPCR analysis of five genes were found to be comparable to the RNA-seq approach (Additional file 8). The values obtained by RT qPCR and RNA-seq were highly similar and correlated with statistical significance (r = 0.523, p = 0.060; Additional file 8).
Discussion
Large amounts of antibiotics and chemicals are used to prevent pathogens in fish farms, which can induce the antibiotic resistance and damage the surrounding aquatic environment [32]. New approaches to combat infectious diseases need to be considered. AMPs, known as nature’s defenders, have been identified in almost all living organisms. [6, 33]. One group of AMPs is the piscidin family, which has recently been found to have potent antiparasitic activities [9, 34,35,36]. In our previous study, Lc-pis was proved to have strong antiparasitic activities against the parasite C. irritans [17], which affects many teleosts and was responsible for significant economic losses to the aquaculture industry [1, 37]. The minimum lethal oxygen concentration of Lc-pis for theronts is 1.5 μM after 90 min (less than 60 min for 2 μM and non-efficent for 1 μM) while tomont, with a thick cyst wall, were non-susceptible at such low concentration (data not show). Here, we explored, for the first time, the cytoskeleton structure of C. irritans after Lc-pis treatment and identified putative mechanisms using transcriptome data.
The majority of unigenes were shorter than 500 bp, and this phenomenon existed in some nonmodel organisms including the transcriptomes of C. irritans from tomont stage [22, 38, 39]. These short unigenes might come from assembly errors, fragmented transcripts of low expressed genes, as well as noncoding RNA. Of the short unigenes, 69.95% were annotated in at least one database. Part of the annotated unigenes were matched to the large yellow croaker in the trophont group, while few fish genes were found in the theront group. This indicates that the trophonts were fed with host cells, which seemed to proceed throughout the trophont stage. Cryptocaryon irritans cannot be cultured without host, and contamination by host RNA cannot be completely eliminated when preparing samples. This was also observed in C. irritans and Ichthyophthirius multifiliis [20, 23]. In addition, previous researches showed parasite genes have revealed high degrees of identity between the nucleotide sequences of parasite and mammalian genes [40, 41]. These genes might help parasites escape host immunosurveillance. Other unigenes revealed high conservation between protozoa which could be conjectured to participate in similar mechanisms in protozoa like basic cellular mechanisms.
As C. irritans progresses through its lifecycle, it must adapt to different environments with differing energy requirements and this is reflected in differential gene expressions. A previous transcriptional analysis of C. irritans demonstrated that metabolic enzymes were highly expressed in the trophont stage [23]. Cytoskeletal proteins such as actin and tubulins were discovered to be persistent throughout the whole life cycle of C. irritans in an immunoproteomics analysis, and studies in mammals indicated they could result to the interplay of signaling pathways and cytoskeletal dynamics [24, 42]. Rab family proteins and dynein were both proved to take part in regulating exocytosis and endocytosis [43]. As many protozoa are incapable of synthesizing purines de novo, enzymes involved in purine salvage are of particular importance [44,45,46]. ATP synthases highly expressing in the trophont stage were observed in our study. For parasites, the infection stage is the only stage to get nutrition from their hosts. Recently, the ingestion of host cells, occurred in the trophont and the early tomont stages, was investigated by using in situ hybridization [47]. The present analysis of theronts and tomonts also demonstrated that metabolic related and cytoskeleton associated genes were enriched especially in vesicular transport pathways. Thus, it was reasonable to hypothesize that phagosome functions occurred prior to the trophont stage and were shed by host tissues and the typical genes demonstrated the mechanism of phagosome formation of endoparasitic ciliates might similar to other eukaryotes.
In the present study, the comparative transcriptome profiling allowed us to clarify the mechanism by which ways the Lc-pis effect on C. irritans and provide more potential targets for vaccine development against C. irritans. Piscidins have potent antimicrobial activity against a broad spectrum of pathogens in vitro. While knowledge of AMPs mechanisms of action remained unclear, and debate continues as to the relative contributions of proposed pore formation or internal killing strategies [48]. Several studies revealed that amphipathic character seemed to play an essential role in the various mechanisms of membrane permeabilization [48,49,50]. Piscidins were found to interact with various membrane-mimicking interfaces, suggesting that its interaction with them was not only electrostatically mediated [12].
In the theront group, the ribosome pathway was the first of the most DEGs-enriched pathways. This result was similar to the result of the previous study on C. irritans transcriptome [22, 25]. Several cell death pathways were present after stimulation and significant alteration transcripts were found. For example, histones, especially H2A, were associated with pathogenesis in some protozoan parasites [51, 52]. The transcripts annotated for histones (e.g. H2A, H2B, H3 and H4) in present study displayed altered transcription profiles in the theront group treated by Lc-pis or without treatment, which might participate in infection of C. irritans. The tenascin family is a large extracellular matrix glycoprotein which transduces and integrates intracellular signals via identifying cell surface receptors. The tenascin-C promoted cell migration upon injury in mice and contributed to migration of fibroblast [53, 54]. However, tenascin was rarely studied in parasites, the roles in C. irritans exposed to Lc-pis remains to be demonstrated. Protein kinases are found to play a central role in the cellular signaling pathways and was involved in biological processes as diverse as gene expression, metabolism, apoptosis, and cellular proliferation through phosphorylation of substrate proteins [55]. The schistosome cAMP-dependent protein kinase (PKA) was suggested to be essential for maintaining parasite viability in Schistosoma mansoni and was proved to affect flagellar wave in Leishmania [56, 57]. A putative PKA was up regulated after stimulation when theronts were observed moving slower than the control group and a large scale of cilia were shed, which indicated that PKA also may take part in the movement of theronts. Further work is required to discover exact killing mechanisms by Lc-pis.
Expression patterns were different between treated trophont and treated theront, observed in K-means analysis. In the present study, DEGs were involved mainly in cell structure and migration after stimulation. Ultrastructural observation of the trophont showed mucus and fibrous materials mixture could be observed surrounding C. irritans in vivo [58]. Two tubulin genes, α tubulin and β tubulin (their nucleotide sequences shared over 81% similarity with Dugesia japonica and Lottia gigantean, respectively), were identified with significant down-regulation. Tubulin is a fundamental constituent of kinetoplastid cytoskeletons, cell division machinery and motile organelle [59]. Studies about tubulin in protozoa suggested that the expression of tubulins varied during the life cycles of Trypanosoma and Leishmania spp., possibly as the relative demand for these structures fluctuates [60, 61]. The suppression of genes by Lc-pis indicates that this peptide inhibits the trophont from developing into protomont. Until now, research into the response of C. irritans to antiparasitics was insufficient. Above all, we noticed that the pore formation might not be the only reason which led to cell death at such a low concentration and the transcriptome analysis after exposed to Lc-pis helped to better understand the killing mechanisms.
Conclusions
Cryptocaryon irritans is an obligate ectoparasitic ciliate pathogen of marine fishes that has caused heavy economic losses in the aquaculture sector. Here, we generated transcriptome sequencings of theronts and trophonts exposed to Lc-pis using a next generation sequencing technology, conducted comprehensive analysis of the stimuli-responsive genes and explored the putative mechanisms of Lc-pis resistance for the first time. The results enlarged the genomic resources of C. irritans and the different developmental stage specific transcripts illustrated that vesicular transports were continuously generated throughout the trophont stage. The differences in gene expression pattern between theronts and trophonts might be due to the pellicular swells underneath the pellicle of trophonts. Our transcriptome analyses provide valuable information for further investigation into the molecular mechanisms of ciliates treated by cationic antimicrobial peptides and facilitate the identification of potential drug targets and vaccines candidates for the control of this devastating fish pathogen.
Abbreviations
- AMPs:
-
Antimicrobial polypeptides
- DEGs:
-
Differentially expressed genes
- HPLC:
-
High-performance liquid chromatography
- PKA:
-
cAMP-dependent protein kinase
- SEM:
-
Scanning electron microscopy
- TFA:
-
Trifluoroacetic acid
References
Colorni A, Burgess P. Cryptocaryon irritans Brown 1951, the cause of ‘white spot disease’in marine fish: an update. Aquar Sci Conserv. 1997;1(4):217–38.
Dickerson HW, Dawe DL. Ichthyophthirius multifiliis and Cryptocaryon irritans (phylum Ciliophora). Fish Diseases and Disorders. 2006;1:116–53.
Dan XM. The propagation, preservation of Cryptocaryon irritans and control of cryptocaryonosis [D]: Jinan University; 2006.
Bo F. China fishery statistical yearbook. Beijing: Agriculture Press; 2017.
Wang C, Wang Y. Disease prevention and control of Cryptocaryon irritans in large yellow croaker Pseudosciaena crocea. Fish Science Technology Inforation. 2002;29(60):e62.
Silphaduang U, Noga EJ. Antimicrobials: peptide antibiotics in mast cells of fish. Nature. 2001;414(6861):268–9.
Huang HW. Action of antimicrobial peptides: two-state model. Biochemistry. 2000;39(29):8347–52.
Salger SA, Cassady KR, Reading BJ, Noga EJ. A diverse family of host-defense peptides (piscidins) exhibit specialized anti-bacterial and anti-protozoal activities in fishes. PLoS One. 2016;11(8):e0159423.
Colorni A, Ullal A, Heinisch G, Noga E. Activity of the antimicrobial polypeptide piscidin 2 against fish ectoparasites. J Fish Dis. 2008;31(6):423–32.
Papo N, Shai Y. Can we predict biological activity of antimicrobial peptides from their interactions with model phospholipid membranes? Peptides. 2003;24(11):1693–703.
Matsuzaki K, Sugishita K-i, Miyajima K. Interactions of an antimicrobial peptide, magainin 2, with lipopolysaccharide-containing liposomes as a model for outer membranes of gram-negative bacteria. FEBS Lett. 1999;449(2–3):221–4.
Campagna S, Saint N, Molle G, Aumelas A. Structure and mechanism of action of the antimicrobial peptide piscidin. Biochemistry. 2007;46(7):1771–8.
Sung WS, Lee J, Lee DG. Fungicidal effect and the mode of action of piscidin 2 derived from hybrid striped bass. Biochem Biophys Res Commun. 2008;371(3):551–5.
Sung WS, Lee J, Lee DG. Fungicidal effect of piscidin on Candida albicans: pore formation in lipid vesicles and activity in fungal membranes. Biol Pharm Bull. 2008;31(10):1906–10.
Papo N, Seger D, Makovitzki A, Kalchenko V, Eshhar Z, Degani H, Shai Y. Inhibition of tumor growth and elimination of multiple metastases in human prostate and breast xenografts by systemic inoculation of a host defense-like lytic peptide. Cancer Res. 2006;66(10):5371–8.
Wang C, Tian LL, Li S, Li HB, Zhou Y, Wang H, Yang QZ, Ma LJ, Shang DJ. Rapid cytotoxicity of antimicrobial peptide tempoprin-1CEa in breast cancer cells through membrane destruction and intracellular calcium mechanism. PLoS One. 2013;8(4):e60462.
Niu SF, Jin Y, Xu X, Qiao Y, Wu Y, Mao Y, Su YQ, Wang J. Characterization of a novel piscidin-like antimicrobial peptide from Pseudosciaena crocea and its immune response to Cryptocaryon irritans. Fish and Shellfish Immunology. 2013;35(2):513–24.
Costa V, Angelini C, De Feis I, Ciccodicola A. Uncovering the complexity of transcriptomes with RNA-Seq. Biomed Res Int. 2010;2010
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–52.
Abernathy J, Xu DH, Peatman E, Kucuktas H, Klesius P, Liu ZJ. Gene expression profiling of a fish parasite Ichthyophthirius multifiliis: insights into development and senescence-associated avirulence. Comp Biochem Physiol Part D: Genomics and Proteomics. 2011;6(4):382–92.
Ehrenkaufer GM, Weedall GD, Williams D, Lorenzi HA, Caler E, Hall N, Singh U. The genome and transcriptome of the enteric parasite Entamoeba invadens, a model for encystation. Genome Biol. 2013;14(7):R77.
Yin F, Sun P, Wang J, Gao Q. Transcriptome analysis of dormant tomonts of the marine fish ectoparasitic ciliate Cryptocaryon irritans under low temperature. Parasites and Vectors. 2016;9(1):280.
Mo ZQ, Li YW, Wang HQ, Wang JL, Ni LY, Yang M, Lao GF, Luo XC, Li AX, Dan XM. Comparative transcriptional profile of the fish parasite Cryptocaryon irritans. Parasites and Vectors. 2016;9(1):630.
Mai YZ, Li YW, Li RJ, Li W, Huang XZ, Mo ZQ, Li AX. Proteomic analysis of differentially expressed proteins in the marine fish parasitic ciliate Cryptocaryon irritans. Vet Parasitol. 2015;211:1):1–11.
Lokanathan Y, Mohd-Adnan A, Wan KL, Nathan S. Transcriptome analysis of the Cryptocaryon irritans tomont stage identifies potential genes for the detection and control of cryptocaryonosis. BMC Genomics. 2010;11(1):76.
Chen RN, Su YQ, Wang J, Liu M, Qiao Y, Mao Y, Ke QZ, Han KH, Zheng WQ, Zhang JS. Molecular characterization and expression analysis of interferon-gamma in the large yellow croaker Larimichthys crocea. Fish and Shellfish Immunol. 2015;46(2):596–602.
Dan X, Li A, Lin X, Teng N, Zhu X. A standardized method to propagate Cryptocaryon irritans on a susceptible host pompano Trachinotus ovatus. Aquaculture. 2006;258(1):127–33.
Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999;27(1):29–34.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Gu F, Ni B: The exploration of preparing protozoan specimen for scanning electron microscopy. J Chin Elec Soc 1993, 12:525–529. (In chinese).
Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talón M, Dopazo J, Conesa A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36(10):3420–35.
Smith P, Hiney MP, Samuelsen OB. Bacterial resistance to antimicrobial agents used in fish farming: a critical evaluation of method and meaning. Annu Rev Fish Dis. 1994;4:273–313.
Magnadóttir B. Innate immunity of fish (overview). Fish and Shellfish Immunol. 2006;20(2):137–51.
Ruangsri J, Salger SA, Caipang CM, Kiron V, Fernandes JM. Differential expression and biological activity of two piscidin paralogues and a novel splice variant in Atlantic cod (Gadus morhua L.). Fish and Shellfish Immunol. 2012;32(3):396–406.
Park N, Silphaduang U, Moon H, Seo J, Corrales J, Noga E. Structure-activity relationships of piscidin 4, a piscine antimicrobial peptide. Biochemistry. 2011;50(16):3288–99.
Peng KC, Lee SH, Hour AL, Pan CY, Lee LH, Chen JY. Five different piscidins from Nile tilapia, Oreochromis niloticus: analysis of their expressions and biological functions. PLoS One. 2012;7(11):e50263.
Cheung P, Nigrelli R, Ruggieri G. Studies on cryptocaryoniasis in marine fish: effect of temperature and salinity on the reproductive cycle of Cryptocaryon irritans Brown, 1951. J Fish Dis. 1979;2(2):93–7.
Xiang LX, He D, Dong WR, Zhang YW, Shao JZ. Deep sequencing-based transcriptome profiling analysis of bacteria-challenged Lateolabrax japonicus reveals insight into the immune-relevant genes in marine fish. BMC Genomics. 2010;11(1):472.
Zhou Z, Dong Y, Sun H, Yang A, Chen Z, Gao S, Jiang J, Guan X, Jiang B, Wang B. Transcriptome sequencing of sea cucumber (Apostichopus japonicus) and the identification of gene-associated markers. Mol Ecol Resour. 2014;14(1):127–38.
Shoemaker CB, Ramachandran H, Landa A, dos Reis MG, Stein LD. Alternative splicing of the Schistosoma mansoni gene encoding a homologue of epidermal growth factor receptor. Mol Biochem Parasitol. 1992;53(1–2):17–32.
Salzet M, Capron A, Stefano G. Molecular crosstalk in host–parasite relationships: Schistosome- and leech-host interactions. Parasitol Today. 2000;16(12):536–40.
Schappi JM, Krbanjevic A, Rasenick MM. Tubulin, actin and heterotrimeric G proteins: coordination of signaling and structure. Biochimica et Biophysica Acta (BBA)-Biomembranes. 2014;1838(2):674–81.
Jordens I, Marsman M, Kuijl C, Neefjes J. Rab proteins, connecting transport and vesicle fusion. Traffic. 2005;6(12):1070–7.
Abernathy JW, Xu P, Li P, Xu DH, Kucuktas H, Klesius P, Arias C, Liu Z. Generation and analysis of expressed sequence tags from the ciliate protozoan parasite Ichthyophthirius multifiliis. BMC Genomics. 2007;8(1):176.
Sullivan WJ, Dixon SE, Li C, Striepen B, Queener SF. IMP dehydrogenase from the protozoan parasite Toxoplasma gondii. Antimicrob Agents Chemother. 2005;49(6):2172–9.
Yao JY, Xu Y, Yuan XM, Yin WL, Yang GL, Lin LY, Pan XY, Wang CF, Shen JY. Proteomic analysis of differentially expressed proteins in the two developmental stages of Ichthyophthirius multifiliis. Parasitol Res. 2017;116(2):637–46.
Watanabe Y, Nishida S, Zenke K, Hui HK, Itoh N, Yoshinaga T. Development of the macronucleus of Cryptocaryon irritans, a parasitic ciliate of marine teleosts, and its ingestion and digestion of host cells. Fish Pathol. 2016;51(3):112–20.
Brogden KA. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol. 2005;3(3):238–50.
Hwang PM, Vogel HJ. Structure-function relationships of antimicrobial peptides. Biochem Cell Biol. 1998;76(2–3):235–46.
Shai Y. Mode of action of membrane active antimicrobial peptides. Pept Sci. 2002;66(4):236–48.
Lowell JE, Kaiser F, Janzen CJ, Cross GA. Histone H2AZ dimerizes with a novel variant H2B and is enriched at repetitive DNA in Trypanosoma brucei. J Cell Sci. 2005;118(24):5721–30.
Sullivan WJ, Naguleswaran A, Angel SO. Histones and histone modifications in protozoan parasites. Cell Microbiol. 2006;8(12):1850–61.
Jones FS, Jones PL. The tenascin family of ECM glycoproteins: structure, function, and regulation during embryonic development and tissue remodeling. Dev Dyn. 2000;218(2):235–59.
Trebaul A, Chan E, Midwood K. Regulation of fibroblast migration by tenascin-C. Portland Press Limited. 2007:695–7.
Hanks SK. Genomic analysis of the eukaryotic protein kinase superfamily: a perspective. Genome Biol. 2003;4(5):111.
Swierczewski BE, Davies SJ. A schistosome cAMP-dependent protein kinase catalytic subunit is essential for parasite viability. PLoS Negl Trop Dis. 2009;3(8):e505.
Biswas A, Bhattacharya A, Das PK. Role of cAMP signaling in the survival and infectivity of the protozoan parasite, Leishmania donovani. Mol Biol Int. 2011;2011
Ma R, Fan XP, Yin F, Ni B, Gu FK. Ultrastructural features of the tomont of Cryptocaryon irritans (Ciliophora: Prostomatea), a parasitic ciliate of marine fishes. Parasitology. 2017;144(6):720–9.
Gull K. Protist tubulins: new arrivals, evolutionary relationships and insights to cytoskeletal function. Curr Opin Microbiol. 2001;4(4):427–32.
Bartholomeu DC, Silva RA, Galvão LM, El-Sayed NM, Donelson JE, Teixeira SM. Trypanosoma cruzi: RNA structure and post-transcriptional control of tubulin gene expression. Exp Parasitol. 2002;102(3):123–33.
Coulson RM, Connor V, Chen JC, Ajioka JW. Differential expression of Leishmania major β-tubulin genes during the acquisition of promastigote infectivity. Mol Biochem Parasitol. 1996;82(2):227–36.
Funding
This study was financially supported by National Natural Science Foundation of China (Grant Nos. 31372504 and 41476118), Major Projects of Science and Technology Department of Fujian Province (Grant No. 2016NZ0001–4), and the Fujian Science and Technology Department (Grant Nos. 2015I11010045 and 2015 N2002–3). Funding bodies had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
Raw Illumina sequences were deposited in the National Center for Biotechnology Information (NCBI) and can be accessed in the Short Read Archive (SRA) database (http://trace.ncbi.nlm.nih.gov/Traces/sra/) under accession numbers: SRX2417025, SRX2417144, SRX2417145, and SRX2417163.
Author information
Authors and Affiliations
Contributions
RC, YS, YM and JW conceived and designed the experiments; QK and WZ participated in sample collection; RC, YQ and LZ performed the experiments; RC analyzed the data; RC, YS and ML wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Primer sets used in RT qPCR validation. (DOCX 14 kb)
Additional file 2:
The length distribution of the transcripts (red) and unigenes (blue). (JPEG 447 kb)
Additional file 3:
BLAST analysis of non-redundant unigenes against public databases. (XLSX 10 kb)
Additional file 4:
Summary of the GO classifications of assembled unigenes. (XLSX 12 kb)
Additional file 5:
Eighteen regulatory patterns determined by K-means clustering of all the DEGs expressed in theront and trophont groups of Cryptocaryon irritans with Lc-pis treatment. (JPEG 929 kb)
Additional file 6:
GO enrichment bar graphs of DEGs between untreated theronts and untreated trophonts of Cryptocaryon irritans. (JPEG 504 kb)
Additional file 7:
GO classification of the DEGs. A: YC-t vs YC-c, B: ZYT-t vs ZYT-c. (JPEG 641 kb)
Additional file 8:
Expression profiles of five genes from four developmental stages of Cryptocaryon irritans from RNA-Seq and RT-qPCR. Log2-fold changes from RNA-Seq analysis were highly correlated with log2-fold change values from RT-qPCR. A, B, C, D and E represented heat shock protein 90, GTP-binding protein, serine/threonine kinase, serum/glucocorticoid regulated kinase, Rab 5, respectively. Log2-fold changes are relative to YC c (A, B, C, D and E for ZYT-c/ YC-c; A1, B1, C1, D1 and E1 for vs YC-t/ YC-c). (JPEG 673 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Chen, R., Mao, Y., Wang, J. et al. Molecular mechanisms of an antimicrobial peptide piscidin (Lc-pis) in a parasitic protozoan, Cryptocaryon irritans. BMC Genomics 19, 192 (2018). https://doi.org/10.1186/s12864-018-4565-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-4565-5