
Abstract
Background
Salvia diterpenes have been found to have health promoting properties. Among them, carnosic acid and carnosol, tanshinones and sclareol are well known for their cardiovascular, antitumor, antiinflammatory and antioxidant activities. However, many of these compounds are not available at a constant supply and developing biotechnological methods for their production could provide a sustainable alternative. The transcriptome of S.pomifera glandular trichomes was analysed aiming to identify genes that could be used in the engineering of synthetic microbial systems.
Results
In the present study, a thorough metabolite analysis of S. pomifera leaves led to the isolation and structure elucidation of carnosic acid-family metabolites including one new natural product. These labdane diterpenes seem to be synthesized through miltiradiene and ferruginol. Transcriptomic analysis of the glandular trichomes from the S. pomifera leaves revealed two genes likely involved in miltiradiene synthesis. Their products were identified and the corresponding enzymes were characterized as copalyl diphosphate synthase (SpCDS) and miltiradiene synthase (SpMilS). In addition, several CYP-encoding transcripts were identified providing a valuable resource for the identification of the biosynthetic mechanism responsible for the production of carnosic acid-family metabolites in S. pomifera.
Conclusions
Our work has uncovered the key enzymes involved in miltiradiene biosynthesis in S. pomifera leaf glandular trichomes. The transcriptomic dataset obtained provides a valuable tool for the identification of the CYPs involved in the synthesis of carnosic acid-family metabolites.
Similar content being viewed by others


Background
Salvia species have attracted great attention due to their biologically active constituents [1]. Two major groups of secondary metabolites are produced in Salvia species; terpenoids and polyphenolics. Diterpenes is the largest group comprising 545 of 791 presently identified Salvia sp. constituents, with labdanes being the main metabolites [1]. Carnosol and carnosic acid, two diterpenes naturally found in sage and rosemary, have been evaluated for their anti-inflammatory [2], antioxidant [3, 4] and anticancer properties [5, 6]. Tanshinones, naturally found in S. miltiorrhiza roots, are widely used in the treatment of cardiovascular diseases [7–9]. They are a mix of chemical compounds mainly consisting of tanshinone I, tanshinone IIA, cryptotanshinone and dihydrotanshinone having various biological activities, such as anti-inflammatory [10], antibacterial [11], antioxidant [12] and antineoplastic [13–15]. So far, extracts of S. miltiorrhiza roots have been successfully enrolled in clinical trials for coronary heart disease [16], pulmonary hypertension [17] and polycystic ovary syndrome [18].
In plants, terpenes are synthesized via two pathways: the mevalonate pathway in the cytosol and the 2-C-methyl-D- erythritol-4-phosphate (MEP) pathway in the plastids. The main building block of terpenes is an isoprene unit that is derived from isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP). Then, by the action of prenyltransferases on IPP and DMAPP, the higher building blocks of terpenes are generated: geranyl diphosphate (GPP) for monoterpene synthesis, (E, E)-farnesyl diphosphate (FPP) for sesquiterpene and (E, E, E)-geranylgeranyl diphosphate (GGPP) for diterpene synthesis [19]. Diterpenes are mainly synthesized via the MEP pathway; nevertheless a crosstalk between the pathways has been reported [20].
Tanshinones and carnosic acid, derive from the universal diterpenoid precursor GGPP that is subsequently converted to copalyl diphosphate (CDP) by the action of CDP synthase. Additional cyclization catalyzed by a class I terpene synthase (SmKSL) results in the formation of miltiradiene [21]. Subsequently, the action of cytochrome P450 monooxygenases, such as CYP76AH1 from S. miltiorrhiza [22] or CYP76AH4 from R. officinalis [23], catalyzes the synthesis of ferruginol, a potential precursor of carnosic acid which in turn may be an intermediate to tanshinone biosynthesis [22]. Due to the biological significance of carnosol, carnosic acid and tanshinones, there is increasing need for improved or alternative methods of production of these metabolites. As a result, efforts aiming to elucidate the related biosynthetic pathways has led to the identification of several enzymatic steps, not only in S. miltiorrhiza [24, 25] but also in other species [26, 27]. Specifically, diterpene synthases catalyzing the same enzymatic reactions with SmCDS and SmKSL have been identified in Rosmarinus officinalis [26] and in Salvia fruticosa [27]. Identification of ferruginol synthase in S. miltiorrhiza, R. officinalis and S. fruticosa enabled the synthesis of ferruginol in yeast cells [22, 27]. Aiming to reconstruct tanshinone biosynthesis in yeast, expression of S. miltiorrhiza ferruginol synthase CYP76AH1 in a dedicated strain resulted in 10.5 mg/L of ferruginol [22].
In the current study, we conducted a combined metabolome and transcriptome analysis of S. pomifera leaves and leaf trichomes, respectively, in order to provide an insight into isoprenoid pathway and identify key enzymes involved in terpene biosynthesis. Metabolite analysis of S. pomifera leaf extracts led to the identification and isolation of several carnosic acid-related metabolites, suggested that transcriptomic analysis may yield important insights into the biosynthesis of these compounds. Two genes that appeared to be likely involved in miltiradiene synthesis, a copalyl diphosphate synthase (SpCDS) and a miltiradiene synthase (SpMilS) were cloned and expressed in S. cerevisiae and their products were characterized. Furthermore, from the full transcriptomic profile of S. pomifera’s, we identified the CYPs providing a useful insight on the subsequent biosynthetic steps.
Results
Isolation and characterization of diterpenes from S.pomifera leaves
Specimens of S. pomifera from the areas of Topolia (Western Crete, Greece) and Aptera (North-western Crete, Greece) were collected and the constituents of their aerial parts were extracted. A series of chromatographic separations of the organic extracts resulted in the isolation of one new (1) and five previously reported metabolites, which were identified as pisiferic acid (2), O-methyl-pisiferic acid (3), 12-methoxycarnosic acid (4), carnosol (5), and salviol (6) (Fig. 1a), by comparison of their spectroscopic and physical characteristics with those reported in the literature [28–33].

Structures and biosynthesis of the main diterpenes found in S. pomifera leaves. a The names of the isolated compounds from S. pomifera leaves are: (1) 2α-hydroxy-O-methyl-pisiferic acid, (2) pisiferic acid, (3) O-methyl-pisiferic acid, (4) 12-methoxycarnosic acid, (5) carnosol and (6) salviol. b The proposed biosynthetic pathway of carnosic acid-family metabolites: Geranylgeranyl diphosphate (GGPP) is subjected to cyclization by a class II diterpene synthase to form copalyl diphosphate ((+)-CPP). Then, by the action of a class I diterpene synthase, miltiradiene is formed. Subsequent oxidation and cytochrome P450 activity results in the formation of ferruginol. Figure was produced using Chemescketch v 14.
The molecular formula of compound 1, isolated as a white solid, was determined to be C21H30O4 on the basis of its HR-ESI-MS data. The spectroscopic characteristics of metabolite 1 were rather similar to those of compounds 2–6, indicating an abietane skeleton. Its 1H NMR spectrum included signals for two aromatic protons resonating at δ 6.72 and 6.91, one oxygenated methine at δ 4.26, one oxymethyl at δ 3.73, two singlet methyls at δ 1.00 and 0.82 and two doublet methyls at δ 1.16 and 1.14 (Table 1). Analysis of the correlations observed in the HSQC, COSY and HMBC spectra led to the complete assignment of the protons and carbons of the molecule, while the relative configuration was proposed on the basis of NOE enhancements. In particular, the heteronuclear correlations of C-20 with H-1α and H-5 indicated the position of the carboxylic acid at C-20, while the correlations of C-12 with the protons of the oxymethyl and H-15 indicated the position of the methoxy group at C-12. Furthermore, the heteronuclear correlations of C-8 and C-13 with H-11 and of C-9 and C-12 with H-14 indicated the position of the aromatic protons. The NOE interactions of H-11 with the protons of the oxymethyl and H-1α and of H-14 with H2-7, H3-16 and H3-17 further confirmed the position of the aromatic protons. Additionally, the NOE interactions of H-2 with H-1β, H-3β and H3-19, in combination with the coupling constants of H-2 determined the equatorial orientation of the hydroxyl group at C-2. Thus, compound 1 was identified as 2α-hydroxy-O-methyl-pisiferic acid. The 1H-NMR and 13C-NMR spectra of 2α-hydroxy-O-methyl-pisiferic acid are provided as Additional file 1: Figure S2 and Additional file 2: Figure S3 along with the 1H-NMR spectra of pisiferic acid (Additional file 3: Figure S4), O-methyl-pisiferic acid (Additional file 4: Figure S5), 12-methoxycarnosic acid (Additional file 5: Figure S6), carnosol (Additional file 6: Figure S7) and salviol (Additional file 7: Figure S8).
These structures of carnosic acid- family metabolites seem to be derived from ferruginol via miltiradiene and abietatriene (Fig. 1b). The presence of small amounts of ferruginol in extracts of the aerial parts of S. pomifera (data not shown) supports this hypothesis. In order to elucidate the biosynthesis of these compounds, we proceeded with the analysis of the transcriptome of S. pomifera glandular trichomes.
Transcriptome sequencing, de novo assembly and sequence clustering of S. pomifera trichome transcripts
Sequencing of S. pomifera leaf trichomes resulted in approximately 54,000,000 raw reads from which nearly 85 % were of high quality. The results of the RNA-Seq regarding the throughput and the quality of S. pomifera leaf’s trichome libraries are included in Table 2. High quality reads were assembled into contigs using the standard pipeline of the Trinity suite software [34]. A K-mer catalogue was produced by jellyfish software and used as input for inchworm to assemble the contigs. Subsequently, reads were mapped back to the assembled contigs to improve assembly using bowtie. In total, 125,371 contigs were assembled with a mean length of 321 nt (Additional file 8: Figure S1A). These contigs were re-assembled into 66,051 larger contigs of 640 nt mean length; however the majority of the contigs was less than 600 nt in length (Additional file 8: Figure S1B). From these, 22,395 were clustered in clusters of contigs (given the prefix cl) and 43,656 remained as singletons (given the prefix unigene) in the final Trinity output produced by the coordinated action of Chrysalis and Butterfly software. Contigs with less than 600 nt in length had in average 600 total reads mapped in each contig.
Functional annotation and transcript expression in trichomes
S. pomifera contigs were employed in BLAST searches against several biological databases for their annotation. 70 % of the S. pomifera contigs had a non-redundant (nr) NCBI BLAST hit, while the percentage of the sequences that were annotated based on Gene Ontology (GO) was 50.4 %. The percentages of the sequences annotated based on BLAST searches against the Nucleotide database (Nt), Swiss-Prot and Kyoto Encyclopedia of Genes and Genomes (KEGG), were similar; 49.74, 44.5 and 41.9 %, respectively. Much lower was the percentage of S. pomifera contigs that were assigned to Clusters of Orthologous Groups (COG), 26.2 %.
Most of the contigs that had a hit against the nr protein database shared significant similarity, 37 %, with Vitis vinifera sequences, 13 % were similar to Ricinus communis sequences, 11.4 % to Populus trichocarpa, 8 % to Glycine max and 23.8 % to other plants. The majority of the contigs, 40.4 %, showed a similarity level of 60–80 % with proteins from the nr database and 21 % of the contigs had a similarity with proteins that extended beyond 80 %.
Among the 33,332 transcripts with at least one GO-term assigned, the highest percentage of transcripts in the cellular component ontology were annotated in the cell/cell part class (21,126) in the molecular function ontology the majority of the transcripts were in binding (18,119) while in the biological process ontology most of the transcripts were grouped in the cellular process class (17,210). The detailed classification of the unigenes in the individual GO-terms of the three GO ontology domains is depicted in Fig. 2. Similar results were obtained from the GO-analysis of transcripts sequenced from glandular and non-glandular tomato stem trichomes [35].

The GO annotation of S. pomifera leaf trichomes contigs. Assignment of S. pomifera contigs on Gene Ontologies: biological process, cellular component and molecular function and their sub-categories
Furthermore, 17,303 S. pomifera transcripts were classified into 25 COG functional categories. For most of the transcripts only a general function prediction was made while the next two most abundant categories were transcription and post-translational modification, protein turnover and chaperones. Extracellular and nuclear structures are the two COG categories with the smallest number of assigned transcripts. The complete classification of S. pomifera transcripts according to COG is depicted in Fig. 3. The identity of the most expressed transcripts in S. pomifera leaf trichomes, their top nr BLAST hits, and their GO annotation are presented in Table 3.

Classification of S. pomifera transcripts in the 25 COG categories
Most of the S. pomifera contigs that are strongly expressed in leaf trichomes share significant similarity with annotated nr sequences. Among them, two abundantly expressed S. pomifera contigs are involved in secondary metabolism; cl182.contig3 (3830 nt) encoding the previously characterized sabinene synthase [36] with one amino acid difference, and cl.contig1 (2282 nt) coding for a full length protein that is highly similar to cinnamoyl CoA reductase (GenBank: AFE84656.1). Apart from the annotated transcripts, unigene5960, cl2955.contig2, unigene34506, cl1963.contig2 and cl5332.contig1 produced no hits versus nr NCBI database and they were used in further BLAST searches in the European Nucleotide Archive (ENA) database. Unigene 5960 (length 726 nt) is highly similar to mRNA sequence FE536539 extracted from leaf trichomes of another Greek Salvia species, S. fruticosa. Transcript cl2955.contig2 (677 nt) has only one nucleotide change to mRNA sequence FE536032.1 from leaf trichomes of S. fruticosa cl5332. contig1 (791 nt) blasts against another S. fruticosa leaf trichome cDNA clone of ENA database (FE536766.1) with almost perfect identity. All the above sequences lack annotation which was also observed for a large number of transcripts in the trichomes of Mentha spicata [37].
Expression analysis of genes involved in the isoprenoid biosynthesis
Several S. pomifera contigs were predicted to code for proteins that participate in the terpenoid precursor biosynthesis pathway (KEGG entry 00900). Terpenoid biosynthesis in plants is carried out via two pathways: the mevalonate pathway (MVA) in the cytosol and the non-mevalonate or 2-C-methyl-D-erythritol 4-phosphate/1-deoxy-D-xylulose 5-phosphate pathway (MEP/DOXP pathway) in the plastids. In Fig. 4, the two terpenoid biosynthetic pathways are depicted with their identified corresponding enzymes and the number of contigs that putatively code for these enzymes in the present transcriptomic analysis. The first protein/enzyme of the plastid MEP pathway, 1-deoxy-D-xylulose-5-phosphate synthase (DXS, EC: 2.2.1.7) is encoded by seven contigs; 6 contigs that contain a partial and 1 contig with a full ORF. Another important enzyme of the pathway, the 4-hydroxy-3-methylbut-2-enyl diphosphate reductase (HDR, EC: 1.17.1.2) is encoded by 9 S. pomifera contigs. On the other hand, enzyme 3-hydroxy-3-methylglutaryl-CoA reductase (HMGR, EC: 1.1.1.34) of the MVA pathway is encoded by 8 contigs.

Graphical representation of the two pathways, MVA and MEP, by which plant terpenes are produced. The KEGG entries of the specific enzymes that participate in the pathways are shown in red as alongside the number of contigs identified that putatively encode for these enzymes
The expression levels of the full or nearly full S. pomifera ORFs involved in terpenoid biosynthesis are presented in Fig. 5. The most expressed transcript of the MEP pathway is unigene32091 (2054 nt) which encodes for a DXS protein and shares high similarity (>90 %) with DXS2 from S. miltiorrhiza (GenBank: ACQ66107.1). The FPKM value for unigene32091 - putative DXS2 is 688.42. In the MVA pathway, the highest expressed transcript is cl1563.contig2 (2263 nt) that encodes for a putative HMGR protein and has 87 % similarity with HMGR1 from S. miltiorrhiza (GenBank: ADC44451.1). The FPKM value for cl1563.contig2 - putative HMGR1 is 1273.02, nearly double than that of unigene32091 of the MEP pathway. The second most abundant is unigene10436 (1978 nt) - FPKM value is 985.72 - encodes for a HMGS protein almost identical to HMGS1 from S. miltiorrhiza (GenBank: ACV65039.1). In general, the transcripts that are predicted to participate in the MEP pathway are present at lower levels than the transcripts of the MVA pathway. Finally, 16 S. pomifera contigs are putative GPP, GGPP and FPP synthases. Only one full length putative GGPPS gene, cl9704.contig1, is included in Fig. 5 with a rather moderate expression. Α putative FPPS gene i.e., unigene28420 that synthesizes FPP, the precursor of sesquiterpenes and triterpenes in the cell cytoplasm, is the most expressed among the prenyltransferases; its FPKM value is 337.79 (Fig. 5).

Expression heatmap of S. pomifera contigs. Contigs that encode full or almost full length proteins and are putatively involved in the MEP and MVA pathways of the terpenoid backbone biosynthesis in the leaf trichomes. Color bar at the bottom of the figure denotes color intensity according to global expression levels. Abbreviations; DXS: 1-deoxy-D-xylulose-5-phosphate synthase, DXR: 1-deoxy-D-xylulose-5-phosphate reductoisomerase, MCT: MEP cytidyltransferase, CMK: 4-(cytidine 5- diphospho)-2-C-methyl-D-erythritol kinase, MDS: 2-C-methyl-D-erythritol 2,4-cyclodiphosphate, HDS: 1-hydroxy-2-methyl-2-butenyl 4-diphosphate synthase, HDR: 1-hydroxy-2-methyl-2-butenyl 4-diphosphate reductase, HMG: 3-hydroxy-3-methylglutaryl, MVK: mevalonate kinase, PMK: phosphomevalonate kinase, MVD: diphosphomevalonate decarboxylase, IPPI: isopentenyl diphosphate, IPPSs: isoprenyl pyrophosphate synthases
Diterpene biosynthesis typically consists of an initial cyclization of GGPP by a class II diTPS to produce a cyclic diphosphate intermediate, followed by conversion of this intermediate to the final diterpene skeleton by a class I diTPS [38]. S. pomifera unigene32268 that is abundantly expressed in trichomes (FPKM = 210.2), encodes for a protein that shares 97 % similarity (identities) to S. fruticosa copalyl diphosphate synthase (CDS) (GenBank: AJA38250.1), 90 % similarity to R. officinalis CDS (GenBank: AHL67261.1) and 87 % similarity to S. miltiorrhiza CDS (GenBank: AHJ59322.1). S. pomifera cl463 cluster of contigs code for a diterpene synthase that was previously characterized as miltiradiene synthase (GenBank: AJQ30185.1) [27]. CL463.contig4 that contains a full ORF identical to miltiradiene synthase is sufficiently expressed in trichomes of S. pomifera (FPKM =140.3).
Identification, cloning and characterization of genes related to miltiradiene biosynthesis
Expression analysis of unigene32268 and cl463 transcripts was performed by quantitative real time PCR (qPCR). Elongation factor 1-A was used as reference gene to normalize cDNA quantity. Expression analysis for the isolated genes was performed on leaves, root and shoot samples of S. pomifera. As a reference tissue sample leaves were selected. The expression levels of all the other tissues were expressed as relatively fold-differences to leaves. As shown in Fig. 6, unigene32268 is expressed only in leaves and more than 20 times less in the shoot of S. pomifera, while no expression was detected in the root. Cl463 expression levels are 100 times lower in the roots compared to leaves and more than 20 times lower in the shoot compared to the leaves (Fig. 6). High expression in leaves is anticipated; however the complete absence of expression of unigene32268 in roots is surprising considering that in extractions from root tissues we have identified sufficient amount of ferruginol (data not shown). It is likely that an alternative (tissue-specific) CDS gene may be responsible for labdane diterpene synthesis in the root.

Relative expression of S. pomifera genes related to miltiradiene biosynthesis. Relative expression of miltiradiene synthase (SpMilS) and copalyl diphosphate synthase (SpCDS) in shoot, root and leaves of S. pomifera. Expression levels are depicted relatively in shoot and root compared to leaves expression
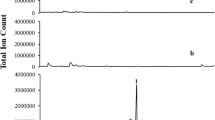
The S. pomifera unigene 32268 encoding for a putative copalyl diphosphate synthase (SpCDS) was PCR amplified using gene specific primers and introduced into the yeast expression vector pUTDH3myc, while the class I terpene synthase gene cl463 was amplified and cloned into pHTDH3myc [39]. For in vivo functional characterization, pseudomature protein was expressed in the yeast strain AM238 (MATα his3, ura3, trp1, rox1, dos2, yer134c, vba5, ynr063w, ygr259c) [40] together with the Erg20p (F96C) variant protein capable of improved production of GGPP [41]. Production of manool and copalol was observed in the yeast strain expressing the putative SpCDS. As proposed [41, 42], formation of manool and copalol is likely the result of acid or phosphatase-mediated hydrolysis, respectively, of excess of copalyl diphosphate (Fig. 7a). The identity of manool and copalol was verified by comparison of the retention time and mass-spectrum with authentic standard (Fig. 7b). Co-expression of the putative miltiradiene synthase (cl463) with SpCDS in AM238 yeast cells expressing the Erg20p(F96C) variant resulted in the production of miltiradiene (Fig. 7c), confirmed by comparison of the retention time and mass-spectrum with authentic standard (Fig. 7d). Miltiradiene titers under these conditions reached 32 mg/L (Fig. 7c), comparable with the previously reported ones [27, 43, 44]. Therefore, biosynthesis of miltiradiene was reproduced in heterologous yeast by employing the corresponding class II diterpene synthase, SpCDS, responsible for biosynthesis of copalyl diphosphate and the class I diterpene synthase, SpMilS (Fig. 7e).

a Characterization of S. pomifera genes related to miltiradiene biosynthesis. S. pomifera Unigene 32268 encoding for a copalyl diphosphate synthase (SpCDS) expressed in AM238 yeast cell enabled production of manool and copalol as result of (+)-CPP hydrolysis. b Mass-spectrum of manool and copalol products and standards. c Production of miltiradiene in yeast cells co-expressing copalyl diphosphate synthase (SpCDS) and miltiradiene synthase (SpMilS). d Mass-spectrum of miltiradiene product and standard. e Biosynthetic pathway of miltiradiene in S. pomifera employ a class II diterpene synthase responsible for cyclization of the linear precursor geranylgeranyl diphosphate (GGPP) to form copalyl diphosphate ((+)-CPP), which is further converted to miltiradiene by a class I diterpene synthase
Cytochrome P450s
Cytochrome P450s is one of the largest families in plant metabolism and are responsible for the high chemical diversity of plant metabolites [45]. They are heme containing enzymes that catalyze the introduction of atmospheric oxygen into non-activated carbon-hydrogen bonds in a stereo- and regio-selective manner [46]. The transcriptomic analysis of S. pomifera trichomes revealed a number of CYPs either of full or partial length (Table 4). All CYPs were sent to Professor David Nelson for annotation [47]. The expression levels along with the annotation of S. pomifera CYPs are presented on Table 4. The highest transcribed CYPs are SpCYP76AH24 ortholog (GenBank: KT157044) (FPKM = 2026.30), SpCYP76AK6 (GenBank: KT157045) (FPKM = 1892.16), SpCYP71BE52 (GenBank: KT157042) (FPKM = 1746.15) and Sp716A96 (cv1) (GenBank: KT157037) (FPKM = 1326.01). Furthermore, several additional CYPs were identified having high expression levels such as SpCYP86A92 ortholog (GenBank: KT157046) (FPKM = 868.07), SpCYP71D455 (FPKM = 791.26), Sp73A120 ortholog (FPKM = 760.08) and SpCYP93B25 ortholog (FPKM = 721.56).
The highest transcribed CYP, SpCYP76AH24 ortholog (FPKM = 2026.30), was found to be homologous (identity 80 %) to S. miltiorrhiza CYP76AH1 (GenBank: AGN04215.1) that catalyzes the synthesis of ferruginol [22]. SpCYP76AH24 ortholog encodes a predicted protein of 492 amino acids that is almost identical (identity 99 %) to the ferruginol synthase from S. fruticosa (GenBank: AJQ30186.1) [27] and highly homologous (identity 91 %) to CYP76AH4, the ferruginol synthase from R. officinalis (GenBank: AJQ30187.1) [23]. It is suggested that miltiradiene spontaneously aromatizes to abietatriene, which is then hydroxylated by ferruginol synthase to yield ferruginol, the precursor of other labdane-related terpenes like carnosol and tanshinones [23].
A phylogenetic analysis of CYPs revealed that the majority belong to the CYP71 clan while the rest of transcripts are categorized to the clans CYP72, CYP85 and CYP86. Clans CYP51 and CYP74 have one member each while CYP97 has two members as demonstrated on the phylogenetic tree (Fig. 8). The clustering of families of S. pomifera’s CYPs on the phylogenetic tree comes to verify the rooted relationships of clans. For instance, the proximity of CYP97 with CYP86 and CYP51 with CYP85 implies a possible evolution from a common ancestor [45]. Members of the CYP71 clan were shown to be close phylogenetically with enzymes involved in isoprenoid biosynthesis. Two members of the CYP76 family, Sp76AK6 and Sp76G16 ortholog are phylogenetically close with Sm76AK3 and Sm76G16, respectively, suggesting an involvement in diterpenoid biosynthesis [48]. On the other hand, SpCYP71D456 showed high similarity with CYP71D55 from Hyoscyamus muticus that is designated as premnaspirodiene oxidase [49] in sesquiterpenoid biosynthesis. Regarding monoterpenes, SpCYP71AU53 ortholog (GenBank: KT157041) and SpCYP81B62 ortholog are probably involved in the biosynthesis of monoterpenoids [48]. Along with clan CYP71, one member from CYP85 clan, SpCYP728D17 ortholog (GenBank: KT157038) is probably involved in diterpenoid biosynthesis [48]. From the CYP85 clan, according to the computational expression analysis, the highest expressed transcripts are Sp716A96 (1) (FPKM = 1326.01) and SpCYP716A97 (FPKM = 451.34); both belonging to CYP716A subfamily which have been shown to be involved in diterpenoid [48] and triterpenoids biosynthesis [50]. In general, members of the CYP85 clan have been shown to be involved in the metabolism of medium to large isoprenoids [45]. The majority of clan CYP86 members are orthologous to their counterparts from S. miltiorrhiza [48] with SpCY96A92 ortholog, SpCYP94A49 ortholog and SpCYP96A107 having the highest expression among them.

A neighbor-joining tree of S. pomifera’s CYPs. Phylogenetic analysis of S. pomifera cytochrome P450s. Green labels represents S. pomifera CYPs and red labels represent the different clans. Among gene variants the full length one (cv) was chosen when possible otherwise the most transcipted partial variant (pv)
Besides isoprenoid metabolism, specific transcripts were identified to be orthologous to S. miltiorrhiza’s CYPs that are probably involved in rosmarinic acid pathway. Sp73A120 ortholog and SpCYP98A75 ortholog have been identified as cinnamate 4-hydroxylase and β-coumaryl-CoA 3′-hydroxylase respectively, two key enzymes in rosmarinic acid biosynthesis in S. miltiorrhiza [48, 51].
Discussion
Labdane diterpenes have important health promoting properties. Carnosic acid-related compounds from rosemary and sage have attracted attention not only as potent antioxidants but also as antibacterial, antiparasitic and cytotoxic compounds [33, 52–56] while tanshinones from S. miltiorrhiza roots have a plethora of bioactive properties [7, 9, 10, 12, 57]. The biosynthesis of carnosic acid and tanshinones is still unclear. Although ferruginol appears to be a common intermediate, the following steps and the point where these pathways disentangle are not yet known. Interestingly, it has been proposed that tanshinone biosynthesis may proceed through carnosic acid [22], although this claim has not since been validated. Elucidating the biosynthesis of these compounds will be an important step towards their biotechnological production in engineered microorganisms.
In the present study, thorough metabolite analysis of S. pomifera leaves led to the isolation and identification of several metabolites from the carnosic acid family (Fig. 1). To elucidate their biosynthesis, the transcriptome of S. pomifera glandular trichomes was sequenced to obtain approximately 54,000,000 raw reads that were assembled into contigs and annotated. Expression analysis of the transcripts involved in the isoprenoid biosynthesis showed higher expression of the genes participating in the MVA pathway in comparison to the MEP pathway. These findings are in contrast to previous observations from S. miltiorrhiza where it was shown that MEP related transcripts were mainly expressed in the leaf while MVA-related transcripts were mainly expressed in the root [24].
In addition to isoprenoid biosynthesis, several highly expressed transcripts that were identified in the trichomes of S. pomifera are highly similar to S. fruticosa ones indicating that they probably play important roles in this specialized tissue which could be investigated further. Other contigs that code for photosynthetic related proteins such as ribulose biphosphate carboxylase (Rubisco) and chlorophyll-binding proteins are among the most expressed in trichomes. Similarly, such ESTs coding for proteins involved in photosynthesis have also been found among the most expressed genes in the trichomes of Artemisia annua, the Asian medicinal plant that produces the sesquiterpene lactone artemisinin [58]. The abundant expression of photosynthesis related genes in the trichomes transcriptome of S. pomifera is also in accordance with data from other plant species such as several Solanum species - both wild and domesticated - in which photosynthetic genes were also highly expressed in glandular trichomes [59]. S. pomifera trichomes are possibly capable of synthesizing secondary metabolites without the need for carbon transport from leaves. The opposite has been suggested by Jin et al. [37] for the peltate glandular trichomes of spearmint, M. spicata, described as poor in genes related to photosynthesis and chlorophyll formation. We proceeded to reassemble and analyze the deposited M. spicata transcriptome data and compared them to the S. pomifera ones. Data analysis of the M. spicata transcriptome sequencing showed that two genes coding for Rubisco and chlorophyll binding proteins are also highly expressed in the trichomes of M. spicata. The common high expressed genes between S. pomifera and M. spicata are indicated in bold in the Additional file 9: Table S1 that includes the complete list of M. spicata most expressed transcripts as a result of our analysis. With regard to photosynthetic competence, the possibility of leaf cells contaminating trichome samples cannot be ruled out, though no such tissue could be seen by visual and microscopic observation of the trichomes destined for S. pomifera NGS sequencing.
The transcriptomic analysis pointed two highly expressed diterpene synthases, SpCDS and SpMilS, that were cloned and expressed in the appropriate yeast platform yielding 32 mg/L miltiradiene, that is in accordance with previously reported ones [27, 43, 44] (Fig. 7c). The abundance of SpCDS and SpMilS was further supported from the expression analysis by qPCR in three different tissue organs of S. pomifera (Fig. 6) that showed high expression in leaves and poor or zero expression in other tissues. This might be explained by the fact that carnosic acid is mainly stored in the glandular trichomes of the leaves [26, 27], thus a higher expression of the relevant enzymes is expected. On the contrary, their homologues in S. miltiorrhiza, SmCPS and SmKSL, were shown to be abundantly expressed in roots, which is expected since tanshinones accumulation takes place in that specific tissue [24].
Along with terpene synthases, CYPs are the key enzymes contributing to the diversification of terpenoids. They are a highly diverse family of enzymes catalyzing various reactions [60] responsible for the chemical diversity of secondary metabolites of the plant kingdom. Terpene synthases create the terpene scaffold giving rise to a variety of metabolites [61], while CYPs undertake further modification of the scaffold increasing the diversity of the resulting terpenes. Study of terpene synthases along with CYPs is essential since a non random functional association of these two families has been revealed [62]. Thus, having available the whole transcriptomic profile of CYPs is of a great significance.
The transcriptomic analysis of S. pomifera trichomes revealed a significant number of highly transcribed CYPs, with SpCYP76AH24 ortholog being the highest among them. SpCYP76AH24 ortholog showed high similarity to ferruginol synthases from other Lamiaceae species; 99 % identity to S. fruticosa ferruginol synthase, 91 % identity to R. officinalis CYP76AH4 and almost 80 % homology to S. miltiorhhiza CYP76AH1. The predicted protein from SpCYP76AH24 ortholog has a full pP450 domain (Pfam family: PF00067) that extends from position 28 to position 463 and due to its similarity with the known Lamiaceae CYP76AHs, it is highly possible to produce ferruginol in S. pomifera.
Phylogenetic analysis of cytochrome P450s showed that the majority of CYPs belong to CYP71, CYP72, CYP85 and CYP86 clans as was anticipated [50]. It was previously shown that CYPs involved in the isoprenoid biosynthesis likely originate from CYP71 and CYP85 clan. CYP71 clan members are mainly involved in the metabolism of specialized metabolites like monoterpenes and sesquiterpenes substrates [50] as well as in the metabolism of aromatic and aliphatic amino acids, alkaloids, fatty acids and precursors of hormones [45]. CYP71 clan includes the majority of the CYP450 families that are involved in the plant secondary metabolism [50, 60]. CYP71A family proteins participate in monoterpenoids oxidation even though it has been suggested that may be highly specialized in their roles in diverse plant species [50]. On the other hand, CYP76 family is mainly involved in isoprenoid metabolism [60] although members of this family have also been implicated in herbicide metabolism in Arabidopsis [63] and in the biosynthesis of defense compounds in Thuja plicata [64]. Clans CYP85 and CYP86 are involved in the metabolism of both general and specialized metabolites [50]. Besides isoprenoid metabolism, two member enzymes were annotated as orthologous to SmCYP98A75 and SmCYP73A120 that are - a β-coumaryl-CoA 3′-hydroxylase and a cinnamate 4-hydroxylase respectively, key enzymes in rosmarinic acid biosynthesis [48, 51].
Conclusions
Over the past years, tanshinones and carnosic family metabolites, have gained great attention due to their unique properties. Thus, elucidation of their biosynthetic pathway is of great importance as it will unlock the key enzymes involved in their synthesis. The transcriptome analysis of S. pomifera trichomes revealed two genes involved in miltiradiene synthesis; a copalyl diphosphate synthase (SpCDS) and a miltiradiene synthase (SpMilS). Furthermore, the phylogenetic analysis of several cytochrome P450s provided useful insights in the biosynthetic mechanism responsible for the production of carnosic acid-family metabolites in S. pomifera. Setting up a resource of CYP450 genes from S. pomifera not only provides information on terpenoid biosynthesis but also assists in mining genes involved in the production of other natural products.
Methods
Chemicals
n-Hexane (≥99 %) and diethyl ether (≥99.8 %) used for plant extract and GC/MS analysis respectively were both purchased from Sigma-Aldrich (St. Louis, MO, USA). Cyclohexane, CH2Cl2, acetone and acetic acid ethyl ester (EtOAc) were all purchased from Sigma-Aldrich (St. Louis, MO, USA).
MyTaq DNA polymerase (BIO-21105, Bioline, Taunton, USA), and Accuzyme DNA polymerase (BIO-21051, Bioline, Taunton, USA) were used in PCR amplifications; NucleoSpin Plasmid Kit (REF 740588.250, Macherey-Nagel, Düren, Germany) was used for plasmid DNA purification; QIAquick Gel Extraction Kit (#28704, Qiagen, Hilden, Germany) was used for gel extraction and DNA purification.
Yeast media: D (+)-Glucose monohydrate (Sigma-Aldrich, St. Louis, MO, USA); Yeast Nitrogen Base w/o AA, carbohydrate & w/AS (Y2025, US Biologicals, Life Sciences, Texas, USA); Complete Minimal (CM) medium is composed of 0.13 % (w/v) dropout powder (all essential amino acids), 0.67 % (w/v) yeast nitrogen base w/o AA, 2 % glucose; TOPO TA Cloning Kit Dual Promoter (K4610-20, Life Technologies, Carlsbad, CA, USA). All yeast transformations were done by lithium acetate transformation.
Plant material
S. pomifera plants were grown from seeds collected from the wild populations of Crete, Greece (Topolia and Aptera area). Plants were grown in the research greenhouse in the farm of Aristotle University of Thessaloniki, Greece, under controlled temperature (25/18 °C, day/night, winter, 32/20 °C, day/night, summer) and natural photoperiod.
Plant extraction and isolation of metabolites
The aerial parts of S. pomifera plants grown from seeds collected from the areas of Topolia and Aptera, Grete, Greece were separately extracted with EtOAc at room temperature. Evaporation of the solvent under vacuum afforded green brownish organic residue.
The organic residue of Aptera (SP1, 607 mg) was subjected to vacuum column chromatography on Sephadex LH-20, using cyclohexane/CH2Cl2 (1:4 v/v), CH2Cl2/acetone (3:2 v/v) and CH2Cl2/acetone (1:4 v/v) as mobile phase, to yield 11 fractions (SP1f1–SP1f11). Fraction SP1f7 (cyclohexane/CH2Cl2 1:4 v/v, 16.3 mg) was further fractionated by gravity column chromatography on silica gel, using cyclohexane with increasing amounts of EtOAc as the mobile phase, to afford 4 fractions (SP1f7a–SP1f7d). Fraction SP1f7c (15 % v/v EtOAc in cyclohexane, 5.2 mg) was purified by normal phase HPLC, using cyclohexane/acetone (85:15 v/v) as eluent, to yield metabolite 2 (1.3 mg). Fraction SP1f8 (CH2Cl2/acetone 3:2 v/v, 64.9 mg) was further fractionated by gravity column chromatography on silica gel, using cyclohexane with increasing amounts of EtOAc as the mobile phase, to afford 5 fractions (SP1f8a–SP1f8e). Fraction SP1f8b (15 % v/v EtOAc in cyclohexane, 39.6 mg) was purified by normal phase HPLC, using cyclohexane/acetone (85:15 v/v) as eluent, to yield metabolites 4 (12.2 mg) and 5 (1.6 mg). Fraction SP1f9 (CH2Cl2/acetone 3:2 v/v, 57.6 mg) was further fractionated by gravity column chromatography on silica gel, using cyclohexane with increasing amounts of EtOAc as the mobile phase, to afford 6 fractions (SP1f9a–SP1f9f). Fraction SP1f9d (25 % v/v EtOAc in cyclohexane, 27.9 mg) was purified by normal phase HPLC, using cyclohexane/acetone (83:17 v/v) as eluent, to yield metabolites 1 (17.0 mg) and 6 (10.0 mg).
The organic residue of Topolia (SP4, 223 mg) was subjected to vacuum column chromatography silica gel, using cyclohexane with increasing amounts of EtOAc as mobile phase, to yield 10 fractions (SP4a–SP4b). Fraction SP4f (12 % v/v in EtOAc, 50 mg) was subjected to normal phase HPLC, using cyclohexane with increasing amounts of EtOAc as mobile phase, to yield 9 fractions (SP4f1–SP4f9). Fraction SP4f5 (26.0 mg) was further fractionated by gravity column chromatography on silica gel, using cyclohexane with increasing amounts of EtOAc as the mobile phase, to afford 5 fractions (SP4f5a–SP4f5e). Fraction SP4f5b (10 % v/v EtOAc in cyclohexane, 18.5 mg) was purified by normal phase HPLC, using cyclohexane/EtOAc (86:14 v/v) as eluent, to yield metabolite 3 (1.6 mg). Fractions SP4f6 to SP4f9 (12, 15, 20 and 30 % v/v EtOAc in cyclohexane, respectively) were separately subjected to normal phase HPLC using a gradient mobile phase as described below. The peaks corresponding to the elution times of the isolated compounds were collected and were further purified by normal phase HPLC using the same conditions as described for SP1 to afford metabolites 1 (0.6 mg), 2 (4.1 mg), 4 (0.7 mg) and 6 (2.3 mg). The isolated compounds (1–6) were subjected to normal phase HPLC analysis using a gradient elution system. Their elution times were determined and their characteristic UV spectra were also recorded, both serving as reference data for the identification of the compounds within the extracts. The mobile phase of the gradient system consisted of cyclohexane (A) and EtOAc (B). A flow rate of 1.5 mL/min was applied, using the following gradient profile: 10 % B up to 100 % B within 50 min and then the solvent was kept constant to 100 % B for 10 min. All organic residues were qualitatively analyzed according to the described method.
2α-hydroxy-O-methyl-pisiferic acid (1)
White solid; [α] \( \begin{array}{c}\hfill 20\hfill \\ {}\hfill \mathrm{D}\hfill \end{array} \) +93.0 (c 0.17, CHCl3); UV (CHCl3) λ max (log ε) 243 (2.62), 284 (2.41) nm; IR (film) ν max 3316, 2966, 22918, 2852, 1679, 1508, 1459, 1247, 1027; NMR data, see Table 1; EIMS (70 eV) m/z (rel. int. %) 346 (76), 301 (13), 283 (100), 241 (12), 159 (7); HR-ESIMS m/z 345.2066 [Μ-H]− (calcd for C21H29O4, 345.2071).
General experimental procedures
Optical rotations were measured on a Perkin-Elmer model 341 polarimeter with a 1 dm cell. UV spectra were recorded on a Perkin Elmer Lambda 40 spectrophotometer. IR spectra were obtained on a Bruker Tensor 27 spectrometer. NMR spectra were recorded on Bruker AC 200 and Bruker DRX 400 spectrometers. Chemical shifts are given on a δ (ppm) scale using TMS as internal standard. The 2D experiments (HSQC, HMBC, COSY, NOESY) were performed using standard Bruker pulse sequences. High resolution ESI mass spectra were measured on a Thermo Scientific LTQ Orbitrap Velos mass spectrometer. Column chromatography separations were performed with Kieselgel 60 (Merck, Kenilworth, USA) and Sephadex LH-20 (Pharmacia & Upjohn Company LLC, USA). HPLC separations were conducted using a Pharmacia LKB 2248 liquid chromatography pump equipped with a RI-102 Shodex refractive index detector, using a Supelcosil SPLC-Si 5 μm (250 × 10 mm i.d.; Supelco) column. HPLC gradient elution separations for qualitative analysis were conducted using a Waters 600 Controller pump equipped with a Waters 996 Photodiode Array detector, using Kromasil Silica 8 μm (250 × 10 mm i.d.; AkzoNobel) column. TLC was performed with Kieselgel 60 F254 (Merck aluminum-backed plates) and spots were detected after spraying with 15 % H2SO4 in MeOH reagent and heating at 100 °C for 1 min.
RNA extraction from S. pomifera leaf trichomes
Leaf material from S. pomifera Topolia was frozen in liquid nitrogen immediately after collection and was kept in −80 °C until processing. Leaf trichomes were isolated with the dry ice abrasion method [65]. High quality total RNA from the plant trichomes was isolated using the commercial Spectrum Plant Total RNA Kit (Sigma Aldrich, St. Louis, MO, USA), according to the manufacturers’ specifications. Quality control of RNA was performed using an Agilent 2100 Bioanalyzer.
For Illumina sequencing, at least 20 μg of RNA was used satisfying the following requirements: concentration ≥ 400 ng/μl, OD260/280 = 1.8 ~ 2.2; OD260/230 ≥ 2.0; RNA 28S:18S > 1.0 and RIN ≥ 7.0.
Transcriptome sequencing, de novo assembly and gene annotation
cDNA synthesis and transcriptome sequencing was performed by the Beijing Genomic Institute, Shenzen, China, using an Illumina Hiseq™ 2000 platform.
The obtained raw reads were cleaned from low quality reads, reads that contained adaptors and unknown nucleotides larger than 5 % using fastqc and cutadapt coordinated by the wrapper Trim_Galore (Version 0.3.7). De novo assembly of high quality reads into unigenes was carried out using the Trinity suite [34] using the default parameters for paired end sequenced libraries.. The resulted output from Trinity contained two classes of contigs; the first class, contained clusters of unigenes with similarity higher than 70 % (prefix cl) and the second class contained singletons (prefix unigene).
In order to decide sequence direction of unigenes, a BlastX alignment (e value < 10−5) between unigenes and protein databases like nr (non redundant nucleotide database, NCBI), Swiss-Prot, KEGG (Kyoto Encyclopedia of Genes and Genomes) and COG (Clusters of Orthologous Groups) was performed and the best alignment result was chosen. If there was a confliction between results, the following order of databases was followed for the sequence direction of unigenes: nr, Swiss-Prot, KEGG and COG. When a unigene was not aligned to any of the above databases, the software ESTScan was employed [66].
Functional annotation of unigenes was performed using nr, Swiss-Prot, KEGG and COG databases. For the GO annotation of unigenes based on nr database, the annotation software Blast2GO [67] was used while the GO functional classification of all unigenes was performed with WEGO software [68]. Transcript quantification was estimated from RNA-Seq data using the RSEM software package contained in the Trinity suite. Unigenes were used as reference to estimate the abundance of expression based on the paired-end RNA-Seq data using the standard instructions and parameters as described in http://deweylab.biostat.wisc.edu/rsem/README.html and in http://trinityrnaseq.sourceforge.net/analysis/abundance_estimation.html.
Phylogenetic analysis
The evolutionary history was inferred using the Neighbor-Joining method [69]. The bootstrap consensus tree inferred from 1000 replicates [70] is taken to represent the evolutionary history of the taxa analyzed [70]. Branches corresponding to partitions reproduced in less than 50 % bootstrap replicates are collapsed. The evolutionary distances were computed using the p-distance method [71] and are in the units of the number of amino acid differences per site. The analysis involved 109 amino acid sequences of CYPs. All ambiguous positions were removed for each sequence pair. There were a total of 788 positions in the final dataset. Evolutionary analyses were conducted in MEGA6 [72].
Yeast strain, expression vectors and miltiradiene analysis
The S. pomifera unigene 32268 encoding for a putative copalyl diphosphate synthase (SpCDS) was PCR amplified using gene specific primers and introduced into the yeast expression vector pUTDH3myc [39]. For in vivo functional characterization, pseudomature SpCDS protein was expressed in the yeast strain AM238 [Mat α his3, ura3, trp1, rox1, dos2, yer134c, vba5, ynr063w, ygr259c] [40] also expressing the Erg20p(F96C) variant protein capable of improved production of GGPP [41]. The SpMils construct into the yeast expression vector has been described previously [44].
For the determination of SpCDS and SpMilS products, 5 ml cultures of the relevant yeast strain were grown overnight at 30 °C in the appropriate selection media rotating in a roller drum. The next day 500 μl of decane (1:10 v/v) was added to each tube, and the cultures were incubated for two additional days. At the end of the incubation period the organic layer was transferred into eppendorf tubes, centrifuged to separate any residual medium left, and 200 μl were loaded on GC vials and analyzed by GC-MS for product identification and GC-FID for quantification.
Expression analysis in different tissues of S. pomifera
Total RNA was extracted from leaves, root and shoot samples of S. pomifera using the commercially available kit Spectrum™ Plant Total RNA Kit (Sigma-Aldrich, St. Louis, MO, USA) according to the manufacturer’s instructions. Total RNA from all tissues were subjected to DNase I treatment (Life Technologies, Carlsbad, CA, USA) in order to remove any traces of simultaneously isolated DNA. One μg of total RNA from all tissues were used in a reverse transcription reaction using random hexamers and SuperScript II-RT (Life Technologies, Carlsbad, CA, USA) following the manufacturers protocol. For every tissue an identical NO-RT reaction was included in which Supercript II reverse transcriptase was replaced with DEPC-treated water. Real-time quantitative PCR was performed on the Rotorgene-Q 5plex Platform (using the Kapa SYBR® Fast qPCR Kit Master Mix (2X) Universal KapaBiosystems, Capetown, South Africa) with 1/20 of the synthesized cDNAs as template and the corresponding primers at 0.4 μM. The cycling conditions were: 3 min 95 °C, 35 cycles of 5 s/95 °C, 30 s/64 °C and a melting step of 65–95 °C with a ramp rate of temperature increase of 0.1 °C with a hold of 2 s. The primers for miltiradiene synthase (MilS) were P1326-F: 5′-GGCGCAGTGTCAAAGACTTTCTAA-3′ and P1326-R: 5′-CCATCGAAACATGCCTACACAAAT-3′, for copalyl diphosphatase synthase (CDS) SpCDS-F: 5′- CCGAGTTGAGGCTGCCTATTATCT-3′ and SpCDS-R: 5′-CGAATTCACTAACGCTGCTTCTGT-3′ were designed using the Primer 3.0 online tool (http://biotools.umassmed.edu/bioapps/primer3_www.cgi). A fragment of the Elongation Factor 1a gene was also amplified in order to validate successful cDNA synthesis and for normalization purposes. The primers used were designated SpEF1a-F: 5′- GTTGCCTCTAACTCCAAGGACGAT-3′ and SpEF1a-R: 5′- CCAGCATCACCATTCTTCAAAAAC-3′. All reactions were performed in triplicates. Relative quantification was calculated using the 2-ΔΔCt algorithm a convenient method to analyze the relative changes in gene expression which requires the assignment of one or more housekeeping genes (assumed to be uniformly and constantly expressed in all samples, as well as one or more reference samples). The expression of other samples is then compared to that in the reference sample [73].
Availability of data
Illumina Hiseq 2000 raw transcriptome sequences and Transcriptome Shotgun Assembly are available at NCBI Bioproject database under the accession number PRJNA292070 and the SRA database under the Run Experiment accession number SRR2136651.
References
Wu YB, Ni ZY, Shi QW, Dong M, Kiyota H, Gu YC, et al. Constituents from Salvia species and their biological activities. Chem Rev. 2012;112(11):5967–6026.
Poeckel D, Greiner C, Verhoff M, Rau O, Tausch L, Hornig C, et al. Carnosic acid and carnosol potently inhibit human 5-lipoxygenase and suppress pro-inflammatory responses of stimulated human polymorphonuclear leukocytes. Biochem Pharmacol. 2008;76(1):91–7.
Lo A-HH, Liang Y-CC, Lin-Shiau S-YY, Ho C-TT, Lin J-KK. Carnosol, an antioxidant in rosemary, suppresses inducible nitric oxide synthase through down-regulating nuclear factor-kappaB in mouse macrophages. Carcinogenesis. 2002;23(6):983–91.
Carvalho AC, Gomes AC, Pereira-Wilson C, Lima CF. Redox-dependent induction of antioxidant defenses by phenolic diterpenes confers stress tolerance in normal human skin fibroblasts: insights on replicative senescence. Free Radic Biol Med. 2015;83:262–72.
Johnson JJ, Syed DN, Heren CR, Suh Y, Adhami VM, Mukhtar H. Carnosol, a dietary diterpene, displays growth inhibitory effects in human prostate cancer PC3 cells leading to G2-phase cell cycle arrest and targets the 5′-AMP-activated protein kinase (AMPK) pathway. Pharm Res. 2008;25(9):2125–34.
Bauer J, Kuehnl S, Rollinger JM, Scherer O, Northoff H, Stuppner H, et al. Carnosol and carnosic acids from Salvia officinalis inhibit microsomal prostaglandin E2 synthase-1. J Pharmacol Exp Ther. 2012;342(1):169–76.
Gao S, Liu Z, Li H, Little PJ, Liu P, Xu S. Cardiovascular actions and therapeutic potential of tanshinone IIA. Atherosclerosis. 2012;220(1):3–10.
Tan X, Li J, Wang X, Chen N, Cai B, Wang G, et al. Tanshinone IIA protects against cardiac hypertrophy via inhibiting calcineurin/NFATc3 pathway. Int J Biol Sci. 2011;7(3):383–9.
Chen M, Liu Y, Yi D, Wei L, Li Y, Zhang L. Tanshinone IIA promotes pulmonary artery smooth muscle cell apoptosis in vitro by inhibiting the JAK2/STAT3 signaling pathway. Cell Physiol Biochem. 2014;33(4):1130–8.
Robertson AL, Holmes GR, Bojarczuk AN, Burgon J, Loynes CA, Chimen M, et al. A zebrafish compound screen reveals modulation of neutrophil reverse migration as an anti-inflammatory mechanism. Sci Transl Med. 2014;6(225):225–9.
Zhao JL, Lou JF, Mou Y, Li PQ, Wu JY, Zhou LG. Diterpenoid tanshinones and phenolic acids from cultured hairy roots of salvia miltiorrhiza Bunge and their antimicrobial activities. Molecules. 2011;16(3):2259–67.
Tao S, Zheng Y, Lau A, Jaramillo MC, Chau BT, Lantz RC, et al. Tanshinone I activates the Nrf2-dependent antioxidant response and protects against As(III)-induced lung inflammation in vitro and in vivo. Antioxid Redox Signal. 2013;19(14):1647–61.
Mosaddik MA. In vitro cytotoxicity of tanshinones isolated from Salvia miltiorrhiza Bunge against P388 lymphocytic leukemia cells. Phytomedicine. 2003;10(8):682–5.
Wang C, Du X, Yang R, Liu J, Xu D, Shi J, et al. The prevention and treatment effects of tanshinone IIA on oestrogen/androgen-induced benign prostatic hyperplasia in rats. J Steroid Biochem Mol Biol. 2015;145:28–37.
Chen W, Lu Y, Chen G, Huang S. Molecular evidence of cryptotanshinone for treatment and prevention of human cancer. Anticancer Agents Med Chem. 2013;13(7):979–87.
ClinicalTrials.gov. Coronary heart disease. A service of the U.S. National Institutes of Health. https://clinicaltrials.gov/ct2/show/NCT01502943?term=NCT01502943&rank=1. Accessed 3 Apr 2015.
ClinicalTrials.gov. Pulmonary hypertension. A service of the U.S. National Institutes of Health. https://clinicaltrials.gov/ct2/show/NCT01637675?term=NCT01637675&rank=1. Accessed 3 Apr 2015.
ClinicalTrials.gov. Polycystic Ovary Syndrome. A service of the U.S. National Institutes of Health. https://clinicaltrials.gov/ct2/show/NCT01452477?term=NCT01452477&rank=1. Accessed 3 Apr 2015.
McGarvey DJ, Croteau R. Terpenoid metabolism. Plant Cell. 1995;7(7):1015–26.
Laule O, Furholz A, Chang HS, Zhu T, Wang X, Heifetz PB, et al. Crosstalk between cytosolic and plastidial pathways of isoprenoid biosynthesis in Arabidopsis thaliana. Proc Natl Acad Sci U S A. 2003;100(11):6866–71.
Gao W, Hillwig ML, Huang L, Cui G, Wang X, Kong J, et al. A functional genomics approach to tanshinone biosynthesis provides stereochemical insights. Org Lett. 2009;11(22):5170–3.
Guo J, Zhou YJ, Hillwig ML, Shen Y, Yang L, Wang Y, et al. CYP76AH1 catalyzes turnover of miltiradiene in tanshinones biosynthesis and enables heterologous production of ferruginol in yeasts. Proc Natl Acad Sci U S A. 2013;110(29):12108–13.
Zi J, Peters RJ. Characterization of CYP76AH4 clarifies phenolic diterpenoid biosynthesis in the Lamiaceae. Org Biomol Chem. 2013;11(44):7650–2.
Yang L, Ding G, Lin H, Cheng H, Kong Y, Wei Y, et al. Transcriptome analysis of medicinal plant Salvia miltiorrhiza and identification of genes related to tanshinone biosynthesis. PLoS One. 2013;8(11):e80464.
Ma Y, Yuan L, Wu B, Li X, Chen S, Lu S. Genome-wide identification and characterization of novel genes involved in terpenoid biosynthesis in Salvia miltiorrhiza. J Exp Bot. 2012;63(7):2809–23.
Bruckner K, Bozic D, Manzano D, Papaefthimiou D, Pateraki I, Scheler U, et al. Characterization of two genes for the biosynthesis of abietane-type diterpenes in rosemary (Rosmarinus officinalis) glandular trichomes. Phytochemistry. 2014;101:52–64.
Bozic D, Papaefthimiou D, Bruckner K, de Vos RC, Tsoleridis CA, Katsarou D, et al. Towards elucidating carnosic acid biosynthesis in lamiaceae: functional characterization of the three first steps of the pathway in salvia fruticosa and Rosmarinus officinalis. PLoS One. 2015;10(5):e0124106.
Yatagai M, Shirato T, Hayashi Y, Fukuhara N. Pisiferic acid, a new phenolic diterpene carboxylic acid from Chamaecyparis pisifera S. et Z. Mokuzai Gakkaishi. 1978;24:3.
Yatagai M, Takahashi T. New diterpenes from chamaecyparis-pisifera. Phytochemistry. 1980;19(6):1149–51.
Dimayuga RE, Garcia SK, Nielsen PH, Christophersen C. Traditional medicine of Baja California Sur (Mexico). III. Carnosol: a diterpene antibiotic from Lepechinia hastata. J Ethnopharmacol. 1991;31(1):43–8.
Hayashi T, Handa T, Ohashi M, Kakisawa H, Hsu H-Y, Chen YP. The structure of salviol, a new phenolic diterpene. J Chem Soc D. 1971;11:541.
Al-Hazimi HMG, Miana GA, Deep MSH. Terpenoids from Salvia lanigera. Phytochemistry. 1987;26(4):1091–3.
Fischedick JT, Standiford M, Johnson DA, Johnson JA. Structure activity relationship of phenolic diterpenes from Salvia officinalis as activators of the nuclear factor E2-related factor 2 pathway. Bioorg Med Chem. 2013;21(9):2618–22.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–52.
Spyropoulou EA, Haring MA, Schuurink RC. RNA sequencing on Solanum lycopersicum trichomes identifies transcription factors that activate terpene synthase promoters. BMC Genomics. 2014;15:402.
Kampranis SC, Ioannidis D, Purvis A, Mahrez W, Ninga E, Katerelos NA, et al. Rational conversion of substrate and product specificity in a Salvia monoterpene synthase: structural insights into the evolution of terpene synthase function. Plant Cell. 2007;19(6):1994–2005.
Jin J, Panicker D, Wang Q, Kim MJ, Liu J, Yin JL, et al. Next generation sequencing unravels the biosynthetic ability of spearmint (Mentha spicata) peltate glandular trichomes through comparative transcriptomics. BMC Plant Biol. 2014;14:292.
Peters RJ. Two rings in them all: the labdane-related diterpenoids. Nat Prod Rep. 2010;27(11):1521–30.
Ignea C, Trikka FA, Kourtzelis I, Argiriou A, Kanellis AK, Kampranis SC, et al. Positive genetic interactors of HMG2 identify a new set of genetic perturbations for improving sesquiterpene production in Saccharomyces cerevisiae. Microb Cell Fact. 2012;11:162.
Trikka FA, Nikolaidis A, Athanasakoglou A, Andreadelli A, Ignea C, Kotta K, et al. Iterative carotenogenic screens identify combinations of yeast gene deletions that enhance sclareol production. Microb Cell Fact. 2015;14(1):60.
Ignea C, Trikka FA, Nikolaidis AK, Georgantea P, Ioannou E, Loupassaki S, et al. Efficient diterpene production in yeast by engineering Erg20p into a geranylgeranyl diphosphate synthase. Metab Eng. 2015;27:65–75.
Muramatsu M, Ohto C, Obata S, Sakuradani E, Shimizu S. Alkaline pH enhances farnesol production by Saccharomyces cerevisiae. J Biosci Bioeng. 2009;108(1):52–5.
Zhou YJ, Gao W, Rong Q, Jin G, Chu H, Liu W, et al. Modular pathway engineering of diterpenoid synthases and the mevalonic acid pathway for miltiradiene production. J Am Chem Soc. 2012;134(6):3234–41.
Ignea C, Ioannou E, Georgantea P, Loupassaki S, Trikka FA, Kanellis AK, et al. Reconstructing the chemical diversity of labdane-type diterpene biosynthesis in yeast. Metab Eng. 2015;28:91–103.
Nelson D, Werck-Reichhart D. A P450-centric view of plant evolution. PlantJ. 2011;66(1):194–211.
Lamb DC, Waterman MR. Unusual properties of the cytochrome P450 superfamily. Philos Trans R Soc Lond B Biol Sci. 2013;368(1612):20120434.
Nelson DR. The cytochrome p450 homepage. Hum Genomics. 2009;4(1):59–65.
Chen H, Wu B, Nelson DR, Wu K, Liu C. Computational identification and systematic classification of novel cytochrome P450 genes in salvia miltiorrhiza. PLoS One. 2014;9(12):e115149.
Takahashi S, Yeo YS, Zhao Y, O’Maille PE, Greenhagen BT, Noel JP, et al. Functional characterization of premnaspirodiene oxygenase, a cytochrome P450 catalyzing regio- and stereo-specific hydroxylations of diverse sesquiterpene substrates. J Biol Chem. 2007;282(43):31744–54.
Hamberger B, Bak S. Plant P450s as versatile drivers for evolution of species-specific chemical diversity. Philos Trans R Soc Lond B Biol Sci. 2013;368(1612):20120426.
Ehlting J, Hamberger B, Million-Rousseau R, Werck-Reichhart D. Cytochromes P450 in phenolic metabolism. Phytochem Rev. 2006;5(2–3):239–70.
Kobayashi K, Nishino C. Biological activities of pisiferic acid and O-methyl-pisiferic acid. Agric Biol Chem. 1986;50(9):3.
Kobayashi K, Nishino C, Tomita H, Fukushima M. Antifungal activity of pisiferic acid-derivatives against the rice blast fungus. Phytochemistry. 1987;26(12):3175–9.
Aburai N, Yoshida M, Ohnishi M, Kimura K. Pisiferdiol and pisiferic acid isolated from Chamaecyparis pisifera activate protein phosphatase 2C in vitro and induce caspase-3/7-dependent apoptosis via dephosphorylation of Bad in HL60 cells. Phytomedicine. 2010;17(10):782–8.
Mokoka TA, Peter XK, Fouche G, Moodley N, Adams M, Hamburger M, et al. Antileishmanial activity of 12-methoxycarnosic acid from Salvia repens Burch. ex Benth. (Lamiaceae). S Afr J Bot. 2014;90:93–5.
Campo JD, Nguyen-The C, Sergent M, Amiot MJ. Determination of the most bioactive phenolic compounds from rosemary against Listeria monocytogenes: influence of concentration, pH, and NaCl. J Food Sci. 2003;68(6):2066–71.
Munagala R, Aqil F, Jeyabalan J, Gupta RC. Tanshinone IIA inhibits viral oncogene expression leading to apoptosis and inhibition of cervical cancer. Cancer Lett. 2015;356(2 Pt B):536–46. doi:10.1016/j.canlet.2014.09.037.
Wang W, Wang Y, Zhang Q, Qi Y, Guo D. Global characterization of Artemisia annua glandular trichome transcriptome using 454 pyrosequencing. BMC Genomics. 2009;10:465.
McDowell ET, Kapteyn J, Schmidt A, Li C, Kang JH, Descour A, et al. Comparative functional genomic analysis of Solanum glandular trichome types. Plant Physiol. 2011;155(1):524–39.
Bak S, Beisson F, Bishop G, Hamberger B, Hofer R, Paquette S, et al. Cytochromes p450. Arabidopsis Book. 2011;9:e0144.
Chen F, Tholl D, Bohlmann J, Pichersky E. The family of terpene synthases in plants: a mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 2011;66(1):212–29.
Boutanaev AM, Moses T, Zi J, Nelson DR, Mugford ST, Peters RJ, et al. Investigation of terpene diversification across multiple sequenced plant genomes. Proc Natl Acad Sci U S A. 2015;112(1):E81–8.
Hofer R, Boachon B, Renault H, Gavira C, Miesch L, Iglesias J, et al. Dual function of the cytochrome P450 CYP76 family from Arabidopsis thaliana in the metabolism of monoterpenols and phenylurea herbicides. Plant Physiol. 2014;166(3):1149–61.
Bohlmann J, Gesell A, Blaukopf M, Madilao L, Yuen MM, Withers SG, et al. The gymnosperm cytochrome P450 CYP750B1 catalyzes stereospecific monoterpene hydroxylation of (+)-sabinene in thujone biosynthesis in Thuja plicata. Plant Physiol. 2015;168(1):94–106. doi:10.1104/pp.15.00315.
Yerger EH, Grazzini RA, Hesk D, Cox-Foster DL, Craig R, Mumma RO. A rapid method for isolating glandular trichomes. Plant Physiol. 1992;99(1):1–7.
Iseli C, Jongeneel CV, Bucher P. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol. 1999;7:138–48.
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–6.
Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34(Web Server issue):W293–7.
Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4(4):406–25.
Felsenstein J. Confidence-limits on phylogenies - an approach using the bootstrap. Evolution. 1985;39(4):783–91.
Nei M, Kumar S. Molecular evolution and phylogenetics. Oxford: Oxford University Press; 2000.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2- CT method. Methods. 2001;25(4):402–8.
Acknowledgements
This work was funded by the General Secretariat of Research and Technology (GSRT) grant 09-SYN-23-879, grant SEE-ERA. NET PLUS: ERA 64/01 and grant KRIPIS (grant no. MIS 448840). The authors would like to thank Prof. David Nelson for kindly annotating S. pomifera cytochrome P450s.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
FAT conducted some experimental work and is responsible for the drafting of the paper. AN, CI, LAT, EAS conducted the experimental work, AT conducted the bioinformatics analysis. VR, EI and AKK assisted in the data analysis and review of the manuscript. DB assisted with the development of yeast expression vectors. AA and SCK assisted in the data analysis, in experimental design and review of the manuscript. AMM is responsible for the design and data analysis as well as review of the paper. All authors read and approved the manuscript.
Additional files
Additional file 1: Figure S2.
1H-NMR spectrum of 2α-hydroxy-O-methyl-pisiferic acid (compound 1). (PDF 114 kb)
Additional file 2: Figure S3.
13C-NMR spectrum of 2α-hydroxy-O-methyl-pisiferic acid (compound 1). (PDF 271 kb)
Additional file 3: Figure S4.
1H-NMR spectrum of pisiferic acid (compound 2). (PDF 116 kb)
Additional file 4: Figure S5.
1H-NMR spectrum of O-methyl-pisiferic acid (compound 3). (PDF 144 kb)
Additional file 5: Figure S6.
1H-NMR spectrum of 12-methoxycarnosic acid (compound 4). (PDF 111 kb)
Additional file 6: Figure S7.
1H-NMR spectrum of carnosol (compound 5). (PDF 116 kb)
Additional file 7: Figure S8.
1H-NMR spectrum of salviol (compound 6). (PDF 113 kb)
Additional file 8: Figure S1.
(A) Distribution of S. pomifera contigs according to their length and (Β) Distribution of S. pomifera clusters of contigs and unigenes according to their length. (PDF 111 kb)
Additional file 9: Table S1.
Thirty-five most expressed genes/contigs in M. spicata trichomes based on their FPKM values produced by the RSEM software. In the last column their top hit is referred, produced by BLAST searches in the NR database. The length of the contigs is sometimes not an integer number due to the fact that one contig may correspond to several transcripts of diverse length. The RSEM software was run on the data produced by the transcriptome sequencing of M. spicata trichomes by Jin et al. [37]. (PDF 103 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Trikka, F.A., Nikolaidis, A., Ignea, C. et al. Combined metabolome and transcriptome profiling provides new insights into diterpene biosynthesis in S. pomifera glandular trichomes. BMC Genomics 16, 935 (2015). https://doi.org/10.1186/s12864-015-2147-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-015-2147-3