
Abstract
Background
Vitiligo is an auto-immune progressive depigmentation disorder of the skin due to loss of melanocytes. Genetic risk is one of the important factors for development of vitiligo. Preponderance of vitiligo in certain ethnicities is known which can be analysed by understanding the distribution of allele frequencies across normal populations. Earlier GWAS identified 108 risk alleles for vitiligo in Europeans and East Asians. In this study, 64 of these risk alleles were used for analysing their enrichment and depletion across populations (1000 Genomes Project and IndiGen) with reference to 1000 Genomes dataset. Genetic risk scores were calculated and Fisher’s exact test was performed to understand statistical significance of their variation in each population with respect to 1000 Genomes dataset as reference. In addition to SNPs reported in GWAS, significant variation in allele frequencies of 1079 vitiligo-related genes were also analysed. Two-tailed Chi-square test and Bonferroni’s multiple adjustment values along with fixation index (≥ 0.5) and minimum allele frequency (≥ 0.05) were calculated and used to prioritise the variants based on pairwise comparison across populations.
Results
Risk alleles rs1043101 and rs10768122 belong to 3 prime UTR of glutamate receptor gene SLC1A2 are found to be highly enriched in the South Asian population when compared with the ‘global normal’ population. Intron variant rs4766578 (ATXN2) was found to be deleted in SAS, EAS and AFR and enriched in EUR and AMR1. This risk allele is found to be under positive selection in SAS, AMR1 and EUR. From the ancillary vitiligo gene list, nonsynonymous variant rs16891982 was found to be enriched in the European and the Admixed American populations and depleted in all others. rs2279238 and rs11039155 belonging to the LXR-α gene involved in regulation of metalloproteinase 2 and 9 (melanocyte precursors) were found to be associated with vitiligo in the North Indian population (in earlier study).
Conclusion
The differential enrichment/depletion profile of the risk alleles provides insight into the underlying inter-population variations. This would provide clues towards prioritisation of SNPs associated with vitiligo thereby elucidating its preponderance in different ethnic groups.
Similar content being viewed by others

Background
Vitiligo is an acquired pigmentation disorder characterised by the loss of functional melanocytes resulting in the development of depigmented macules and patches on the skin [1]. It affects ∼ 0.5-2% of the world’s population and is observed to have the highest prevalence of ∼ 8.8% in few states of India [2,3,4]. The exact etiology of vitiligo remains unknown and involves multiple factors such as genetic, immunological, and environmental [5]. The autoimmune nature of this polygenic disorder, combined with involvement of multiple susceptibility loci with different degrees of penetrance cannot be explained by Mendelian genetics [6]. Vitiligo is found to be commonly associated with other autoimmune diseases viz., atopic dermatitis, alopecia areata and psoriasis [7]. Genome wide association studies (GWAS) have the potential to identify multiple loci associated with such complex diseases [8]. Polygenic risk scores aggregate the cumulative effects of multiple identified variants from GWAS responsible for a particular disease in a sampled population, and estimate an individual’s genetic predisposition towards a complex trait [9, 10]. Genetic risk score calculates the probability of an individual for predisposition towards developing the disease [11].
Several susceptibility genes associated with vitiligo, including HLA class I and II genes, NLRP1, PTPN22, and FOXP3 have been identified using cohort-based GWAS [6]. Studies of allele frequency distribution of SNPs belonging to genes related to pigmentation, viz., HERC2, MC1R have also been found to be associated with susceptibility to vitiligo [12].
Majority of GWAS studies for vitiligo are limited to Caucasian and East Asian populations [6, 13,14,15]. The risk alleles identified in GWA studies for diseases such as age-related macular disorders, obesity and psoriasis have been used to probe prevalence in normal individuals belonging to other population groups [16,17,18,19]. Hence, genetic variation of risk alleles across populations can be one of the factors to understand disease prevalence [20].
National Human Genome Research Institute-European Bioinformatics Institute (NHGRI-EBI) GWAS catalogue lists 108 loci to be associated with the prevalence of vitiligo [21]. In this study we aim to investigate the association of genetic risk scores of vitiligo risk alleles in normal individuals across different ethnic populations. As most GWAS pertain to European and East Asian ethnicities, the objective of the current work is to estimate genetic variation of these vitiligo associated risk alleles across 5 super-populations and 26 ethnic populations belonging to the Phase 3 data of the 1000 Genomes Project (1KGP) in addition to 1029 individuals from the IndiGen Project [22]. Such comparative analysis may aid in prioritisation of risk alleles to analyse diseased cohorts belonging to other ethnicities. Additionally, allele frequency variation of prioritised SNPs belonging to vitiligo associated genes were also analyzed across populations in order to obtain a comprehensive understanding of disease prevalence as GWAS for vitiligo are scarce.
Materials and methods
Datasets
High coverage (30x) data belonging to 3202 samples (∼ 2 TB) from 1000 Genome Project comprising of 5 super-populations (African (AFR), Ad Mixed American (AMR), East Asian (EAS), European (EUR), and South Asian (SAS)) was used for the study [23]. Ad Mixed Americans were categorized into two sub-populations, ‘AMR1 (European derived, consisting of CLM and PUR populations) and AMR2 (Latino, comprising MXL and PEL populations) based on ancestry [24]. Allele frequency variations across populations pertaining to each super-population were also calculated using bcftools v1.9 [25]. To improve the representation of the genetic diversity from the Indian subcontinent, data representing genetic variation from 1029 healthy individuals across 27 states of the Indian subcontinent were extracted from the IndiGen database [22, 26]. Henceforth, these super-populations would be referred to as “7 super-populations” (Supplementary Table 01).

Flow-chart for population-wide estimation of enrichment and depletion of risk alleles and vitiligo associated genes
Risk alleles from GWAS catalog
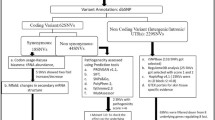
A total of 108 risk alleles associated with vitiligo from the NHGRI-EBI Catalog [21] of human GWAS were obtained. These risk alleles were obtained from eight studies belonging to European [13, 15, 27,28,29,30,31] and four studies belonging East Asian [32,33,34,35] ethnicities. Filtering based on odds ratio, risk allele frequency, redundant alleles and alleles absent in 1KGP, resulted in a total of 64 SNPs (62 and 2 SNPs belonging to European and East Asian GWAS respectively) belonging to 55 genes and were termed as ‘risk alleles’ (Supplementary Table 01) (Fig. 1).
Compilation of vitiligo-associated genes
Databases such as Open Targets [36, 37] and VitVar [38, 39] were used to fetch 1079 vitiligo-associated genes and their corresponding 3,318,351 SNPs were derived from the 1KGP data.
Genetic risk score calculation
Genetic risk score quantifies an individual’s genetic predisposition to a particular trait or disease based on their genetic information [11]. The following equation was used to calculate the genetic risk score for vitiligo, as described by [16].
where, I is the number of vitiligo SNPs and Xi is copies of risk alleles at ith SNP.
Bias analysis
Bias analysis was performed to eliminate concerns regarding genetic risk scores being biased towards the European and East Asian populations as the risk alleles used in this study have been derived from these populations. Hence, in order to understand the risk allele bias towards the said populations, three different sets of risk alleles (pertaining to the GWAS) were used for determining bias. Set1 includes 2 risk alleles from EAS and 8 random risk alleles from EUR; Set2 has 10 risk alleles from Set1 along with 5 risk alleles from EUR and Set3 has 10 random risk alleles from only EUR.
Enrichment and depletion analysis
Fisher’s exact test was carried out to assess the effect of risk alleles in 7 super-populations and 26 sub-populations which were compared against the global 1KGP (referred to as ‘global normal’). Fisher’s exact test was used to determine the enrichment and depletion of risk alleles in each population belonging to 1KGP and IndiGen as compared to the global normal risk allele score. This test is carried out by constructing a 2 × 2 contingency table where rows represent two different populations and columns represent presence and absence of alleles at given loci in two populations which are being compared. Log10 transformed adjusted p-value (≤ 0.05) was used for calculating depletion, whereas, to calculate enrichment, negative of the log10 transformed adjusted p-value (≤ 0.05) was used [17]. Seaborn and Matplotlib were used for generating heat maps [40, 41].
Estimation of significant SNPs
Additional SNPs belonging to vitiligo-associated genes were prioritised using two-tailed Chi-square test and Bonferroni’s multiple adjustment values along with fixation index (Fst≥0.5) and Minimum Allele Frequency (MAF ≥ 0.05) [42]. Chi-square test of significance quantifies the difference between observed and expected frequencies using the following formula:
Fixation Index (Fst) is a conventional metric used to detect allele frequency variation at population level [43]. Fst estimates the proportion of genetic variation at a specific locus between two populations. Fst is governed by common alleles (MAF) and size of each population.
Annotation of risk alleles
Vannoportal, that provides in-depth annotation of variants (risk alleles) was used to analyse enriched/depleted alleles in order to ascertain if they are under positive selection [44]. The tests used for positive selection include χ2 test for Hardy-Weinberg equilibrium, Difference of Derived Allele Frequency, Fixation Index (Cockerham & Weir method), Integrated Haplotype score and Tajima’s D. Criterion of ≥ 3 tests indicating positive selection was used.
Results
SNPs identified from GWAS catalogue
A non-redundant set of 64 risk alleles (from EUR and EAS) belonging to 55 genes (Supplementary Table 01) were analysed. Vitiligo-associated genetic risk scores based on these risk alleles were calculated and their distribution across different super-populations were studied (Supplementary Fig. 01). As evident from this figure, the genetic risk scores (from GWAS) showed a similar range of distribution across the global population (normal samples). Hence, this suggests that the risk alleles obtained from the European and Han Chinese GWAS are suitable to investigate vitiligo prevalence in other population groups (Supplementary Fig. 01). Further in SAS, EUR, AMR1 and IndiGen the range of quartiles (Q1-Q3) possess similar values and EAS, AMR2 and AFR have marginally wider range.

(A) Genetic Risk score obtained from GWAS for vitiligo across super-populations of 1000 Genomes and IndiGen projects. (B) Genetic Risk score obtained from GWAS for vitiligo across sub-populations of 1000 Genomes project. The dotted line represents the median of ‘global normal’ risk score
Genetic risk scores for SNPs related to vitiligo
The distribution of genetic risk scores for the six super-populations and 26 sub-populations belonging to 1KGP as well as IndiGen are shown in Fig. 2. Similar trends in the distribution of genetic risk scores were observed in SAS and IndiGen. The genetic risk score distribution of the AFR super-population tends towards upper quartile (Q2 and Q3) as compared to ‘global normal’, whereas, AMR2 tends towards lower quartile (Q1 and Q2). In case of SAS, EUR, EAS, AMR1 and IndiGen, genetic risk score was found to be in the range of ‘global normal’ distribution (Fig. 2).
The genetic risk score is observed to follow a similar distribution for every sub-population pertaining to each super-population (Fig. 3). The score varies from 0.01 (rs117744081 belonging to gene CPVL) to 0.988 (rs6059655 belonging to gene RALY) (Fig. 3).

Heat map of genetic risk scores of vitiligo-associated risk alleles plotted for 1000 Genome and IndiGen projects
Bias analysis
Comparison of genetic risk score distribution (based on all 64 risk alleles) of EUR with EAS revealed an enriched median of genetic risk score in EUR (Fig. 2).

Distribution of genetic risk scores between EUR and EAS populations with different risk allele sets
In order to further validate this observation, three random sub-sets of risk alleles were sampled as described in the Methods section. The genetic risk score (median value) of these sub-sets for EAS and EUR along with the complete 64 risk alleles ranges between 0.35 and 0.39 and 0.41–0.46 respectively (Fig. 4; Table 1).
Enriched and depleted risk alleles across super-populations
Based on enrichment and depletion values of the risk alleles, two distinct clusters were observed with AFR being the outermost branch (cluster 1) (Fig. 5). In cluster 2, all other populations group together, in which EAS forms a separate branch (sub-cluster 2.1). EUR, AMR1, AMR2, SAS along with IndiGen cluster together (sub-cluster 2.2). AMR1 was found to group with both EUR and SAS, whereas, AMR2 clustered along with the South Asian populations (Fig. 5). Of the 64 risk alleles, 57 are either enriched or depleted or satisfy both conditions in at least one population, whereas the remaining seven risk alleles were found to have no significant change and hence were excluded from the heatmap (Fig. 5).

Heatmap of vitiligo-associated risk alleles from 1000 Genomes and IndiGen projects. Red and blue colours represent enriched and depleted effect alleles respectively
The risk alleles which contribute to the clustering include, rs10876864 (TF binding site variant of IKZF4, SUOX and involved in disease like asthma, allergic diseases, squamous cell carcinoma, vitiligo), rs2687812 (intron variant of TG and involved in vitiligo) and rs34346645 (intron variant of FOXP1 involved in vitiligo), rs9611565 (intron variant of TEF involved in vitiligo) which are found to be highly enriched only in AFR and EAS populations respectively. Similarly, rs71508903 (intron variant of ARID5B involved in rheumatoid arthritis, hypothyroidism and vitiligo), rs2304206 (5 prime UTR variant of BCL2L12, IRF3 involved in vitiligo), rs4409785 (intron variant of LNCRNA-IUR, FAM76B and involved in rheumatoid arthritis, multiple sclerosis, autoimmune thyroid disease, basal cell carcinoma, vitiligo), rs1043101, rs10768122 (3 prime UTR variant of SLC1A2 involved in vitiligo) are found to be depleted in AFR. rs9926296 (intron variant of FANCA and involved in vitiligo) is found to be depleted only in EAS.
Risk alleles rs1043101 and rs10768122 are found to be highly enriched in SAS (BEB, GIH and ITU) while depleted in AFR. Risk alleles rs301807 and rs4908760 (intron variants of RERE) were observed to be highly enriched in SAS (GIH and ITU) and depleted in EAS. Risk alleles rs1393350 (intron variant of TYR) and rs1126809 (missense variant of TYR) are observed to be highly enriched in ITU (SAS) and moderately enriched in AFR and EAS while being depleted in EUR. rs1129038 (3 prime UTR variant from HERC2) is found to be enriched in SAS (STU, ITU) along with all sub-populations of EAS and AFR, however it was found to be depleted in EUR. rs4409785 (intron variant of FAM76B) is observed to be enriched only in STU (SAS) and depleted in AFR. rs5952553 (intergenic variant GAGE1 - VDAC1P2) is found to be enriched in SAS (STU, BEB) and EUR while it was depleted in IndiGen and AFR.
Conversely, risk alleles rs4766578 and rs10774624 (intron variants of ATXN2) were found to be depleted in STU (SAS), EAS and AFR while these were enriched in EUR. Risk allele rs10774624 was found to be under positive selection in EUR (Supplementary Table 01). Risk allele rs2017445 (intron variant of IKZF4) is depleted in IndiGen, AFR and found to be enriched in IBS (EUR). rs8083511 (intron variant of TNFRSF11A) is depleted in PJL (SAS) and enriched in EAS and AFR.
Enrichment/depletion patterns of risk alleles belonging to cluster 2.2 (EUR, SAS, AMR1, AMR2 and IndiGen) have been analysed in detail as higher prevalence of vitiligo has been reported earlier in South Asian populations [2] (Table 2). The details of the enrichment/depletion of risk alleles in SAS are given below.
Risk alleles found to be depleted in SAS
Risk allele rs4766578 (intron variant of ATXN2) is found to be depleted in SAS, EAS and AFR and enriched in EUR and AMR1. This risk allele is found to be under positive selection in SAS, AMR1 and EUR (Supplementary Table 01). Apart from being associated with vitiligo susceptibility, this SNP is also found to be a risk allele for diastolic blood pressure and coronary artery disease in Europeans [6, 28, 45, 46]. Recently this SNP has been associated with a role in positive selection by regulating ALDH2 gene expression that protects cells from acetaldehyde toxicity in the European population [47]. rs10774624 is found to be depleted in SAS, EAS and AFR and enriched in EUR and AMR1. This risk allele is found to be associated with rheumatoid arthritis in Pakistanis and in severe COVID-19 patients from the UK Biobank cohort [48, 49].
Risk alleles found to be enriched in SAS
Risk alleles rs1043101, rs10768122 (3 prime UTR variants of SLC1A2) and rs4409785 (intron variant of FAM76B) were found to be enriched in SAS and depleted in AFR. These SNPs have been associated with other disorders apart from vitiligo susceptibility. Risk allele rs1043101, is found to be associated with bipolar disorder and schizophrenia in European populations [50]. rs4409785 has been found to be associated with Graves’ disease in Europeans, multiple autoimmune diseases in European Americans, rheumatoid arthritis in Pakistanis and myasthenia gravis in cohort of UK Biobank [48, 51,52,53].
rs1129038 (3 prime UTR variants of HERC2) is found to be enriched in SAS, EAS, AFR and depleted in EUR. This risk allele is predicted to be under positive selection in EUR (Supplementary Table 01). It is found to be associated with eye colour in Europeans, skin pigmentation in Brazilians, susceptibility to myopia in UK Biobank, uveal melanoma risk in Americans [54,55,56,57,58,59,60]. rs4908760 (intron variant of RERE) is found to be enriched in SAS whereas it is depleted in EAS, AFR and has association with smoking behaviour-related traits [61]. rs301807 (intron variant of RERE) is found to be enriched in SAS and depleted in EAS. rs5952553 (intergenic variant of GAGE1 - VDAC1P2) is found to be enriched in SAS and EUR while being depleted in AFR. A non-synonymous variant rs1126809 (TYR) is found to be highly enriched in ITU (SAS) and moderately enriched in AFR and EAS while being depleted in EUR. This SNP has correlation with longitude, latitude, sunshine hours in the Chinese population, cancer among the South-East Asians, brown eye colour in Europeans and increased risk of melanoma in south Brazilians [57, 62,63,64]. rs1393350 (TYR) is found to be highly enriched in ITU (SAS) and moderately enriched in AFR and EAS while depleted in EUR. It is found to be associated with eye and hair colour in the Slovenians, skin colour in the Europeans and eye colour in the Pakistanis [65,66,67,68,69].
Similarly, the details of enriched and depleted risk alleles in EUR, EAS, AMR1, AMR2 and AFR are provided in Supplementary document S1.
Risk alleles under positive selection
Apart from the above-mentioned risk alleles that are found to be enriched/depleted in SAS and under positive selection, few more risk alleles are also observed to meet the selection criterion. rs1635168 (intron variant of HERC2) in EUR, rs9926296 (intron variant of FANCA) in EAS, rs2304206 (5 prime UTR variant of IRF3) and rs2111485 (intron variant of IFIH1) in AFR are depleted and found to be under positive selection (Supplementary Table 01).
rs2111485 in EUR and rs10200159 (non-coding transcript exon variant of PPP4R3B), rs10876864 (TF binding site variant of IKZF4), rs2687812 (intron variant of TG), rs12771452 (intron variant of CASP7) and rs11079035 (intron variant of RAB5C) in AFR are enriched and found to be under positive selection (Supplementary Table 01).
Overall, the highest number of enriched risk alleles are observed in AFR super-population followed by EAS and EUR (Table 3). It is interesting to note that none of the risk alleles are enriched in IndiGen. The highest percentage of depleted risk alleles are observed in EAS followed by AMR2 and AFR.
Analysis of ancillary vitiligo-associated genes
A comprehensive list of 1079 genes associated with vitiligo, were compiled from two databases as mentioned in the Methods section. These genes include 3,318,351 SNPs from the 1KGP data with GRCh38 as reference. Among these SNPs, 3,195,407 were found to be biallelic. Of these, 143,562 SNPs satisfied the MAF ≥ 0.05 in all populations. To understand the preponderance of vitiligo in SAS, 197,543 SNPs with MAF ≥ 0.05 were further extracted (Supplementary Table 02). Significant allele frequency variation was observed in 117 SNPs belonging to 44 genes (Supplementary Fig. 02). MAF ≥ 0.05 ensured that the selected SNPs were relatively common in the population with Fst ≥ 0.5. To account for multiple testing, Bonferroni’s correction, a stringent method for controlling the family-wise error rate was used, which further validated the significance of prioritised SNPs. It is envisaged that these SNPs represent a subset of genetic variants that warrant special attention, as they are likely to play a critical role in vitiligo. Enrichment and depletion analysis of these SNPs was performed as described previously in the Methods section for which allele frequencies were retrieved from 1KGP (Supplementary Fig. 03). Both the distribution patterns (allele frequency distribution and enrichment/depletion of alleles) reveal distinct population-specific clustering (Supplementary Figs. 02 and 03).
Functional annotation of the obtained SNPs
Of the 117 SNPs, only one SNP is non-synonymous, nine are downstream, 11 are upstream and 96 are intron variants (Supplementary Table 01). All 117 SNPs are either enriched or depleted for one or more populations. Missense variant rs16891982 belonging to SLC45A2 gene is enriched in EUR and AMR1 super-populations and depleted in all others. This SNP has been reported to be associated with skin pigmentation disorder [70]. Enrichment analysis shows that the maximum enriched SNPs are in EAS followed by AFR, AMR2, EUR, SAS, AMR1 and IndiGen (Supplementary Fig. 03).
Discussion
There exist varying reports pertaining to the preponderance of vitiligo across populations [71, 72]. In this study, we compared the allele frequencies of vitiligo-associated risk alleles across different populations to gain insight into the role of genetic variation in estimating disease risk. The enrichment and depletion patterns of 64 vitiligo-associated risk alleles quantifies genetic variations and its prevalence in populations (from 1KGP and IndiGen) as compared to ‘global normal’. Several studies have reported variants identified through GWAS to be multi-ethnically reproducible [17, 73]. GWAS pertaining to vitiligo are majorly confined to European populations [6]. The calculated genetic risk scores in this study obtained across super-populations of 1KGP reveal similar distribution, thereby suggesting the relevance of these risk alleles to be analysed across ethnicities (Supplementary Fig. 01). Further, bias analysis unveiled the effect of allele frequencies to be a significant factor that determines the outcome of genetic risk scores and not the mere occurrence of vitiligo-associated risk alleles.
Population-specific genetic risk scores of few alleles (which are also associated with other autoimmune diseases (Table 2)) were observed to be higher in normal populations when compared against the ‘global normal’. For instance, rs4766578 belonging to gene ATXN2 (contributing towards the pathogenesis of vitiligo, cardiovascular diseases and involved in haematological parameters governing platelet counts and volume) and rs10774624 (involved in rheumatoid arthritis, preeclampsia, heart diseases) was found to be highly enriched in EUR and AMR1 populations and depleted in AFR and EAS thereby indicating the frequent occurrence of these alleles in the populations belonging to European ancestry [6, 24, 45, 46, 74, 75]. Both these risk alleles are also found to be under positive selection (Supplementary Table 01). Conventionally in complex diseases minor alleles are associated with disease risk [76]. Many of the risk alleles chosen in this study have reported odds-ratio of ∼[1-1.1] and hence their allele frequencies are close to normal population (Supplementary Table 01).
The higher proportion of enriched alleles in AFR obtained in this study corroborates with meta-analysis of vitiligo prevalence [77]. Lower percentage of enriched risk alleles in SAS and IndiGen observed in our study does not substantiate reports of higher prevalence of vitiligo in this region [2]. Further, enrichment and depletion analysis revealed AFR and EAS to form separate clusters indicating their ethnicity-specific genetic variation. This clustering can be explained by the ‘Out of Africa’ hypothesis, which proposes East Africa being the cradle for the origin of modern humans [78]. Earlier studies have shown the African populations to be genetically most diverse [79]. Genetic diversity studies predicted the origin of humans along with their migration routes to subsequent human expansion from East Africa [80]. The heterozygosities and the observed patterns of genetic diversities existing in the global populations are explained due to this expansion originating from Africa. With the increasing geographic distance, a decrease in genetic similarity is observed between the populations due to geographic isolation (measured as FST), genetic drift and natural selection [81]. The ‘trellis model’ based on genetic distances existing among human populations proposes Africans and Asians to be genetically distant, which is also reiterated in our study, wherein AFR and EAS are found to cluster independently (Fig. 5) [78]. These prioritized risk alleles may be investigated further while performing population-specific vitiligo related GWA studies.
Clustering of AMR1 with EUR explains its European ancestry [74]. The observed grouping of CLM (belonging to AMR1 - Admixed Americans from the Colombian/Peruvian/Mexican group) with AMR2 supports the reports suggesting CLM to be closely related to the Peruvian (PEL) population [74]. IndiGen and PJL (belonging to SAS) cluster together and their enrichment and depletion profiles were found to be similar to the ‘global normal’. SAS sub-populations (except for PJL) are observed to cluster together and are in close proximity to AMR2 along with CLM (belonging to AMR1). This clustering may be due to a large number of risk alleles having similar scores as ‘global normal’ (Fig. 5). Overall, the clustering of SAS, IndiGen with EUR can be attributed to majority of the gene pool belonging to the ancient north Indian ancestry which is known to be genetically close to Europeans [75, 82].
It should be mentioned that as vitiligo is a polygenic disorder, enrichment/depletion of individual risk alleles may have limited scope to explain aetiology. Estimation of polygenic risk score may provide a better insight into disease susceptibility. Additionally, the pleiotropic effect of risk alleles may also play a role in their selection especially in auto immune diseases [83].
As limited GWA studies are available for populations belonging to other ethnicities apart from EAS and EUR, additional SNPs belonging to vitiligo-associated disorder were also studied. Allele frequency distribution pattern along with enrichment and depletion patterns revealed distinct population-specific clustering (Supplementary Figs. 02 and 03). Individuals from the IndiGen cohort are observed to cluster along with the South Asian population from 1KGP indicating similar trends in allele frequencies (Supplementary Figs. 02 and 03). rs2279238 and rs11039155 belonging to the LXR-α gene involved in regulation of metalloproteinase 2 and 9 (melanocyte precursors) are associated with vitiligo risk in the North Indian population [84]. The allele frequency distribution of rs2279238 and rs11039155 was found to be 0.2 and 0.1 respectively in populations belonging to the SAS and IndiGen cohort, which is in agreement to earlier reports from India (Supplementary Table 02) [84]. This elucidates the importance of identification and study of other functional SNPs related to genes involved in vitiligo. Hence, such large-scale comparisons of allele frequencies across different population groups can provide additional markers for population-specific GWA studies.
It needs to be noted that interpretation of the role of GWAS risk alleles across population may lead to over/under-estimation of disease risk and hence adequate caution has to be taken [85]. Overall, the prioritized risk alleles and additional SNPs identified in this study can play a role in designing precision public health initiatives for tackling vitiligo. Such population-specific variants may help in screening for vitiligo prevalence.
Conclusion
The comprehensive analysis of vitiligo-associated risk alleles for enrichment and depletion across diverse populations reveals intriguing patterns. Notably, many variants are observed to be differentially enriched/depleted in various populations which is indicative of intricate inter-population variations. The risk alleles were obtained primarily from the EUR population, highlighting the need for expanded investigations across varied ethnicities to gain comprehensive insights. These risk alleles, linked to vitiligo, are not only associated with the disease but are also implicated in other autoimmune conditions, emphasising their role in a broader disease spectrum. Additional sets of vitiligo-associated SNPs identified based on allele frequency variation can complement GWAS. These findings collectively emphasise the importance of considering genetic diversity and population-specific factors when evaluating disease risk.
Data availability
Data analyzed in this study is provided as additional Supplementary material.
Abbreviations
- SNPs:
-
Single nucleotide polymorphisms
- MAF:
-
Minimum Allele Frequency
- GWAS:
-
Genome wide association studies
- 1KGP:
-
1000 Genomes Project
- NHGRI-EBI:
-
National Human Genome Research Institute-European Bioinformatics Institute
- EAS:
-
East Asian Ancestry
- EUR:
-
European Ancestry
- AFR:
-
African Ancestry
- AMR:
-
Admixed American Ancestry
- SAS:
-
South Asian Ancestry
- CHB:
-
Han Chinese in Beijing, China
- JPT:
-
Japanese in Tokyo, Japan
- CHS:
-
Han Chinese South
- CDX:
-
Dai in Xishuangbanna, China
- KHV:
-
Kinh in Ho Chi Minh City, Vietnam
- CEU:
-
Utah residents (CEPH) with Northern and Western European ancestry
- TSI:
-
Toscani in Italia
- GBR:
-
British in England and Scotland
- FIN:
-
Finnish in Finland
- IBS:
-
Iberian populations in Spain
- YRI:
-
Yoruba in Ibadan, Nigeria
- LWK:
-
Luhya in Webuye, Kenya
- GWD:
-
Gambian in Western Division, The Gambia
- MSL:
-
Mende in Sierra Leone
- ESN:
-
Esan in Nigeria
- ASW:
-
African Ancestry in Southwest US
- ACB:
-
African Caribbean in Barbados
- MXL:
-
Mexican Ancestry in Los Angeles, California
- PUR:
-
Puerto Rican in Puerto Rico
- CLM:
-
Colombian in Medellin, Colombia
- PEL:
-
Peruvian in Lima, Peru
- GIH:
-
Gujarati Indian in Houston, TX
- PJL:
-
Punjabi in Lahore, Pakistan
- BEB:
-
Bengali in Bangladesh
- STU:
-
Sri Lankan Tamil in the UK
- ITU:
-
Indian Telugu in the UK
References
Ezzedine K, Eleftheriadou V, Whitton M, van Geel N. Vitiligo Lancet. 2015;386(9988):74–84. https://doi.org/10.1016/S0140-6736(14)60763-7
Shajil EM, Chatterjee S, Agrawal D, Bagchi T, Begum R. Vitiligo: pathomechanisms and genetic polymorphism of susceptible genes. Indian J Exp Biol. 2006;44(7):526–39.
Alikhan A, Felsten LM, Daly M, Petronic-Rosic V. Vitiligo: a comprehensive overview part I. introduction, epidemiology, quality of life, diagnosis, differential diagnosis, associations, histopathology, etiology, and work-up. J Am Acad Dermatol. 2011;65(3):473–91. https://doi.org/10.1016/j.jaad.2010.11.061
Ezzedine K, Lim HW, Suzuki T, Katayama I, Hamzavi I, Lan CC et al. Vitiligo Global Issue Consensus Conference Panelists. Revised classification/nomenclature of vitiligo and related issues: the Vitiligo Global Issues Consensus Conference. Pigment Cell Melanoma Res. 2012;25(3):E1-13. https://doi.org/10.1111/j.1755-148X.2012.00997.x
Spritz RA. The genetics of generalized vitiligo. Curr Dir Autoimmun. 2008;10:244–57. https://doi.org/10.1159/000131501
Shen C, Gao J, Sheng Y, Dou J, Zhou F, Zheng X, et al. Genetic susceptibility to Vitiligo: GWAS approaches for identifying Vitiligo susceptibility genes and loci. Front Genet. 2016;7:3. https://doi.org/10.3389/fgene.2016.00003
Mohr N, Petersen J, Kirsten N, Augustin M. Epidemiology of Vitiligo - A Dual Population-Based Approach. Clin Epidemiol. 2021;13:373–82. https://doi.org/10.2147/CLEP.S304155
Uffelmann E, Huang QQ, Munung NS, et al. Genome-wide association studies. Nat Rev Methods Primers. 2021;1:59. https://doi.org/10.1038/s43586-021-00056-9
Kachuri L, Chatterjee N, Hirbo J, et al. Principles and methods for transferring polygenic risk scores across global populations. Nat Rev Genet. 2024;25:8–25. https://doi.org/10.1038/s41576-023-00637-2
Corpas M, Megy K, Metastasio A, Lehmann E. Implementation of individualised polygenic risk score analysis: a test case of a family of four. BMC Med Genomics. 2022;15(Suppl 3):207. https://doi.org/10.1186/s12920-022-01331-8
Igo RP Jr, Kinzy TG, Cooke Bailey JN. Genetic risk scores. Curr Protoc Hum Genet. 2019;104(1):e95. https://doi.org/10.1002/cphg.95
Aponte JL, Chiano MN, Yerges-Armstrong LM, Hinds DA, Tian C, Gupta A, et al. Assessment of rosacea symptom severity by genome-wide association study and expression analysis highlights immuno-inflammatory and skin pigmentation genes. Hum Mol Genet. 2018;27(15):2762–72. https://doi.org/10.1093/hmg/ddy184
Jin Y, Andersen G, Yorgov D, Ferrara TM, Ben S, Brownson KM, et al. Genome-wide association studies of autoimmune vitiligo identify 23 new risk loci and highlight key pathways and regulatory variants. Nat Genet. 2016;48(11):1418–24. https://doi.org/10.1038/ng.3680
Cheng L, Liang B, Tang XF, Cai XY, Cheng H, Zheng XD, et al. Validation of susceptibility loci for Vitiligo identified by GWAS in the Chinese Han Population. Front Genet. 2020;11:542275. https://doi.org/10.3389/fgene.2020.542275
Birlea SA, Gowan K, Fain PR, Spritz RA. Genome-wide association study of generalized vitiligo in an isolated European founder population identifies SMOC2, in close proximity to IDDM8. J Invest Dermatol. 2010;130(3):798–803. https://doi.org/10.1038/jid.2009.347
Mao L, Fang Y, Campbell M, Southerland WM. Population differentiation in allele frequencies of obesity-associated SNPs. BMC Genomics. 2017;18(1):861. https://doi.org/10.1186/s12864-017-4262-9
Shin HT, Yoon BW, Seo JH. Comparison of risk allele frequencies of single nucleotide polymorphisms associated with age-related macular degeneration in different ethnic groups. BMC Ophthalmol. 2021;21(1):97. https://doi.org/10.1186/s12886-021-01830-9
Lee D, Koo T, Park J, Shin HT. Comparison of risk allele frequencies of Psoriasis-Associated single-nucleotide polymorphisms in different Population groups. Ann Dermatol. 2023;35(1):32–7. https://doi.org/10.5021/ad.22.110
Carlson CS, Matise TC, North KE, Haiman CA, Fesinmeyer MD, Buyske S, et al. PAGE Consortium. Generalization and dilution of association results from European GWAS in populations of non-european ancestry: the PAGE study. PLoS Biol. 2013;11(9):e1001661. https://doi.org/10.1371/journal.pbio.1001661
Prohaska A, Racimo F, Schork AJ, Sikora M, Stern AJ, Ilardo M, et al. Human Disease Variation in the light of Population Genomics. Cell. 2019;177(1):115–31. https://doi.org/10.1016/j.cell.2019.01.052
Sollis E, Mosaku A, Abid A, Buniello A, Cerezo M, Gil L et al. The NHGRI-EBI GWAS catalog: knowledgebase and deposition resource. Nucleic Acids Res 2022 Nov 9:gkac1010. https://doi.org/10.1093/nar/gkac1010
Jain A, Bhoyar RC, Pandhare K, Mishra A, Sharma D, Imran M, et al. IndiGenomes: a comprehensive resource of genetic variants from over 1000 Indian genomes. Nucleic Acids Res. 2021;49(D1):D1225–32. https://doi.org/10.1093/nar/gkaa923
Byrska-Bishop M, Evani US, Zhao X, Basile AO, Abel HJ, Regier AA, et al. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell. 2022;185(18):3426–e344019. https://doi.org/10.1016/j.cell.2022.08.004
Gómez R, Vilar MG, Meraz-Ríos MA, Véliz D, Zúñiga G, Hernández-Tobías EA, et al. Y chromosome diversity in Aztlan descendants and its implications for the history of Central Mexico. iScience. 2021;24(5):102487. https://doi.org/10.1016/j.isci.2021.102487
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10(2):giab008. https://doi.org/10.1093/gigascience/giab008
IndiGen Project. https://clingen.igib.res.in/indigen/. Accessed 6 June 5 2024.
Jin Y, Roberts GHL, Ferrara TM, Ben S, van Geel N, Wolkerstorfer A, et al. Early-onset autoimmune vitiligo associated with an enhancer variant haplotype that upregulates class II HLA expression. Nat Commun. 2019;10(1):391. https://doi.org/10.1038/s41467-019-08337-4
Jin Y, Birlea SA, Fain PR, Ferrara TM, Ben S, Riccardi SL, et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat Genet. 2012;44(6):676–80. https://doi.org/10.1038/ng.2272
Jin Y, Birlea SA, Fain PR, Gowan K, Riccardi SL, Holland PJ, et al. Genome-wide analysis identifies a quantitative trait locus in the MHC class II region associated with generalized vitiligo age of onset. J Invest Dermatol. 2011;131(6):1308–12. https://doi.org/10.1038/jid.2011.12
Jin Y, Birlea SA, Fain PR, Gowan K, Riccardi SL, Holland PJ, et al. Variant of TYR and autoimmunity susceptibility loci in generalized vitiligo. N Engl J Med. 2010;362(18):1686–97. https://doi.org/10.1056/NEJMoa0908547
Yu X, Chen Y, Chen J, Fan Y, Lu H, Wu D, et al. Shared genetic architecture between autoimmune disorders and B-cell acute lymphoblastic leukemia: insights from large-scale genome-wide cross-trait analysis. BMC Med. 2024;22(1):161. https://doi.org/10.1186/s12916-024-03385-0
Quan C, Ren YQ, Xiang LH, Sun LD, Xu AE, Gao XH, et al. Genome-wide association study for vitiligo identifies susceptibility loci at 6q27 and the MHC. Nat Genet. 2010;42(7):614–8. https://doi.org/10.1038/ng.603
Cheong KA, Kim NH, Noh M, Lee AY. Three new single nucleotide polymorphisms identified by a genome-wide association study in Korean patients with vitiligo. J Korean Med Sci. 2013;28(5):775–9. https://doi.org/10.3346/jkms.2013.28.5.775
Tang XF, Zhang Z, Hu DY, Xu AE, Zhou HS, Sun LD, et al. Association analyses identify three susceptibility loci for vitiligo in the Chinese Han population. J Invest Dermatol. 2013;133(2):403–10. https://doi.org/10.1038/jid.2012.320
Okamura K, Abe Y, Naka I, Ohashi J, Yagami A, Matsunaga K, et al. Genome-wide association study identifies CDH13 as a susceptibility gene for rhododendrol-induced leukoderma. Pigment Cell Melanoma Res. 2020;33(6):826–33. https://doi.org/10.1111/pcmr.12904
Open Targets Platform. https://platform.opentargets.org/. Accessed 6 June 5 2024.
Ochoa D, Hercules A, Carmona M, Suveges D, Baker J, Malangone C, et al. The next-generation open targets platform: reimagined, redesigned, rebuilt. Nucleic Acids Res. 2023;51(D1):D1353–9. https://doi.org/10.1093/nar/gkac1046
VitiVar. https://vitivar.igib.res.in/. Accessed 6 June 5 2024.
Gupta I, Narang A, Singh P, Manchanda V, Khanna S, Indian Genome Variation Consortium, Mukerji M, Natarajan VT, Dash D. VitiVar: A locus specific database of vitiligo associated genes and variations. Gene X. 2019;3:100018. https://doi.org/10.1016/j.gene.2019.100018
Han S, Kwak IY. Mastering data visualization with Python: practical tips for researchers. J Minim Invasive Surg. 2023;26(4):167–75. https://doi.org/10.7602/jmis.2023.26.4.167
Hunter JD, Matplotlib. A 2D graphics environment. Comput Sci Eng. 2007;9(03):90–5. https://doi.org/10.1109/MCSE.2007.55
Bharti N, Banerjee R, Achalere A, Kasibhatla SM, Joshi R. Genetic diversity of very important pharmacogenes in two South-Asian populations. PeerJ. 2021;9:e12294. https://doi.org/10.7717/peerj.12294
Berner D, Correction. Berner, D. Allele Frequency Difference AFD-An Intuitive Alternative to FST for Quantifying Genetic Population Differentiation. Genes 2019, 10, 308. Genes (Basel). 2019;10(10):810. https://doi.org/10.3390/genes10100810. Erratum for: Genes (Basel). 2019;10(4).
Huang D, Zhou Y, Yi X, Fan X, Wang J, Yao H, et al. VannoPortal: multiscale functional annotation of human genetic variants for interrogating molecular mechanism of traits and diseases. Nucleic Acids Res. 2022;50(D1):D1408–16. https://doi.org/10.1093/nar/gkab853
Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A, et al. Genome-wide association study of blood pressure and hypertension. Nat Genet. 2009;41(6):677–87. https://doi.org/10.1038/ng.384
Soranzo N, Spector TD, Mangino M, Kühnel B, Rendon A, Teumer A, et al. A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat Genet. 2009;41(11):1182–90. https://doi.org/10.1038/ng.467
Schaschl H, Göllner T, Morris DL. Positive selection acts on regulatory genetic variants in populations of European ancestry that affect ALDH2 gene expression. Sci Rep. 2022;12(1):4563. https://doi.org/10.1038/s41598-022-08588-0. Erratum in: Sci Rep. 2023;13(1):17123.
Aslam MM, John P, Fan KH, Bhatti A, Aziz W, Ahmed B, et al. Investigating the GWAS-Implicated loci for rheumatoid arthritis in the Pakistani Population. Dis Markers. 2020;2020:1910215. https://doi.org/10.1155/2020/1910215
Li J, Tian A, Zhu H, Chen L, Wen J, Liu W, et al. Mendelian randomization analysis reveals no causal relationship between nonalcoholic fatty liver disease and severe COVID-19. Clin Gastroenterol Hepatol. 2022;20(7):1553–e156078. https://doi.org/10.1016/j.cgh.2022.01.045
Fiorentino A, Sharp SI, McQuillin A. Association of rare variation in the glutamate receptor gene SLC1A2 with susceptibility to bipolar disorder and schizophrenia. Eur J Hum Genet. 2015;23(9):1200–6. https://doi.org/10.1038/ejhg.2014.261
Khong JJ, Burdon KP, Lu Y, Laurie K, Leonardos L, Baird PN, et al. Pooled genome wide association detects association upstream of FCRL3 with Graves’ disease. BMC Genomics. 2016;17(1):939. https://doi.org/10.1186/s12864-016-3276-z
Restrepo NA, Butkiewicz M, McGrath JA, Crawford DC. Shared Genetic etiology of Autoimmune diseases in patients from a Biorepository Linked to De-identified Electronic Health Records. Front Genet. 2016;7:185. https://doi.org/10.3389/fgene.2016.00185
Chia R, Saez-Atienzar S, Murphy N, Chiò A, Blauwendraat C, International Myasthenia Gravis Genomics Consortium. Identification of genetic risk loci and prioritization of genes and pathways for myasthenia gravis: a genome-wide association study. Proc Natl Acad Sci U S A. 2022;119(5):e2108672119. https://doi.org/10.1073/pnas.2108672119. Erratum in: Proc Natl Acad Sci U S A. 2022;119(23):e2206754119.
Simcoe M, Valdes A, Liu F, Furlotte NA, Evans DM, Hemani G, et al. Genome-wide association study in almost 195,000 individuals identifies 50 previously unidentified genetic loci for eye color. Sci Adv. 2021;7(11):eabd1239. https://doi.org/10.1126/sciadv.abd1239
Ruiz Y, Phillips C, Gomez-Tato A, Alvarez-Dios J, Casares de Cal M, Cruz R, et al. Further development of forensic eye color predictive tests. Forensic Sci Int Genet. 2013;7(1):28–40. https://doi.org/10.1016/j.fsigen.2012.05.009
Cerqueira CC, Hünemeier T, Gomez-Valdés J, Ramallo V, Volasko-Krause CD, Barbosa AA et al. Implications of the admixture process in skin color molecular assessment. PLoS One. 2014;9(5):e96886. https://doi.org/10.1371/journal.pone.0096886. Erratum in: PLoS One. 2014;9(9):e109451.
Reis LB, Bakos RM, Vianna FSL, Macedo GS, Jacovas VC, Ribeiro-Dos-Santos AM, et al. Skin pigmentation polymorphisms associated with increased risk of melanoma in a case-control sample from southern Brazil. BMC Cancer. 2020;20(1):1069. https://doi.org/10.1186/s12885-020-07485-x
Shah RL, Guggenheim JA, UK Biobank Eye and Vision Consortium. Genome-wide association studies for corneal and refractive astigmatism in UK Biobank demonstrate a shared role for myopia susceptibility loci. Hum Genet. 2018;137(11–12):881–96. https://doi.org/10.1007/s00439-018-1942-8
Ferguson R, Vogelsang M, Ucisik-Akkaya E, Rai K, Pilarski R, Martinez CN, et al. Genetic markers of pigmentation are novel risk loci for uveal melanoma. Sci Rep. 2016;6:31191. https://doi.org/10.1038/srep31191
Andersen JD, Pietroni C, Johansen P, Andersen MM, Pereira V, Børsting C, et al. Importance of nonsynonymous OCA2 variants in human eye color prediction. Mol Genet Genomic Med. 2016;4(4):420–30. https://doi.org/10.1002/mgg3.213
Erzurumluoglu AM, Liu M, Jackson VE, Barnes DR, Datta G, Melbourne CA, et al. Meta-analysis of up to 622,409 individuals identifies 40 novel smoking behaviour associated genetic loci. Mol Psychiatry. 2020;25(10):2392–409. https://doi.org/10.1038/s41380-018-0313-0
Wang Y. Association of pigmentation related-genes polymorphisms and geographic environmental variables in the Chinese population. Hereditas. 2021;158(1):24. https://doi.org/10.1186/s41065-021-00189-7
Khoruddin NA, Noorizhab MN, Teh LK, Mohd Yusof FZ, Salleh MZ. Pathogenic nsSNPs that increase the risks of cancers among the Orang Asli and Malays. Sci Rep. 2021;11(1):16158. https://doi.org/10.1038/s41598-021-95618-y
Meyer OS, Lunn MMB, Garcia SL, Kjærbye AB, Morling N, Børsting C, et al. Association between brown eye colour in rs12913832:GG individuals and SNPs in TYR, TYRP1, and SLC24A4. PLoS ONE. 2020;15(9):e0239131. https://doi.org/10.1371/journal.pone.0239131
Andersen JD, Johansen P, Harder S, Christoffersen SR, Delgado MC, Henriksen ST, et al. Genetic analyses of the human eye colours using a novel objective method for eye colour classification. Forensic Sci Int Genet. 2013;7(5):508–15. https://doi.org/10.1016/j.fsigen.2013.05.003
Kastelic V, Drobnic K. A single-nucleotide polymorphism (SNP) multiplex system: the association of five SNPs with human eye and hair color in the Slovenian population and comparison using a bayesian network and logistic regression model. Croat Med J. 2012;53(5):401–8. https://doi.org/10.3325/cmj.2012.53.401
Kastelic V, Pośpiech E, Draus-Barini J, Branicki W, Drobnič K. Prediction of eye color in the Slovenian population using the IrisPlex SNPs. Croat Med J. 2013;54(4):381–6. https://doi.org/10.3325/cmj.2013.54.381
Jacobs LC, Liu F, Pardo LM, Hofman A, Uitterlinden AG, Kayser M, et al. IRF4, MC1R and TYR genes are risk factors for actinic keratosis independent of skin color. Hum Mol Genet. 2015;24(11):3296–303. https://doi.org/10.1093/hmg/ddv076
Rahat MA, Akbar F, Rasool A, Ilyas M, Rakha A, Shams S, et al. Phenotypic classification of Eye Colour and Developmental Validation of the Irisplex System on Population living in Malakand Division. Pakistan Biomedicines. 2023;11(4):1228. https://doi.org/10.3390/biomedicines11041228
Stokowski RP, Pant PV, Dadd T, Fereday A, Hinds DA, Jarman C, et al. A genomewide association study of skin pigmentation in a south Asian population. Am J Hum Genet. 2007;81(6):1119–32. https://doi.org/10.1086/522235. Epub 2007 Oct 15.
Krüger C, Schallreuter KU. A review of the worldwide prevalence of vitiligo in children/adolescents and adults. Int J Dermatol. 2012;51(10):1206–12. https://doi.org/10.1111/j.1365-4632.2011.05377.x
Al Hammadi A, Silva de Castro CC, Parmar NV, Ubogui J, Hatatah N, Ahmed HM, et al. Prevalence and burden of vitiligo in Africa, the Middle East and Latin America. Skin Health Dis. 2023;4(1):e317. https://doi.org/10.1002/ski2.317
Yajnik CS, Wagh R, Kunte P, Asplund O, Ahlqvist E, Bhat D, et al. Polygenic scores of diabetes-related traits in subgroups of type 2 diabetes in India: a cohort study. Lancet Reg Health Southeast Asia. 2023;14:100182. https://doi.org/10.1016/j.lansea.2023.100182
Gaspar HA, Breen G. Probabilistic ancestry maps: a method to assess and visualize population substructures in genetics. BMC Bioinformatics. 2019;20(1):116. https://doi.org/10.1186/s12859-019-2680-1
Yang HC, Chen CW, Lin YT, Chu SK. Genetic ancestry plays a central role in population pharmacogenomics. Commun Biol. 2021;4(1):171. https://doi.org/10.1038/s42003-021-01681-6
Kido T, Sikora-Wohlfeld W, Kawashima M, Kikuchi S, Kamatani N, Patwardhan A, et al. Are minor alleles more likely to be risk alleles? BMC Med Genomics. 2018;11(1):3. https://doi.org/10.1186/s12920-018-0322-5
Zhang Y, Cai Y, Shi M, Jiang S, Cui S, Wu Y, et al. The prevalence of Vitiligo: a Meta-analysis. PLoS ONE. 2016;11(9):e0163806. https://doi.org/10.1371/journal.pone.0163806
Templeton AR. Out of Africa? What do genes tell us? Curr Opin Genet Dev. 1997;7(6):841–7. https://doi.org/10.1016/s0959-437x(97)80049-4
Prugnolle F, Manica A, Balloux F. Geography predicts neutral genetic diversity of human populations. Curr Biol. 2005;15(5):R159–60. https://doi.org/10.1016/j.cub.2005.02.038
Ashraf Q, Galor O. The out of Africa Hypothesis, Human Genetic Diversity, and comparative Economic Development. Am Econ Rev. 2013;103(1):1–46. https://doi.org/10.1257/aer.103.1.1
Ramachandran S, Deshpande O, Roseman CC, Rosenberg NA, Feldman MW, Cavalli-Sforza LL. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc Natl Acad Sci U S A. 2005;102(44):15942–7. https://doi.org/10.1073/pnas.0507611102
Reich D, Thangaraj K, Patterson N, Price AL, Singh L. Reconstructing Indian population history. Nature. 2009;461(7263):489–94. https://doi.org/10.1038/nature08365
Ramos PS, Shedlock AM, Langefeld CD. Genetics of autoimmune diseases: insights from population genetics. J Hum Genet. 2015;60(11):657–64. https://doi.org/10.1038/jhg.2015.94
Agarwal S, Kaur G, Randhawa R, Mahajan V, Bansal R, Changotra H. Liver X Receptor-α polymorphisms (rs11039155 and rs2279238) are associated with susceptibility to vitiligo. Meta Gene. 2016;8:33–6. https://doi.org/10.1016/j.mgene.2016.02.001
Kim MS, Patel KP, Teng AK, Berens AJ, Lachance J. Genetic disease risks can be misestimated across global populations. Genome Biol. 2018;19(1):179. https://doi.org/10.1186/s13059-018-1561-7
Acknowledgements
Authors acknowledge the Bioinformatics Resources and Applications Facility (BRAF) for the compute infrastructure. We extend our sincere thanks to Dr. T. N. Vivek, CSIR-IGIB for his critical inputs. We acknowledge the help provided by team members Sandeep Malviya, EP Ramakrishnan and Renu Gadhari during this project.
Funding
This work was supported by the National Supercomputing Mission, steered jointly by the Ministry of Electronics and Information Technology (MeitY) and Department of Science and Technology (DST), Government of India. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
NB contributed in data acquisition, performed the statistical tests and data analysis. All authors contributed in analysis, data interpretation and drafting of the manuscript. RB, SMK, AA helped prepare the manuscript and jointly developed the structure and arguments for the paper. SMK and RB made critical revisions and contributed to the writing of the manuscript. AA supervised the statistical analysis performed. RJ conceptualized and supervised the study. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
12863_2024_1254_MOESM2_ESM.pdf
Supplementary Figure 01: Comparison of vitiligo associated genetic risk scores from GWAS across super-populations reported in 1000 Genome and IndiGenomes Project
12863_2024_1254_MOESM3_ESM.pdf
Supplementary Figure 02: Distribution of vitiligo associated SNP frequency from prominent databases and literature across super-populations / populations reported in 1000 Genome and IndiGenomes project
12863_2024_1254_MOESM4_ESM.pdf
Supplementary Figure 03: Enriched and depleted pattern of significant variation in 117 SNPs of vitiligo in across populations reported in 1000 Genome and IndiGenomes project
12863_2024_1254_MOESM5_ESM.xlsx
Supplementary Table 01: Details of vitiligo-associated risk alleles, computed risk allele scores in 1KGP and IndiGen populations and their sites under positive selection as predicted by VannoPortal
12863_2024_1254_MOESM6_ESM.xlsx
Supplementary Table 02: SNPs belonging to additional vitiligo-associated genes in 1KGP super-populations (filtered based on MAF ≥ 0.05 in South Asian super-population) and annotation of significant SNPs
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bharti, N., Banerjee, R., Achalare, A. et al. Estimation of genetic variation in vitiligo associated genes: Population genomics perspective. BMC Genom Data 25, 72 (2024). https://doi.org/10.1186/s12863-024-01254-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-024-01254-6