
Abstract
Background
To satisfy an increasing demand for dietary protein, the poultry industry has employed genetic selection to increase the growth rate of broilers by over 400% in the past 50 years. Although modern broilers reach a marketable weight of ~ 2 kg in a short span of 35 days, a speed twice as fast as a broiler 50 years ago, the expedited growth has been associated with several negative detrimental consequences. Aside from heart and musculoskeletal problems, which are direct consequences of additional weight, the immune response is also thought to be altered in modern broilers.
Results
Given that identifying the underlying genetic basis responsible for a less sensitive innate immune response would be economically beneficial for poultry breeding, we decided to compare the genomes of two unselected meat control strains that are representative of broilers from 1957 and 1978, and a current commercial broiler line. Through analysis of genetic variants, we developed a custom prioritization strategy to identify genes and pathways that have accumulated genetic changes and are biologically relevant to immune response and growth performance. Our results highlight two genes, TLR3 and PLIN3, with genetic variants that are predicted to enhance growth performance at the expense of immune function.
Conclusions
Placing these new genomes in the context of other chicken lines, reveal genetic changes that have specifically arisen in selective breeding programs that were implemented in the last 50 years.
Similar content being viewed by others

Background
Poultry production is key to meeting a rising global demand for high quality dietary protein. In efforts to meet this demand, the poultry industry has employed intensive breeding programs aimed at selecting for lines that promote growth and enhance the efficiency of feed conversion ratios [1]. In the last 50 years, such selection has resulted in broilers displaying remarkable gains in growth and efficiency in feed conversion, such that the modern broiler attains a marketable weight in less than half the time, using half the amount of feed relative to breeds produced in the 1950’s. However, such gains can be associated with compromised health; selection for rapid growth without due regard to physiological support systems can lead to musculoskeletal disorders and sudden heart failures, and greater susceptibility to disease compared to older breeds [2, 3]. Of particular concern to the broiler industry is necrotic enteritis (NE), caused by the bacterial pathogen Clostridium perfringens. Global losses due to NE are estimated to be $2B/year and they are expected to rise [4].
To help mitigate against losses due to NE, the industry has relied on the use of sub-therapeutic quantities of antibiotic (antibiotic growth promotants, AGPs) that are added to chicken feed to protect from NE and enhance production efficiency [5]. Unfortunately, with the global emergence of antimicrobial resistance, many countries are seeking to ban the use of AGPs and the incidence of enteric diseases in chicken is expected to rise [6, 7]. Consequently, with selective breeding predicted to maximize growth potential in the next decade, the industry has shifted towards maintaining poultry health to minimize economic losses. To help support these programs, there is a need to identify the genetic modifications that have resulted in declined health and disease susceptibility. In attempts to meet this need, several groups have employed genomic approaches to characterize a diverse set of breeds, including native Taiwanese chickens [8], experimental lines that serve as animal models [9], as well as domestic layer and broiler lines [10, 11]. Aside from identifying genes that dictate physical characteristics such as plumage or skin color, these studies have identified numerous selective sweeps [8, 10, 12], reflecting regions with reduced heterozygosity as a consequence of positive selection. Such sweeps have revealed the fixation of alleles predicted to be beneficial. For example, specific alleles of the gene encoding thyroid stimulating hormone receptor (TSHR), which is critical for proper metabolic regulation and reproduction, have been shown to be fixed in all modern chickens, but not in the genomes of the Red Jungle Fowl [10, 13, 14] or chickens dating from ~ 280 B.C. to ~ 1700 A.D [13]. Beyond allelic variation, several studies have also focused on the role of copy number variation on disease and phenotype diversity [11, 15, 16]. For example, a study of 12 diversified chicken genomes identified a number of copy number variable regions covering genes that include FZD6 and LIMS1, which have been associated with increased resistance to Marek’s disease [11], while a comparison of Leghorn and Fayoumi chickens identified a duplication of BD7, involved in host innate immune response, in the latter [15]. Typically, these studies have focused on pairwise comparisons of individual lines, as opposed to genetic variation that occurs within the context of a breeding program. The availability of lines with shared ancestry that have undergone selective breeding over a number of decades, offers an opportunity to explore how the accumulation of genetic traits might impact phenotype.
In this study, we performed a comprehensive genomic analysis of domestic chicken breeds, focusing on broilers that underwent intensive selective breeding in the last 50 years. We generated genome sequence data for broiler lines generated in 1957, 1978, and 2016, which offer, for the first time, snapshots of genetic changes introduced during the breeding program, as well as insight into the underlying genetics of accelerated growth and changes in immune function. Additionally, we integrate whole genome datasets of other domestic chicken breeds and performed single nucleotide polymorphism calling on all data sets. We have since then identified genetic variants unique and common to different breeds that we suspect may contribute to breed-specific phenotypes. More importantly, we identified genes and pathways involved in metabolism and innate immune responses that have been altered by artificial selection, with some pathways showing potential in modifying modern broiler responses to foreign pathogens, that represent novel avenues for future investigations focusing on improving poultry health.
Results
Genome sequencing reveals the modern broiler to be closely related to the 1957 and 1978 lines
The goal of this study was to identify the genetic basis for traits selected for in modern breeding programs, such as growth and feed efficiency, with the purpose of informing on the selection of otherwise unanticipated traits such as skeletal defects, metabolic disorders and altered immune function [2, 3, 17]. To this end we sequenced the genomes of two University of Alberta Meat Control lines, unselected since 1957 and 1978 and representing synthetic crosses of a large number of contemporary commercial broiler lines (designated 1957 and 1978, respectively), and a 2016 commercial Ross 308 broiler (designated Ross308). The 1957 line is the result of the crossing of 4 (3 commercial, one experimental) contemporary chicken lines, starting in 1955 [18]. The 1978 line is a female parent line synthesized from nine sire lines and seven dam lines from breeding companies that voluntarily responded to a request for the lines, starting in 1978 [19].
After DNA extraction, Illumina sequencing resulted in the generation of 954, 957, and 753 million 150 bp reads for 1957, 1978, and Ross 308 respectively, representing from 76X to 92X coverage – ensuring sufficient coverage to accurately infer single nucleotide polymorphisms (SNPs; Table 1) [20]. To supplement our analyses, we included genome data from 12 additional breeds (Additional file 1: Table S1) obtained from the National Center for Biotechnology Information Sequence Read Archive (NCBI SRA) [8, 10]. These include: two layers – Swedish white layer (WLA) and commercial White Leghorn (WLB); two broilers - Ross 308 (CB1) and an undisclosed commercial broiler (CB2); two dual purpose chickens bred for both meat and egg production - Rhode Island Red (RIR) and a Taiwan heritage L2 line (L2), a Silkie line (Silkie); three experimental lines – high (High) and low (Low) growth lines established from White Plymouth Rock chickens in 1957 [21] and an Obese line (Obese) established in 1955 from a White Leghorn line as a model for autoimmune thyroiditis [22]; and two distinct genome sequences derived from Red Jungle Fowls (RJF and RJFswe).
Alignment of sequence reads to the reference chicken genome assembly (GCA_000002315.3) [23] reveal the number of variants to be broadly consistent with known breeding history. For example, the Red Jungle Fowl, aligned against its own genome, had the fewest (80,000) variants. Further, experimental lines (High, Low and Obese) also tended to have fewer variants than the production lines. Lines sequenced with the Illumina platform (1957, 1978, Ross308, Silkie and L2), which yield greater depth of sequence coverage, each had more variant calls than those sequenced with SOLiD technology. Interestingly, unlike other lines, the three lines sequenced here exhibited greater numbers of intronic variants (~ 3.9–4.3 million) compared to intergenic variants (~ 2.7–2.9 million). Also noteworthy, the second Red Jungle Fowl dataset (RJFswe) exhibited the greatest number of variants amongst those lines sequenced by the SOLiD platform. This increase in variation may reflect both a greater genetic heterogeneity in this line, together with the pooling of DNA from two different sources (8 males were selected from two different zoos in Sweden [10]).
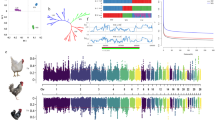
To further examine the potential impact of sequencing platform on variant calls, we performed a phylogenetic analysis on the variants common to lines sequenced by SOLiD and Illumina platforms (Fig. 1). Reassuringly our analysis grouped both Ross 308 datasets (Ross308 and CB1), together with the 1957 and 1978 lines. Furthermore, the clustering of the High and Low lines, as well as the Obese, WLA and WLB lines is consistent with known breeding history [10]. On the other hand our analysis split the CB1 and CB2 lines, which were previously considered close in ancestry. Our analysis also grouped the L2 and the Silkie lines, sequenced by Illumina, with the three broiler lines. The L2 line was established as a closed population from a slow growing Taiwanese line more than 30 years ago and has been selected for both egg and meat production1, while the Silkie line, was originally bred for ornamental purposes in the 19th century2. Of note, the RJFswe line did not group with the RJF line, suggesting that pooling of samples from two sources may have impacted variant calls in this dataset.

Overview of variant results following SNP and CNV calling. a Phylogenetic analysis of chicken lines. The phylogenetic tree was reconstructed using PhyML with 1024 bootstrap replicates. Bootstrap values are indicated at the branch points, the scale bar indicates number of nucleotide substitutions per site. Red Jungle Fowl was used to root the tree. Only SNPs identified in at least one Illumina-sequenced and two SOLiD sequenced datasets and occuring within genomic regions covered by more than 5 reads in all SOLiD datasets and more than 10 reads in all Illumina datasets were used in the analysis. b and c Venn diagrams displaying the number of SNPs (b) and CNVs (c) that are unique and shared between the three genomes sequenced in this study.
Focusing specifically on the three broiler lines (1957, 1978 and Ross308), we found that while each line possessed ~ 1.7–1.8 million SNPs not present in the other two lines, they share a common core of ~ 3.9 million SNPs. Amongst this core were 14,686 non-synonymous SNPs, together with 1796 frameshifts and 80 stop gains – variants that were likely introduced in the common ancestor of the three lines. Further, we found that the 1978 and Ross308 genomes share more SNPs (1,639,814) than the 1957 and the Ross308 genomes (1005,841). In addition to analyzing single-nucleotide polymorphisms (SNPs), we also compared copy number variants (CNVs; see Methods). CNVs that encompass coding regions have the potential to alter gene expression [24], with a concomitant impact on phenotypic traits. Multiple studies have already identified CNVs in diverse chicken breeds [8, 11, 16, 25, 26]. Here we aimed to compare CNVs across our three lines to identify loci with the potential to contribute to phenotypic changes in commercial broilers. Our analyses identified over 2500 CNVs for each line (Table 2), spanning ~ 8% of the chicken genome. Of these, 674 are common to all three lines (Fig. 1), with Ross 308 having the most unique CNVs in terms of number (1395 CNVs) which together account for 30% of the length of all CNV regions found in that line. These regions were associated with copy gains for ~ 150–170 genes and copy losses of ~ 150–160 genes. Gene set enrichment analysis of CNVs for each set of copy gains or losses revealed no enrichment of any Gene Ontology (GO) terms.
Notable genes associated with CNVs include two copies of CD8alpha in Ross308, two copies of tumor necrosis factor superfamily member 13b (TNFS13B) in both 1978 and Ross308, and the loss of one copy of Apolipoprotein-AII (APO-AII) in both 1978 and Ross308. CD8alpha is a co-receptor on CD8+ T cells, a specialized group of T-cells that recognizes antigens presented by MHC class I molecules, then respond to damaged cells by triggering production of cytokines and the release of cytotoxic molecules [27]. Related, TNFS13B (tumor necrosis factor superfamily member 13b; alternate name B cell activating factor, BAFF) is a cytokine that binds its corresponding receptors to induce proliferation and differentiation of B cells [28]. Heightened expression of this ligand has been observed in Crohn’s disease patients compared to healthy controls and has been proposed as a potential marker of inflammatory bowel disease [29]. Hence, copy number gains in CD8alpha and TNFSF13B in the 1978 and Ross308 lines may be linked to increased inflammation in response to infection. APO-AII, on the other hand, coats lipids to form high-density lipoproteins and promote cholesterol efflux [30, 31]; deficiency in this protein results in high serum cholesterol levels, which has been observed in Ross308 broilers relative to chickens indigenous to Iran [32].
Enrichment analysis reveals genetic variants associated with specific biological processes that vary across lines
To achieve a broad understanding of the impact of SNPs in each line, a functional enrichment analysis was performed to reveal biological processes significantly enriched in SNPs in the different chicken lines. First, for each gene we define its SNP density as the number of mutations divided by the coding sequence length. We then constructed a set of SNP dense genes as the 10% of genes displaying the greatest SNP density (and hence most likely impacted by genetic variation) and performed a gene set enrichment analysis for enriched functional categories as defined through GO and pathways defined by the Kyoto Encyclopedia of Genes and Genomes (KEGG; Fig. 2). Across all lines, 17 GO terms were defined as significantly enriched in SNP dense genes, of these 10 were enriched in more than one line, with the four immune related terms: “Herpes simplex infection” (KEGG: 05168); “Cytokine-cytokine receptor interaction” (KEGG: 04060); “Intestinal immune network for IgA production” (KEGG: 04672); and “Phagosome” (KEGG: 04145), being the most widely represented (significant in 7, 7, 5 and 5 lines respectively). Given the association of these terms with a diverse range of lines (broilers, Silkie, Taiwanese heritage chicken), this enrichment highlights the impact of domestication on the chicken immune response [33, 34]. Not all lines had GO terms enriched in SNP dense genes, and while the Ross308 genome sequenced here possessed the most enriched terms (10), only 3 were shared with the previously sequenced genome (CB1), which was additionally enriched in the immune-related term “Defense response to bacterium” (GO:0042742). Of note, Ross308 was uniquely associated with three terms relating to bone morphogenetic proteins (BMPs): “BMP signaling pathway” (GO:0030509); “Cellular response to BMP stimulus” (GO:0071773); and “Response to BMP” (GO:0071772). Such enrichment may reflect changes in pathways contributing to musculoskeletal disorders suffered by modern broilers [35]. For the three broilers that we sequenced, the terms “Defense response to other organism” (GO:0098542) and “Defense response” (GO:0006952) were only enriched in Ross308. Among the genes assigned to these terms are three that potentially link immune response and metabolic stress: TMEM173, a gene that activates type I interferon production and promotes glucose intolerance and tissue inflammation under metabolic stress induced by high fat diets [36]; and the two cathelicidins, CATHB1 and CATH3, antimicrobial peptides that kill bacteria by puncturing their cell membranes. With CATHB1, present in the intestinal tract where it can influence intestinal permeability, potential dysbiosis, and growth performance [37], and CATH3 able to bind lipopolysaccharides and reduce inflammation [38], the association of these categories with Ross308 and not 1957 or 1978, suggest that recent selection may have resulted in compromised immune function. Together these GO enrichment analyses reveal biological processes that may have been altered through breeding programs.

Heat map showing enrichment of GO and KEGG categories of genes containing nonsynonymous SNP densities in the 90th percentile across all breeds. All categories and pathways shown have a FDR-adjusted p-value of less than 0.05 in at least one line.
Combinatorial prioritization strategy identifies candidate genes for future functional characterization
In the previous section we identified sets of biological processes enriched in SNP dense genes associated with 10 lines; many were associated with growth and immune responses. To identify transcripts that have been functionally impacted in the development of the Ross308 line and lay the framework for future experimental investigations, we developed a novel prioritization scheme (Fig. 3) based on combining five complementary strategies (see Methods). Note, we refer here to transcripts rather than genes since four of the five strategies rely on normalization of SNPs and SNP effects by transcript length.

Prioritization strategies used to identify genes associated with selection of fast growing broiler lines. Top panels: Schematic representation of strategies used to prioritize genes (see main text for more explanation). Lower panel, upset plot showing numbers of transcripts identified through each strategy and combination of strategies. Known genes associated with more restricted combinations of strategies are indicated. Genes in red are the 12 manually curated genes mentioned in manuscript.
The first strategy (Strategy 1) defines transcripts that have accumulated the greatest number of SNPs over the past 60 years. Here we compared increases in non-synonymous SNP density between the 1957 and the 1978 lines, and the 1978 and Ross308 lines. Under this strategy, we select transcripts that are in the 95th percentile in terms of increase in SNP density either for the 1957/1978 comparison or the 1978/Ross308 comparison. We additionally specify that both changes in SNP density must be positive (i.e. there are more SNPs in more recently bred lines). In Strategy 2 we define transcripts based on unique SNPs in faster-growing lines. Transcripts are selected based on possessing the highest (90th percentile) density of SNPs that are unique to the 1978 and/or Ross308 lines relative to all other lines. Strategy 3 focuses on transcripts that possess high impact variants (i.e. frameshift, start loss, and stop gain variants) restricted to either the 1978 or Ross308 lines, but not the 1957 line. Similarly, Strategy 4 identifies transcripts based on decreasing scores from application of the Sorting Intolerant From Tolerant (SIFT; a measure of whether a SNP will affect protein function) algorithm [39]. Under this strategy, a list of transcripts in which the mean SIFT score decreases from the 1957 to 1978 lines and the 1978 to Ross308 lines is first generated. Subsequently, transcripts from this list are selected if they are in the lower 50th percentile of SIFT scores for either the 1978 or Ross308 lines. In the final strategy (Strategy 5) we identify transcripts that have accumulated deleterious SNPs. Here we select for transcripts that show an increase in the number of deleterious variants (SNPs with SIFT scores less than 0.05) when comparing the 1957, 1978, and Ross308 broilers and, additionally, possess the highest (99th percentile) density of deleterious variants.
Applying each strategy resulted in the generation of five complementary sets of transcripts (Fig. 3). Strategy 2 resulted in the largest number of transcripts (1477) and strategy 5 the fewest (211). Combining strategies, we find that only 20, of 120 possible combinations of strategies result in unique sets of transcripts. To help prioritize genes for future experimental investigations, we subsequently filtered for transcripts that are identified in at least three strategies (Fig. 3). This final filtering step ensures the selection of transcripts with SNPs that result in functional consequences (use of only one or two strategies might result in the selection of transcripts that have simply accumulated many low-impact variants) and identified a total of 78 transcripts. From this list, manual reviews of GO and domain annotations allowed us to identify 12 transcripts with mammalian homologs involved in immune response and/or promoting growth, that have likely been compromised in the selection of the 1978 and Ross308 lines (CATH3, CYB5R4, GBA2, GSTA2, IL20RB, MYOL7L2, NTHI1, PLA2G2E, PLIN3, SLAMF8 and TAP1). After manual review of the potential functional impact of their associated SNPs, we identified TLR3 and a putative homolog of mammalian PLIN3 (ENSGALT00000003248), for more focused analysis.
TLR3 is part of the Toll-like receptor family that plays a crucial role in innate immunity by recognizing double stranded viral RNA and promoting anti-viral defenses in other cells [40]. In the Ross308 line, TLR3 has acquired nine more variants than the 1978 line and 16 more than the 1957 line. Several of these variants are unique to the Ross308 (relative to the other lines considered in this study) and occur in conserved regions (Fig. 4). These variants include N174H and D466H, both found in the leucine-rich surface that is responsible for recognizing and binding double stranded viral RNA and activating innate immunity. When viewed in the context of other mutations that alter charge (D19V, K43Q, R266S, E283G), we hypothesize that the sensitivity of TLR3 to double stranded viral components could be diminished. This decrease in functional efficacy could be physiologically important for growth performance, for example, a mouse knock out of a toll-like receptor (TLR5) has previously been shown to modulate gut microbiome characteristic of obesity and other metabolic disorders [41].

Domain architectures of TLR3 and PLIN3 showing non-synonymous SNPs identified in Ross308. a Domain architecture of TLR3, showing leucine-rich domain and the Toll/interleukin-1 receptor homology domain. b Domain architecture of PLIN3. Highlighted for both genes are SNPs considered deleterious (SIFT score < 0.05), as well as those that are unique to Ross308 and/or result in a loss or change in charge.
ENSGALT00000003248, a PLIN3 homolog, is the only gene that was identified by all five strategies; of the ten SNPs found in the Ross308 line, five were unique, including the deleterious variant G199R and a stop codon W298X predicted to cause premature termination (Fig. 4). ENSGALT00000003248 is a gene product of ENSGALG00000002083 and identified by Ensembl as a putative paralog of PLIN3. PLIN3 is a member of the perilipin family, a group of proteins known for regulating lipid metabolism in different cell types. PLIN3 has been observed to respond to stimulation by lipopolysaccharides in neutrophils, while knocking down PLIN3 in neutrophils results in an immediate decrease in lipid droplets biogenesis and prostaglandin E2 production [42], both of which are critical for initiating an appropriate innate inflammatory immune response. Moreover, PLIN3 participates in exercise-induced lipolysis by co-localizing with lipid droplets and interacting with adipose triglyceride lipase [43], while its malfunction can lead to dysregulations in fat oxidation often observed in obese or diabetic patients. Given its role in fat deposition, and that it is the only gene to be prioritized by all five strategies, we hypothesize that alterations in this gene have helped to promote growth in the Ross 308 line.
Discussion
In this study, we compared genetic variants in the genomes of three broiler lines: two meat control strains representative of broilers in 1957 and 1978, and a 2005 commercial Ross 308 broiler. Our central hypothesis is that immune pathways were compromised during selective breeding targeting increased growth and efficiency. For example, the selection for growth has likely reduced the systemic effects of inflammation, which in turn means that modern birds use less energy to maintain a high body temperature (fever) for a long period of time in response to the same stimulus. Thus, we may expect production traits are interlinked with immune traits. To address this question, we developed a selection strategy to identify genes that have acquired variants over the course of selective breeding programs, impacting their function. Our initial expectation is that this strategy would result in the identification of genes that contribute to both immune and growth-related processes.
Our combinatorial strategy incorporates three considerations neglected by other studies. First, different genes could experience different levels of selective constraints which has been observed in domestication of cattle [44] and maize [45], where variants that may be considered “deleterious” in natural populations are relaxed under changes in environments (for example as might occur in commercial broiler facilities). Second, while previous studies focused on identification of causal variants [46,47,48], genes can also accumulate multiple mutations which are tolerated individually but may act synergistically to affect gene function. Lastly, this study is relatively unique from a poultry perspective in presenting analyses of genomes that are representative of different time points across commercial broiler genetic selection, offering a novel opportunity to track the accumulation of mutations in genes over time. Consequently, we focused our analyses on longitudinal comparisons of the 1957, 1978, and Ross308 genomes.
It should be appreciated that rather than sampling across each population, only a single individual from each line was sequenced. Consequently, given their heterogeneous genetic backgrounds, we expect that the variants identified here may only represent a small fraction of those that distinguish the three lines. Nevertheless, we are encouraged by the enrichment of variants in genes associated with immune functions, with many of the genes that passed our final filter (prioritized by more than 3 strategies) contributing to both immune response and growth (Figs. 2 and 3). TLR3 already has an established role of recognizing viral dsRNA to elicit an innate immune response. However, Toll-like receptors, the superfamily that encompasses TLR3, have also been shown to indirectly influence metabolism [49, 50]. Knocking out TLR2 [50] and TLR4 [51] can prevent diet-induced obesity and insulin resistance through inflammatory signaling pathways. Conversely, TLR5 knockouts in mice have been shown to induce insulin resistance, hyperphagia, and many other symptoms characteristic of metabolic disorders [41]. Examining the gut microbiome composition in TLR5-deficient mice reveals that it was altered in such a way that it induced low-level inflammatory signaling, this in turn, attenuated insulin signaling to promote insulin resistance and hyperphagia. Similarly, we hypothesize that TLR3, which does not bind fatty acids like TLR2 and TLR4 [52, 53], may also mediate metabolism in broiler chickens. Indeed, TLR3 has already been observed to protect mice from colitis due to its ability to sense dsRNA produced by commensal lactic acid bacteria and suppress inflammatory responses reserved for pathogenic bacteria [54]. By measuring dsRNA amounts and inflammatory response following digestion of dsRNA, it was established that the dsRNA level in commensal Lactobacillales, which is higher than dsRNA amounts in pathogens such as Salmonella enterica serovar Typhimurium and Escherichia coli, is sufficient to induce anti-inflammatory signals via TLR3. Another study that used a high-density SNP array to investigate polymorphisms in a commercial broiler line also found that TLR3 falls within a region of positive selection [12]. This raises the interesting possibility that mutations that result in decreased TLR3 function may result in poor discrimination between commensal and pathogenic flora of the gut, that while resulting in dysbiosis, may nevertheless improve growth performance.
PLIN3 belongs to the perilipin family of proteins that associate with lipid droplets to regulate lipase access to lipid storage. With known roles in exercise-induced lipid degradation [43] and lipopolysaccharide-induced lipid droplet formation in neutrophils [42], the accumulation of genetic variants in PLIN3 during selective breeding of the Ross308 line suggests that its functions may be altered in this line, with potential impact on both lipid accumulation and immune response.
In addition to identifying genes of interest, we observed that all three broilers that we sequenced are over-represented in SNP dense genes from several functional categories such as phagosome, cytokine-cytokine receptor interaction, and intestinal immune network for IgA production. That suggests that commercial broiler immune response, irrespective of lineage, is distinct from the Red Jungle Fowl immune function. The Ross308 line, however, is the only one of the three broilers that we sequenced with SNP dense genes enriched in the GO term: “Defense response to other organisms” (GO:0098542). This result is supported by previous studies; one study found that commercial broilers had a lower febrile response (ability to generate fever) and lower cytokine expression in response to gram-negative bacteria than commercial layers [55]. Comparing the gene expression profiles of two broiler lines and quantifying pathogen burden after infection with Salmonella revealed the fast-growing chickens relied on T-cell activation and were slower at clearing S. enterica serovar Enteritidis infection than slow growing chickens, which induced macrophage activation post infection [56]. Therefore, from our investigations of enriched GO terms (specifically: “Defense response to bacterium” (GO:0042742) and “Defense response to other organisms” (GO:0098542)), these studies support the hypothesis that the Ross308 line may respond differently to pathogenic invasion.
Conclusions
Together this study illustrates how comparisons of the genome sequences derived from temporally related lines can help reveal the genes and biological processes that help drive livestock breeding programs.
Methods
Genome sequencing and data collection of additional datasets
The genomes of two female chickens from each of the University of Alberta Meat Control strains unselected since 1957 and 1978, respectively, and a commercial Ross 308 broiler (Aviagen North America Inc., Huntsville, Alabama) were generated as follows:
Genomic DNA was isolated from 30 ul of chicken blood samples using the Promega Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Genomic DNA was quantified with a Qubit 2.0 fluorometer using the Qubit dsDNA BR assay kit (Life Technologies, Carlsbad, CA) and diluted in buffer EB to the 20 ng/ul input for library preparation. DNA samples were also assessed for quality by gel electrophoresis. Library preparation was performed following the Illumina Truseq DNA PCR-Free LT Library Prep Kit (Illumina, San Diego, CA) protocol using 1μg of isolated genomic DNA. The input DNA was sheared to 350 bp using Covaris microTUBES and the S2 Ultrasonicator (Covaris, Woburn, MA). End repair, A-tailing, adaptor ligation and library nomalization were performed according to Illumina’s instructions. Libraries were quantified for normalization by qPCR using the KAPA Library Quantification Kit for Illumina sequencing platforms (KAPA Biosystems, Boston, MA). The final libraries were also assessed using the DNA1000 kit for the Bioanalyzer 2100 (Agilent Technologies, Palo Alto, CA). The final libraries were denatured and diluted to a final loading volume of 1.3 ml at a concentration of 1.8pM. Sequencing was performed on the NextSeq500 sequencer (Illumina, San Diego, CA), according to the manufacturer’s instructions, using the 300 cycle High Output V2 sequencing kit and a spike-in of 8% PhiX control library, generating 150 bp paired end reads. Sequence data is available through the sequence repository archive, hosted by the NCBI, under BioProject PRJNA561435.
Additional genomes were downloaded from the sequence read archive (SRA) at the National Center for Biotechnology Information (NCBI) [57] and include the genomes of Taiwan L2 and Silkie chickens (SRP022583), together with ten lines generated through pooled blood samples (SRP022583): a White Leghorn line developed at the Swedish University of Agricultural Sciences (WLA), a commercial White Leghorn line (WLB), an Obese line, Rhode Island Red (RIR), Ross 308 (CB1), a second unidentified commercial broiler line (CB2), high- and low-growth lines, Red Jungle Fowl from a Swedish zoo population (RJFswe), and the partly inbred UCD 001 line used to generate the reference chicken genome (RJF).
Alignment and variant detection of Illumina data sets
For lines sequenced using the Illumina platform (1957, 1978, and Ross 308 broilers sequenced here, as well as Taiwan L2 and Silkie), adapters and low quality reads were identified and filtered using FastQC [58] and Trimmomatic [59]. Remaining reads were mapped separately to the Gallus gallus reference genome (galGal5, January, 2016 [23]) with BWA 0.7.5 [60] using default settings. Picard tools (http://broadinstitute.github.io/picard) removed duplicates in the alignment. Genome analysis toolkit (GATK) [61] was used for indel re-alignment and base score recalibration, where high confidence variants were first detected with HaplotypeCaller and used to recalibrate base scores. HaplotypeCaller was subsequently used to call SNPs, with the minimum emission confidence threshold set at a phred score of 15 and the minimum calling confidence threshold set at a phred score of 50. Detected variants were further filtered with Variant Filtration (parameters used were QUAL / DP < 2.0, FS > 60.0, MQ < 40.0, MQRankSum < − 12.5, ReadPosRankSum < − 12.5).
Putative copy number variants (CNVs) of the 1957, 1978, and Ross 308 genomes were detected using CNVnator [62] (bin size set to 45) and further filtered by size (> 1000 bp) and reads mapping location (fraction of reads that can map to 2 or more locations must be less than 0.5). When comparing CNVs between genomes, CNVs overlapping by more than 50% in genomic location were considered shared between breeds. CNV related genes were defined on the basis of their sequence overlapping by at least 50% with a CNV.
Phylogenetic tree construction
Phylogenetic trees were generated using SNPs detected from all breeds. First, reads that mapped to multiple genomic regions were filtered. Since Illumina-sequenced datasets possessed greater depth coverage than SOLiD-sequenced datasets, only regions that were covered by more than 10 uniquely mapped reads in all Illumina-sequenced data sets and more than 5 uniquely mapped reads in all SOLiD-sequenced datasets were used in this analysis. Further, only SNPs that fall within these regions and were present in at least one Illumina-sequenced breed and two SOLiD-sequenced breeds were considered for phylogenetic analysis. SNPs were concatenated to generate multiple sequence alignments, and phylogenetic trees generated by PhyML [63] with 1024 bootstrap replicates, using the Red Jungle Fowl as the outgroup. The final trees were visualized using FigTree [64].
SNP calling of additional SOLiD sequenced datasets
Analyzing reads generated using the SOLiD platform required a different pipeline to detect SNPs. Bfast (v0.7.0) [65] converted fasta files and corresponding quality scores to csfastq files, which are normal outputs of the SOLiD platform and encode DNA sequences in colour space, the shrimp (v2.2.3) [66] aligner then mapped reads to the reference genome, with --max-alignments set to 5 to reduce reads that multi-mapped to many regions in the genome. Normal SNP calling steps were followed as before; Samtools and picard-tools were used again to sort, index, and remove duplicates from the alignment. Freebayes (v0.9.2) [67] was subsequently used to detect SNPs for SOLiD datasets with the flags –genotype-qualities –use-mapping-quality –genotyping-max-iterations 500.
Functional annotation and analysis of genetic variants
SNPs were annotated with both ANNOVAR (v2016-02-01) [68] and Variant Effect Predictor (release 87) [69]. Variant Effect Predictor also provides information regarding the impact of variants on the basis of Sorting Intolerant From Tolerant (SIFT) score [39]. The vcf-isec package from VCFtools [70] was used to determine variants unique or shared across lines, for variants specific to broiler breeds, BCFtools [71] first merged all non-broiler variant files, and vcf-isec applied to compare broiler variants to the merged file to identify unique variants. SNP density for each gene was defined as the number of SNPs normalized by gene length, global SIFT score was defined as the average SIFT score of all variants in a gene normalized by gene length, and number of variants predicted to be deleterious for each gene. Ortholog [72], Gene Ontology [73], and functional annotations were retrieved from Ensembl Biomart [74] and Uniprot [75].
Enrichment analysis
Genes with SNP densities in the 90th percentile in each breed were subject to pathway enrichment analysis based on Gene Ontology (GO) [73] and Kyoto Encyclopedia of Genes and Genomes (KEGG) [76] categories. Only GO and KEGG categories containing at least 3 genes were considered. Enrichment analysis was performed by the R package clusterProfiler [77], and only categories with a p-value < 0.05 after Benjamini-Hochberg false discovery rate [78] correction were considered.
Prioritization strategy
Based on SNP analyses described above, we defined five prioritization strategies to define transcripts of interest (Fig. 3):
Strategy 1. Transcripts that have accumulated most SNPs in the last 50 years. Nonsynonymous SNP density increase between the 1957 and the 1978 broiler was compared to the nonsynonymous SNP density increase between the 1978 and Ross 308 broiler. For a transcript to be prioritized by this strategy, the difference in SNP density between the 1957 and 1978 broiler OR between the 1978 broiler and Ross 308 must be in the 95th percentile. Additionally, all SNP density changes between 1957 and 1978 and 1978 and Ross 308 must be positive (i.e. more SNPs in more recently bred lines).
Strategy 2. Transcripts with the most SNPs unique to fast-growing broilers. This strategy selected for transcripts with the highest number of variants (90th percentile after normalizing by transcript length) that belonged exclusively to the 1978 and/or Ross 308 subset OR the subset of SNPs exclusive only to Ross 308. The former subset (transcripts with high number of unique variants in 1978 and Ross 308 combined) includes transcripts that have not gained unique variants between 1978 and Ross 308 but have accumulated unique variants since the 1950s.
Strategy 3. Transcripts containing high impact variants (i.e. frameshift, start loss, and stop gain variants) only in Ross 308 or the 1978 broiler.
Strategy 4. Transcripts with decreasing SIFT score. For every transcript, the mean SIFT score is calculated, only transcripts that show a decrease in SIFT score when comparing the 1957, 1978, and Ross 308 broilers and are also in the bottom 50th percentile for either the 1978 or Ross 308 broiler are selected.
Strategy 5. Transcripts that have accumulated deleterious SNPs. Transcripts that show an increase in number of deleterious variants (SNPs with SIFT scores less than 0.05) when comparing the 1957, 1978, and Ross 308 broilers and possess the highest number of deleterious variants (99th percentile) after normalizing by transcript length.
Availability of data and materials
Sequence data generated in this study is available through the sequence repository archive, hosted by the NCBI, under BioProject PRJNA561435.
Abbreviations
- AGPs:
-
Antibiotic growth promotants
- CB1:
-
Ross 308, a commercial broiler
- CB2:
-
An undisclosed commercial broiler
- CNV:
-
Copy number variant
- DNA:
-
Deoxyribonucleic acid
- dsRNA:
-
Double stranded ribonucleic acid
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- kg:
-
Kilogram
- L2:
-
A Taiwan heritage L2 line
- NCBI:
-
National Center for Biotechnology Information
- NE:
-
Necrotic enteritis
- qPCR:
-
Quantitative polymerase chain reaction
- RIR:
-
Rhode Island Red
- RJF:
-
Red jungle fowl
- RJFswe:
-
Red jungle fowl from swedish zoos
- RNA:
-
Ribonucleic acid
- SIFT:
-
Sorting Intolerant From Tolerant
- SNP:
-
Single nucleotide polymorphism
- SRA:
-
Sequence Read Archive
- TSHR:
-
Thyroid stimulating hormone receptor
- WLA:
-
A swedish white layer
- WLB:
-
A commercial white leghorn
References
Zuidhof MJ, Schneider BL, Carney VL, Korver DR, Robinson FE. Growth, efficiency, and yield of commercial broilers from 1957, 1978, and 2005. Poult Sci. 2014;93:2970–82.
Yunis R, Ben-David A, Heller ED, Cahaner A. Immunocompetence and viability under commercial conditions of broiler groups differing in growth rate and in antibody response to Escherichia coil vaccine. Poult Sci. 2000;79:810–6.
Cheema MA, Qureshi MA, Havenstein GB. A comparison of the immune response of a 2001 commercial broiler with a 1957 randombred broiler strain when fed representative 1957 and 2001 broiler diets. Poult Sci. 2003;82:1519–29.
Van Immerseel F, Rood JI, Moore RJ, Titball RW. Rethinking our understanding of the pathogenesis of necrotic enteritis in chickens. Trends Microbiol. 2009;17:32–6.
Taylor JH, Gordon WS. Growth-promoting activity for pigs of inactivated penicillin. Nature. 1955;176:312–3.
Liao SF, Nyachoti M. Using probiotics to improve swine gut health and nutrient utilization. Animal Nutrition. 2017;3:331–43.
Gaucher ML, Quessy S, Letellier A, Arsenault J, Boulianne M. Impact of a drug-free program on broiler chicken growth performances, gut health, Clostridium perfringens and campylobacter jejuni occurrences at the farm level. Poult Sci. 2015;94:1791–801.
Fan WL, Ng CS, Chen CF, Lu MY, Chen YH, Liu CJ, Wu SM, Chen CK, Chen JJ, Mao CT, et al. Genome-wide patterns of genetic variation in two domestic chickens. Genome Biol Evol. 2013;5:1376–92.
Jang HM, Erf GF, Rowland KC, Kong BW. Genome resequencing and bioinformatic analysis of SNP containing candidate genes in the autoimmune vitiligo Smyth line chicken model. BMC Genomics. 2014;15:707.
Rubin CJ, Zody MC, Eriksson J, Meadows JR, Sherwood E, Webster MT, Jiang L, Ingman M, Sharpe T, Ka S, et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature. 2010;464:587–91.
Yi G, Qu L, Liu J, Yan Y, Xu G, Yang N. Genome-wide patterns of copy number variation in the diversified chicken genomes using next-generation sequencing. BMC Genomics. 2014;15:962.
Stainton JJ, Charlesworth B, Haley CS, Kranis A, Watson K, Wiener P. Use of high-density SNP data to identify patterns of diversity and signatures of selection in broiler chickens. J Anim Breed Genet. 2017;134:87–97.
Girdland Flink L, Allen R, Barnett R, Malmstrom H, Peters J, Eriksson J, Andersson L, Dobney K, Larson G. Establishing the validity of domestication genes using DNA from ancient chickens. Proc Natl Acad Sci U S A. 2014;111:6184–9.
Gheyas AA, Boschiero C, Eory L, Ralph H, Kuo R, Woolliams JA, Burt DW. Functional classification of 15 million SNPs detected from diverse chicken populations. DNA Res. 2015;22:205–17.
Lee MO, Bornelov S, Andersson L, Lamont SJ, Chen J, Womack JE. Duplication of chicken defensin7 gene generated by gene conversion and homologous recombination. Proc Natl Acad Sci U S A. 2016;113:13815–20.
Yan Y, Yang N, Cheng HH, Song J, Qu L. Genome-wide identification of copy number variations between two chicken lines that differ in genetic resistance to Marek's disease. BMC Genomics. 2015;16:843.
Calenge F, Mignon-Grasteau S, Chanteloup NK, Bree A, Lalmanach AC, Schouler C. Broiler lines divergently selected for digestive efficiency also differ in their susceptibility to colibacillosis. Avian Pathol. 2014;43:78–81.
Merritt ES, Gowe R. Development and genetic properties of a control strain of meat-type fowl. Proc. 12th World’s Poultry Congress, Sydney; 1962. p. 66–70.
Chambers JR, Bernon DE, Gavora JS. Synthesis and parameters of new populations of meat-type chickens. Theor Appl Genet. 1984;69:23–30.
Fang H, Wu Y, Narzisi G, O'Rawe JA, Barron LT, Rosenbaum J, Ronemus M, Iossifov I, Schatz MC, Lyon GJ. Reducing INDEL calling errors in whole genome and exome sequencing data. Genome Med. 2014;6:89.
Dunnington EA, Siegel PB. Long-term divergent selection for eight-week body weight in white Plymouth rock chickens. Poult Sci. 1996;75:1168–79.
Cole RK. Hereditary hypothyroidism in the domestic fowl. Genetics. 1966;53:1021–33.
Warren WC, Hillier LW, Tomlinson C, Minx P, Kremitzki M, Graves T, Markovic C, Bouk N, Pruitt KD, Thibaud-Nissen F, et al. A New Chicken Genome Assembly Provides Insight into Avian Genome Structure. G3 (Bethesda). 2017;7:109–17.
Huang L, Teng D, Wang H, Sheng G, Liu T. Association of copy number variation in the AHI1 gene with risk of obesity in the Chinese population. Eur J Endocrinol. 2012;166:727–34.
Crooijmans RP, Fife MS, Fitzgerald TW, Strickland S, Cheng HH, Kaiser P, Redon R, Groenen MA. Large scale variation in DNA copy number in chicken breeds. BMC Genomics. 2013;14:398.
Rao YS, Li J, Zhang R, Lin XR, Xu JG, Xie L, Xu ZQ, Wang L, Gan JK, Xie XJ, et al. Copy number variation identification and analysis of the chicken genome using a 60K SNP BeadChip. Poult Sci. 2016;95:1750–6.
Gao GF, Jakobsen BK. Molecular interactions of coreceptor CD8 and MHC class I: the molecular basis for functional coordination with the T-cell receptor. Immunol Today. 2000;21:630–6.
Schneider K, Kothlow S, Schneider P, Tardivel A, Gobel T, Kaspers B, Staeheli P. Chicken BAFF--a highly conserved cytokine that mediates B cell survival. Int Immunol. 2004;16:139–48.
Zhang P, Liu X, Guo A, Xiong J, Fu Y, Zou K. B cell-activating factor as a new potential marker in inflammatory bowel disease. Dig Dis Sci. 2016;61:2608–18.
Marzal-Casacuberta A, Blanco-Vaca F, Ishida BY, Julve-Gil J, Shen J, Calvet-Marquez S, Gonzalez-Sastre F, Chan L. Functional lecithin:cholesterol acyltransferase deficiency and high density lipoprotein deficiency in transgenic mice overexpressing human apolipoprotein A-II. J Biol Chem. 1996;271:6720–8.
Remaley AT, Stonik JA, Demosky SJ, Neufeld EB, Bocharov AV, Vishnyakova TG, Eggerman TL, Patterson AP, Duverger NJ, Santamarina-Fojo S, Brewer HB Jr. Apolipoprotein specificity for lipid efflux by the human ABCAI transporter. Biochem Biophys Res Commun. 2001;280:818–23.
Abdi-Hachesoo B, Talebi A, Asri-Rezaei S. Comparative study on blood profiles of indigenous and Ross-308 broiler breeders. Global Veterinaria. 2011;7:238–41.
Li Q, Wang N, Du Z, Hu X, Chen L, Fei J, Wang Y, Li N. Gastrocnemius transcriptome analysis reveals domestication induced gene expression changes between wild and domestic chickens. Genomics. 2012;100:314–9.
Belteky J, Agnvall B, Johnsson M, Wright D, Jensen P. Domestication and tameness: brain gene expression in red junglefowl selected for less fear of humans suggests effects on reproduction and immunology. R Soc Open Sci. 2016;3:160033.
Francis PH, Richardson MK, Brickell PM, Tickle C. Bone morphogenetic proteins and a signalling pathway that controls patterning in the developing chick limb. Development. 1994;120:209–18.
Mao Y, Luo W, Zhang L, Wu W, Yuan L, Xu H, Song J, Fujiwara K, Abe JI, LeMaire SA, et al. STING-IRF3 triggers endothelial inflammation in response to free fatty acid-induced mitochondrial damage in diet-induced obesity. Arterioscler Thromb Vasc Biol. 2017;37:920–9.
Goitsuka R, Chen CL, Benyon L, Asano Y, Kitamura D, Cooper MD. Chicken cathelicidin-B1, an antimicrobial guardian at the mucosal M cell gateway. Proc Natl Acad Sci U S A. 2007;104:15063–8.
Bommineni YR, Dai H, Gong YX, Soulages JL, Fernando SC, Desilva U, Prakash O, Zhang G. Fowlicidin-3 is an alpha-helical cationic host defense peptide with potent antibacterial and lipopolysaccharide-neutralizing activities. FEBS J. 2007;274:418–28.
Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–4.
Brownlie R, Allan B. Avian toll-like receptors. Cell Tissue Res. 2011;343:121–30.
Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT. Metabolic syndrome and altered gut microbiota in mice lacking toll-like receptor 5. Science. 2010;328:228–31.
Nose F, Yamaguchi T, Kato R, Aiuchi T, Obama T, Hara S, Yamamoto M, Itabe H. Crucial role of perilipin-3 (TIP47) in formation of lipid droplets and PGE2 production in HL-60-derived neutrophils. PLoS One. 2013;8:e71542.
Covington JD, Galgani JE, Moro C, LaGrange JM, Zhang Z, Rustan AC, Ravussin E, Bajpeyi S. Skeletal muscle perilipin 3 and coatomer proteins are increased following exercise and are associated with fat oxidation. PLoS One. 2014;9:e91675.
MacEachern S, McEwan J, McCulloch A, Mather A, Savin K, Goddard M. Molecular evolution of the Bovini tribe (Bovidae, Bovinae): is there evidence of rapid evolution or reduced selective constraint in domestic cattle? BMC Genomics. 2009;10:179.
Innan H, Kim Y. Pattern of polymorphism after strong artificial selection in a domestication event. Proc Natl Acad Sci U S A. 2004;101:10667–72.
Bai Y, Sun G, Kang X, Han R, Tian Y, Li H, Wei Y, Zhu S. Polymorphisms of the pro-opiomelanocortin and agouti-related protein genes and their association with chicken production traits. Mol Biol Rep. 2012;39:7533–9.
Han R, Wei Y, Kang X, Chen H, Sun G, Li G, Bai Y, Tian Y, Huang Y. Novel SNPs in the PRDM16 gene and their associations with performance traits in chickens. Mol Biol Rep. 2012;39:3153–60.
Norup LR, Dalgaard TS, Friggens NC, Sorensen P, Juul-Madsen HR. Influence of chicken serum mannose-binding lectin levels on the immune response towards Escherichia coli. Poult Sci. 2009;88:543–53.
Fresno M, Alvarez R, Cuesta N. Toll-like receptors, inflammation, metabolism and obesity. Arch Physiol Biochem. 2011;117:151–64.
Himes RW, Smith CW. Tlr2 is critical for diet-induced metabolic syndrome in a murine model. FASEB J. 2010;24:731–9.
Kim KA, Gu W, Lee IA, Joh EH, Kim DH. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS One. 2012;7:e47713.
Lee JY, Zhao L, Youn HS, Weatherill AR, Tapping R, Feng L, Lee WH, Fitzgerald KA, Hwang DH. Saturated fatty acid activates but polyunsaturated fatty acid inhibits toll-like receptor 2 dimerized with toll-like receptor 6 or 1. J Biol Chem. 2004;279:16971–9.
Lee JY, Ye J, Gao Z, Youn HS, Lee WH, Zhao L, Sizemore N, Hwang DH. Reciprocal modulation of toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J Biol Chem. 2003;278:37041–51.
Kawashima T, Kosaka A, Yan H, Guo Z, Uchiyama R, Fukui R, Kaneko D, Kumagai Y, You DJ, Carreras J, et al. Double-stranded RNA of intestinal commensal but not pathogenic bacteria triggers production of protective interferon-beta. Immunity. 2013;38:1187–97.
Leshchinsky TV, Klasing KC. Divergence of the inflammatory response in two types of chickens. Dev Comp Immunol. 2001;25:629–38.
van Hemert S, Hoekman AJ, Smits MA, Rebel JM. Gene expression responses to a Salmonella infection in the chicken intestine differ between lines. Vet Immunol Immunopathol. 2006;114:247–58.
Leinonen R, Sugawara H, Shumway M. The sequence read archive. Nucleic Acids Res. 2011;39:D19–21.
Andrews S: FastQC A Quality Control tool for High Throughput Sequence Data [https://www.bioinformatics.babraham.ac.uk/projects/fastqc/]. Accessed 2 November 2017.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Li H, Durbin R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics. 2010;26:589–95.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21:974–84.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21.
Rambault A: FigTree [http://tree.bio.ed.ac.uk/software/figtree/]. Accessed 2 November 2017.
Homer N, Merriman B, Nelson SF. BFAST: an alignment tool for large scale genome resequencing. PLoS One. 2009;4:e7767.
Rumble SM, Lacroute P, Dalca AV, Fiume M, Sidow A, Brudno M. SHRiMP: accurate mapping of short color-space reads. PLoS Comput Biol. 2009;5:e1000386.
Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arXiv preprint 2012. arXiv preprint arXiv:12073907; 2018.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP effect predictor. Bioinformatics. 2010;26:2069–70.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8.
Narasimhan V, Danecek P, Scally A, Xue Y, Tyler-Smith C, Durbin R. BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics. 2016;32:1749–51.
Vilella AJ, Severin J, Ureta-Vidal A, Heng L, Durbin R, Birney E. EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009;19:327–35.
Ashburner M, Ball C, Blake J, Botstein D, Butler H, Cherry J, Davis A, Dolinski K, Dwight S, Eppig J, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9.
Kinsella RJ, Kahari A, Haider S, Zamora J, Proctor G, Spudich G, Almeida-King J, Staines D, Derwent P, Kerhornou A, et al. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database (Oxford). 2011;2011:bar030.
Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, et al. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2004;32:D115–9.
Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7.
Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B-Statistical. Methodology. 1995;57:289–300.
Acknowledgements
Not applicable
Funding
This work was funded by grants from the Natural Sciences and Engineering Research Council of Canada (RGPIN-2014-06664 / RGPIN-2019-06852), Alberta Livestock and Meat Agency, Ontario Ministry of Agriculture, Food and Rural Affairs and the Canadian Poultry Research Council. High performance computing was provided by the Centre for Computational Medicine at the SickKids’ Research Institute. The funders had no role in the design of the study, collection of data and analysis, preparation of the manuscript and decision to publish.
Author information
Authors and Affiliations
Contributions
AZ performed analyses and drafted manuscript; KN provided resources; PWW and JYL performed sequencing and contributed to analyses; DSG, SS, DK, JHB helped conceive the project, secure resources, provided input on analyses and edited the manuscript. JP helped conceive the project, secure resources, provided input on analyses, edited the manuscript and supervised the project. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethics approval for collection of blood from poultry for sequencing was obtained from the University of Alberta Animal Care and Use Committee: Livestock, protocol AUP00001401.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Breeds and Naming Convention for the 15 Samples Analyzed in this Study. The main text uses the abbreviation to refer to each breed. Genomes sequenced from pooled blood samples are specified in brackets in the “Name” column.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zou, A., Nadeau, K., Wang, P.W. et al. Accumulation of genetic variants associated with immunity in the selective breeding of broilers. BMC Genet 21, 5 (2020). https://doi.org/10.1186/s12863-020-0807-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-020-0807-z