
Abstract
Background
Amorphophallus albus P. Y. Liu & J. F. Chen (Araceae) is a plant species with extremely small populations (PSESP) and an important economic crop endemic to dry-hot valleys along the Jinsha River. In order to gain information for sustaining the development and conservation of A. albus, we studied the genetic diversity and population structure of this species using microsatellite markers (SSR). In this study, we analysed 364 individuals belonging to 24 populations, including four wild populations and three ex-situ cultivated populations, collected in the provinces Yunnan, Sichuan and Hubei.
Results
The population genetic analyses indicated that A. albus possesses moderate genetic diversity with the percentage of polymorphic loci (PPL) from 69.23 to 100%, an expected heterozygosity (He) of 0.504 and an average Shannon’s Information Index (I) 0.912. Analysis of molecular variance (AMOVA) indicated that most of the variance (71%) resided within populations and the estimated gene flow (Nm) was 0.61. The results of UPGMA cluster tree, STRUCTURE analyses together with the Mantel test (R2 = 0.352, P < 0.01) indicated that geographically closely located populations are clustered together with some exceptions.
Conclusions
Our results showed that A. albus still possesses moderate genetic variation in most of the studied populations, and for now, most cultivated populations were naturally distributed but still some reintroduction exists. For sustaining the present genetic variation, some protections measures are necessary for the wild populations and also for the cultivated ones with high genetic diversity.
Similar content being viewed by others

Background
Amorphophallus albus P. Y. Liu & J. F. Chen (Araceae) is a herbaceous perennial plant species occurring along the Jinsha River in southern Sichuan and northern Yunnan. It is growing in open forests between 800 to 1000 m altitude on arid locations [1]. It’s an economic crop widely used for food, medicine and industry due to the glucomannan (KGM) content in its tubers [2, 3]. The high quality and purity of KGM obtained from A. albus makes this species the second most cultivated Amophophallus species after A. konjac K. Koch in China [4]. At present, the cultivation of A. albus is one of the pillars in agriculture of counties along the Jinsha River. For example, in Jinyang, the cultivation area is more than 3333 ha with commodity production more than 30,000 kg and a production value of about 120 million Yuan every year [5]. Moreover, the resistance against high temperatures and drought tolerance of A. albus are important factors for the breeding of drought-resistant varieties [6]. Since it has been cultivated for hundreds of years, wild populations are almost disappeared. In 2017, A. albus was listed as a potential targeted PSESP (Plant Species with Extremely Small Populations) for the China National Key Program of Survey and Germplasm Conservation of Plant Species with Extremely Small Populations in Southwest China [7].
Genetic diversity is the variation of the genetic material of organisms and the basis for adaptation of species to the natural environment [8]. Characteristics as such provides many useful information about history, adaptive potentials and relationships, and is also basis for phylogeny or classification of taxa [9, 10]. Analyses of molecular markers, especially microsatellites, are widely applied to reveal genetic diversity of threatened species in recent years [11,12,13]. Endangered plant species usually have low genetic variation, like Abies ziyuanensis L.K. Fu & S.L. Mo (He = 0.337) [14], Elaeagnus mollis Diels (He = 0.2683, I = 0.3815) [15, 16]. According to Nybom [17], the average expected heterozygosity (He) of endemic plant species analyzed by microsatellite is 0.42, whilst for species with narrow distribution is 0.56 and 0.62 for widespread species, respectively. At present, studies focusing on genetic diversity of Araceae species were valued mostly by the first generation of molecular markers including RFLP [18], RAPD [19, 20], AFLP [21,22,23], only Amorphophallus paeoniifolius (Dennst.) Nicolson and Xanthosoma sagittifolium (L.) Schott were analysed by microsatellites [24, 25], and inter-simple sequence repeat (ISSR) markers [26,27,28]. Among these molecular markers, microsatellite markers have high mutation rate, large amount of information, large numbers of loci, and low requirements for DNA quantity/purity. Thus, they play an important role in genetic diversity of plant species [29].
In the present study, we used 13 pairs of microsatellite loci to analyse the genetic diversity and population structure of A. albus from 24 populations including four wild populations and 17 in-situ cultivated populations as well as three ex-situ populations for following purposes: 1) to explore the trends of natural formation and evolution; 2) to provide a theoretical basis for conservation; 3) to reveal the net of introduction into the present cultivation area and 4) to determine the origin of this species.
Results
Genetic diversity
In this study, we finally collected 364 individuals from 24 populations including four wild populations and 17 in-situ populations together with three ex-situ populations, each populations’ information are listed in Table 1. In addition, 13 pairs of microsatellite primers are screened to analyse genetic diversity and population structure of A. albus (Table 2). The genetic characters of 13 microsatellite loci are listed in Table 3, according to the results, the polymorphism information content (PIC) range from 0.439 to 0.869 with an average of 0.683, which indicate a high polymorphism and their suitability for genetic analysis, the null allele frequency range from 0.013 to 0.523 with an average of 0.195, meanwhile, only three loci (TR6, TR17, TR54) were detected with null alleles through Micro-checker software [30]. In total, 100 alleles were detected, each locus had 3–13 alleles with an average of 8.7 alleles per locus. The genetic diversity parameters assessed by these microsatellite primers are listed in Table 4. Briefly, the average allele number (Na) was 3.619, with a range from 1.846 (SDC/TWC) to 4.615 (HLX). The average effective allele number (Ne) was 2.372, with a range from 1.541 (SLC) to 3.404 (LIZ). The average Shannon’s Information Index (I), observed heterozygosity (Ho), expected heterozygosity (He) are 0.912, 0.528, 0.504 on average, respectively. The percentage of polymorphic loci (PPL) of each population ranged from 69.23 to 100%. Based on the results, population LIZ (He = 0.667, I = 1.245) and HLX (He = 0.654, I = 1.238) showed the highest genetic diversity, while population SLC (He = 0.293, I = 0.511) and SDC (He = 0.334, I = 0.494) showed the lowest genetic diversity.
Genetic differentiation
According to the results of AMOVA analysis, about 29.23% of the total genetic variation occurred among populations, whereas the remaining 70.77% of the variation occurred within populations (Table 5). The estimated population differentiation coefficient (Fst) and estimated gene flow (Nm) was 0.29 and 0.61, respectively. The results of F-statistics in each locus are shown in Table 3. The results indicated that the inbreeding coefficients (Fis) of most loci were less than zero with an average of − 0.04. The estimated population differentiation coefficient (Fst) of each locus ranged from 0.221 to 0.419, with an average of 0.321, the average gene flow (Nm) of all the loci was 0.560, almost identical to the results calculated by AMOVA.
Population structure
The genetic identities (above diagonal) and genetic distances (below diagonal) of population pairs were listed in Table S1. Among all the populations, the farthest genetic distance and lowest genetic identity existed in SJX and SDC, while MYZ and HB had the nearest genetic distance and highest genetic identity. The dendrogram based on Nei’s genetic distance (Fig. 1) showed that all the populations were clustered in four groups where geographically contiguous populations were more genetically related than distant populations. Specifically, the population SDC alone gathered into IV branch, two populations of Jinyang County (SJX, HLX) and a population of Zhaoyang District (TBC) in the south clustered into III branch, while three populations of Jinyang County (TSC, LGLH, SJX) together with five populations of Yongshan County (ML, HH, LIZ, XP, STC) in central part clustered into II branch. Moreover, the remaining populations from Leibo County, Pingshan County, part of Yongshan County in the north and other three ex-situ cultivated populations gathered into I branch. The Bayesian cluster analysis based on the STRUCTURE software run K from 1 to 24, according to the evaluation criteria and calculation formula of Evanno [31], the relationship of ΔK and K are shown in Fig. 2, the results indicated that ΔK reached the peak when K = 3. Thus, the populations were clustered into three branches (Fig. 3) by Bayesian cluster analysis. Among them, six populations from Jinyang County, one population from Yongshan and one population from Zhaoyang District clustered together. Four populations including three from Yongshan County and one from Suijiang County clustered together, the remaining population formed the biggest branch which includes populations from Yongshan County, Leibo County, Pingshan County and ex-situ cultivation. Lastly, the Mantel test showed that the population genetic distance was positively correlated with geographic distance (R2 = 0.352, Fig. 4).

Dendrogram based on Nei’s genetic distance of Amorphophallus albus. Colors represent different regions

Graph showing the relationship between ΔK and K

Structure dendrogram in clustering analysis among 24 populations of Amorphophallus albus

Mantel test for correlation of genetic and geographic distances in Amorphophallus albus
Discussion
In this study, 13 microsatellite loci were analyzed to reveal the genetic diversity and population structure of A. albus from 24 populations in Sichuan, Yunnan, Hubei Province and they all expressed high polymorphism with an average PPL of 95.19%. According to the results, we observed a moderate genetic diversity of this species (He = 0.504, I = 0.912). In comparison, the genetic diversity observed was lower than in other studied Amorphophallus species using microsatellite markers, e.g., in A. paeoniifolius (He = 0.598, I = 1.172) [32], but higher than the estimated mean of genetic diversity of endemic species (He = 0.42) summarized by Nybom [17]. Genetic diversity of plant species usually depends on their breeding system, distribution or life form [33, 34]. Generally, perennial species with wide distribution, self-incompatible mating system and seed dispersal by animals possess higher genetic diversity [35]. For A. albus, which is a perennial herb with limited distribution showing self-incompatible mating system and endozoochory, it is supposed to have relatively higher genetic diversity. However, as an important economic crop, A. albus was inevitably disturbed by human activities such as habitat destruction and over excavation in recent years similar to A. konjac [23]. Consequently, wild populations of A. albus can hardly be found in nature. Moreover, most farmers, who cultivated this species for commercial purposes, tend to use asexual reproduction to get more corms and shorter life cycles [36]. This finally led to a reduced genetic diversity which is clearly observable in the populations of SDC and JLC. In contrast, some cultivated populations still maintain high genetic diversity, even higher than those wild populations, like HLX and LIZ. Presumably, these populations were transplanted from their native habitats and cultivated without or just little human disturbances. Wild populations comprises of not more than 50 individuals may lose genetic diversity in bottleneck events. Another possible reason is, that the existed wild populations were feral from cultivated populations and did not possess much genetic variation originally. According to our results, the populations with high genetic diversity are almost in or around Jinyang County, whilst the populations with the lowest genetic diversity are present in Pingshan and Suijiang. Based on our results, we assume that Jinyang is the natural origin of A. albus, and the gene flow from Jingyang to Pingshan showed a trend of expanding towards east along the river. This pattern could also be observed from other species native to the dry and hot valleys along the Jinsha River [37, 38].
The genetic analysis of A. albus indicated a high level of differentiation (Fst = 0.29) and low gene flow (Nm = 0.61) among populations. According to Wright [39], populations show high genetic differentiation and low gene flow when Fst > 0.25/Nm < 1. High genetic differentiation may result from heterogeneous environments [40]. Though all the populations distributed along Jinsha River, much differences in temperature, humidity, vegetation form existed between the hot-dry valleys and warm-dry valleys [41]. Additionally, Araceae species commonly pollinated by small insects such as ants, beetles and hover flies [42, 43], and A. albus is pollinated by rove beetles (Tang et al., unpublished data). This small insect pollination mating system and the complex geography may have limited gene flow among populations and therefore promoted genetic differentiation of this species [44]. Moreover, though the fruits of A. albus possess traits for seed dispersal by birds, but this could not be observed.
The observed fixation coefficient (Fis) in most loci were less than zero (Table 3) which indicates a great excess of heterozygosity in this species. This is a common phenomenon resulting from the applied sampling strategy, asexual reproduction, heterosis and too small breeding populations [45,46,47]. Regarding A. albus, sampling may be one of the reasons because quite a number of sampled populations belonged to small populations of less than 50 individuals. Another important reason is asexual reproduction independent whether the plants are cultivated or growing the wild. During cultivation, the farmers usually cut inflorescences in order to get bigger tubers, meanwhile, asexual reproduction allows to harvest commercial konjak faster [23]. In latter case, there are always many ramets around an adult plant, which also could be observed from the related species A. paeoniifolius [32]. As a result, asexual reproduction seems to be the main reason for excess of heterozygosity in A. albus.
In this study, the results of UPGMA cluster tree, Bayesian cluster analysis and Mantel test indicated that the genetic distance was slightly positive correlated with the geographical distance, and geographically close populations are usually clustered together (Figs. 1 and 5). These results showed that most of the cultivated populations nowadays are collected from native populations. But some populations were put in different places between the two clusters analysis like MYZ. Those populations mostly are the important base of their county of A. albus cultivation, every year people buy corms from other counties to increase their own variety. On account of different algorithms of the two software, these populations may be treated differently. Thus, reintroduction was proved to exist in many populations. In addition, the occurrence of three ex-situ cultivated populations in cluster I together with populations of MYZ, BJ and SYC indicated an introduction of these populations either from Yongshan, Leibo or Pingshan County. Reintroduction of plants from MYZ in downstream areas is also conceivable. The occurrence of population SDC in cluster IV (Fig. 1) is may be caused by introgression after hybridization with A. konjac. Spatial proximity to the distribution area of the latter species together with the already proved cross-breeding of both species [6] support this assumption.

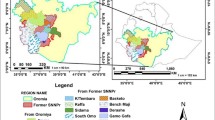
Geographical distribution of the sampled populations of Amorphophallus albus along the Jinsha River. Details of each location are given in Table 1
Conclusion
In conclusion, most populations of A. albus showed moderate genetic diversity due to short domestication history and weak artificial selection. Some of the studied populations showed a fairly low genetic diversity which may resulted from asexual reproduction or bottleneck effects. At present, most populations from the second branch still possess comparatively higher genetic diversity and therefore it is supposed that these populations are the center of genetic diversity of this species. Based on our results, we demand the three wild populations and the four cultivation populations of HH, HLX, XP and LIZ as conservation units to sustain most of the genetic variety of A. albus. As a next step, ex-situ conservation should also be undertaken in case of ongoing habitat destruction due to human activities. To ensure the genetic diversity, the sexual reproduction of this species must be promoted. These measures would counteract against degradation of this plant species.
Methods
Plant collection
Twenty-four populations of A. albus samples were collected in the dry-hot valleys along the Jinsha River in the provinces Yunnan and Sichuan together with three ex-situ cultivation populations from Yunnan and Hubei, China between September 2017 and October 2018. All the wild materials were collected outside at any natural reserves. All the cultivated materials were collected under the owner’s permission. In total, 364 individuals from 24 populations were sampled, 4–17 individuals were collected randomly in each population at intervals of 10 m. The collected plant issues were dried using silica gel. Detailed information about localities and samples are given in Table 1 and Fig. 5. Two voucher specimens were collected for each population and deposited in the herbarium of Kunming Institute of Botany, Chinese Academy of Sciences (code TR201701–TR201724).
DNA extraction, primer selection, PCR procedure, and product detection
The genomic DNA was extracted from approximately 5 g of dried leaves of each collected sample using the modified CTAB method [48]. DNA concentrations were estimated by nano drop spectrophotometer (ND 2000, USA) and the quality was analyzed by electrophoresis on 2% agarose gel. Microsatellite markers were designed and synthesized on the base of Genome Skimming data obtained from sequencing by MiSeq Benchtop Sequencer (Illumina) using MISA software. Totally 180 pairs novel microsatellite markers were developed, from which 80 pairs microsatellite markers were selected to amplify and finally 13 pairs microsatellite markers were successfully amplified with high polymorphism (microsatellite markers information are shown in Table 2). The polymerase chain reactions (PCR) were carried out at a volume of 20 μL containing 50 ng template DNA, 0.5 μL of each primer, 10 μL 2 × Taq PCR MasterMix (Tiangen: 0.1 U Taq Polymerase/μL, 0.5 mM dNTP each, 20 mM Tris-HCl (pH 8.3), 100 mM KCl, 3 mM MgCl2). PCR amplification was performed under the following conditions: 95 °C for 3 min, 32 cycles of 95 °C for 30 s, annealing at 56–60 °C for 30 s, and elongation at 72 °C for 30 s, and a final extension step at 72 °C for 5 min. The PCR products were separated and visualized using the QIAxcel capillary gel electrophoresis system (QLAGEN, Irvine, California, USA).
Data analysis
Data from QIAxcel capillary gel electrophoresis were analyzed by GeneMarker V. 2.2.0 to get allele fragment data. Micro-checker software was used to detect whether null alleles were present [30] and CERVUS software was used to calculate their frequency of each microsatellite loci [49]. Population genetic diversity parameters including average of sample sizes (N), average number of alleles (Na), effective number of alleles (Ne), Shannon’s information index (I), expected heterozygosity (He) and observed heterozygosity (Ho), fixation index (F) and percentage of polymorphic loci (PPL) were detected using GeneAlex version 6.0. F-statistics (Fis, Fit and Fst) were estimated for each locus across all populations using Fstat version 2.9.3.2. Genetic distances and genetic identity between each pair of accessions were measured from shared allele frequencies using PopGene 32. A dendrogram was constructed based on Nei’s genetic distance matrix using the MEGA version 4 software using the unweighted pair group method and the arithmetic averages (UPGMA) algorithm [50]. An analysis of the molecular variance (AMOVA) was used to verify the diversity within and among populations using Arlequin software version 3.5.1.3 [51]. A Mantel test [52] to compare pairwise geographic distance and pairwise genetic distance in terms of Fst / (1-Fst) with 1000 random permutations was conducted using NTSYSpc software version 2.10e [53]. The geographical distances among populations were calculated using the program Franson CoordTrans version 2.3. The population structure (the number of potentially different clusters) was assessed with a Bayesian-based cluster analysis using the program STRUCTURE version 2.3.4 [31]. Admixture model (AD) were tested with 10,000 replicates for burn-in and 10,000 replicates for Markov Chain Monte Carlo (MCMC) processes through five iterations (runs). To obtain the most probable K value (number of genetic groups), values of K from 1 to 24 were tested, with 10 independent runs for each K. The K value with the greatest probability was calculated estimating the maximum value of the ΔK statistic, according to Evanno et al. [54].
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Abbreviations
- AD:
-
Admixture model
- AFLP:
-
Amplified fragment length polymorphism
- AMOVA:
-
Analysis of molecular variance
- CTAB:
-
Cetyltriethylammnonium bromide
- F :
-
Fixation index
- Fis :
-
mean inbreeding coefficient within individuals relative to subpopulation
- Fit :
-
mean inbreeding coefficient within individuals relative to the total population
- F (null) :
-
Null allele frequency
- Fst :
-
mean inbreeding coefficient within subpopulation relative to the total population
- He :
-
The expected heterozygosity
- Ho :
-
The observed heterozygosity
- HW :
-
Hardy-Weinberg equilibrium
- I :
-
Shannon’s information index
- ISSR:
-
Inter-simple sequence repeat
- KGM:
-
Konjac glucomannan
- MCMC:
-
Markov Chain Monte Carlo
- N :
-
The average of sample sizes
- Na :
-
The average number of alleles
- Ne :
-
The effective number of alleles
- Nm :
-
Gene flow
- PCR:
-
The polymerase chain reactions
- PIC :
-
Polymorphism information content
- Pop:
-
Population
- PPL :
-
The percentage of polymorphic loci
- PSESP:
-
Plant species with extremely small populations
- RAPD:
-
Random amplification polymorphic DNA
- RFLP:
-
Restriction fragment length polymorphism
- UPGMA:
-
The unweighted pair-group method of arithmetic
References
Li H, Hetterscheid WLA. Araceae. In: Flora of China. Beijng & St. Louis: Science Press & Missouri Botanical Garden Press; 2000. p. 23–33.
Liu PY. Konjac. Beijing: China Agriculture Press; 2004.
Liu N, Yang F. An overview about konjac glucomannan research progress and its application. J Ankang Teachers Coll. 2011;23:95–8.
Liu EX. Study on identification technology adaptability evaluation and propagation characteristics in Amorphophallus germplasm. Enshi: Hubei Minzu University; 2016.
Li DG, Lei ZG, Jia BHL. Problems and countermeasures of white konjac industry in Jinyang County. Sci Technol Sichuan Agric. 2007;9:38–9.
Liu EX, Mou FG, Pan YN, Teng JX, Jiang QL, Zhao QH, Yang CZ. Study on the biological characteristics and mating ability of flower organs of Amorphophallus. Chinese Agric Sci Bull. 2011;27:126–31.
Yang J, Sun WB. A new programme for conservation of plant species with extremely small populations in south-West China. Oryx. 2017;51:396–7.
Ma KP, Qian YQ. Principles and methods of biodiversity research. Beijing: Science and Technology of China Press; 1994.
Zhang DQ, Yang YP. Statistical analysis of genetic diversity parameters of several commonly used molecular markers. Acta Bot Yunnanica. 2008;30:159–67.
Han JP, Zhang WS, Cao HB, Chen SL, Wang YY. Genetic diversity and biogeography of the traditional Chinese medicine, Gardenia jasminoides, based on AFLP markers. Biochem Syst Ecol. 2007;35:138–45.
Kalia RK, Rai MK, Kalia S, Singh R, Dhawan AK. Microsatellite markers: an overview of the recent progress in plants. Euphytica. 2011;177:309–34.
Zhang X, Zhang L, Schinnerl J, Sun WB, Chen G. Genetic diversity and population structure of Hibiscus aridicola, an endangered ornamental species in dry-hot valleys of Jinsha River. Plant Diversity. 2019;7:1–7.
Wenburg JK, Bentzen P, Foote CJ. Microsatellite analysis of genetic population structure in an endangered salmonid: the coastal cutthroat trout (Oncorhynchus clarki clarki). Mol Ecol. 1998;7:733–49.
Tang SQ, Dai WJ, Li MS, Zhang Y, Geng YP, Wang L, Zhong Y. Genetic diversity of relictual and endangered plant Abies ziyuanensis (Pinaceae) revealed by AFLP and SSR markers. Genetica. 2008;133:21–30.
Wang YL, Qin YY, Du Z, Yan GQ. Genetic diversity and differentiation of the endangered tree Elaeagnus mollis Diels (Elaeagnus L.) as revealed by Simple Sequence Repeat (SSR) Markers. Biochem Syst Ecol. 2012;40:25–33.
Qin YY, Wang YL, Zhang QD, Bi RC, Yan QG. Analysis on the population genetic diversity of an endangered plant (Elaeagnus mollis) by SSR Markers. J Wuhan Bot Res. 2010;28:466–72.
Nybom H. Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol Ecol. 2004;13:1143–55.
Sriboonma D, Hasebe M, Murakami N, Murata J, Iwatsuki K. Phylogeny of Typhonium (Araceae) inferred from restriction fragment analysis of chloroplast DNA. J Plant Res. 1993;106:11–4.
Acharya L, Mukherjee AK, Panda PC, Das P. Molecular characterization of five medicinally important species of Typhonium (Araceae) through random amplified polymorphic DNA (RAPD). Zeitschrift fur Naturforschung C. 2005;60:600–4.
Liao LC, Hsiao JY. Relationship between population genetic structure and riparian habitat as revealed by RAPD analysis of the rheophyte Acorus gramineus Soland. (Araceae) in Taiwan. Mol Ecol. 2002;7:1275–81.
Kreike CM, Van Eck HJ, Lebot V. Genetic diversity of taro, Colocasia esculenta (L.) Schott, in Southeast Asia and the Pacific ok. Theor Appl Genet. 2004;109:761–8.
Santosal E, Sugiyama N, Kawabata S, Hikosaka S. Genetic variations of Amorphophallus variabilis Blume (Araceae) in Java using AFLP. J Agronomi Indonesia. 2012;40:62–8.
Pan C, Gichira A, Chen JM. Genetic variation in wild populations of the tuber crop Amorphophallus konjac (Araceae) in Central China as revealed by AFLP markers. Genet Mol Res. 2015;14:18753–63.
Santosa E, Mine Y, Nakata M, Lian C, Sugiyama N. Genetic diversity of cultivated elephant foot yam (Amorphophallus paeoniifolius) in Kuningan, West Java as revealed by microsatellite markers. J Appl Hortic. 2010;12:125–8.
Cathebras C, Traore R, Malapa R, Risterucci AM, Chaïr H. Characterization of microsatellites in Xanthosoma sagittifolium (Araceae) and cross-amplification in related species. Appl Plant Sci. 2014;2:1–5.
Prakashkumar B, Sivu AR, Pradeep SN. Analysis of genetic diversity of Lagenandra spp. (Araceae) of Kerala (South India) using ISSR markers. Int J Sci Res. 2015;4:775–7.
Teng CZ, Diao Y, Chang FHS, Xie SQ, Han YH, Hu LZ. ISSR analyses of relative relationships among germplasm resources of Amorphophallus Blume from Yunnan Province. Anhui Agric Sci Bull. 2006;12:54–6.
Ren PY, Pan MQ. Population genetic structure of five Amorphophallus species from the south of Yunnan Province by Inter-Sequences ISSR markers. J Wuhan Univ (Nat Sci Ed). 2013;59:99–104.
Powell W, Morgante M, Andre C, Hanafey M, Vogel J, Tingey S, Rafalski A. The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol Breed. 1996;2:225–38.
Oosterhout CV, Hutchinson WF, Wills DPM, et al. Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Resour. 2004;4:535–8.
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14:2611–20.
Santosa E, Lian CL, Sugiyama N, Misra RS, Boonkorkaew P, Thanomchit K. Population structure of elephant foot yams (Amorphophallus paeoniifolius (Dennst.) Nicolson) in Asia. PLoS One. 2017;12:1–18.
Loveless MD, Hamrick JL. Ecological determinants of genetic structure in plant populations. Annu Rev Ecol Syst. 1984;15:65–95.
Hamrick JL, Godt MJW. Effects of life history traits on genetic diversity in plant species. Philos Trans R Soc B. 1996;351:1291–8.
Nybom H, Bartish IV. Effects of life history traits and sampling strategies on genetic diversity estimates obtained with RAPD markers in plants. Perspect Plant Ecol. 2000;3:93–114.
Liu EX, Yang CZ, Zheng XJ, Mou FG, Xun N, Pan YN, Wu P, Wang L, Xiong K, Xie YM. Adaptability evaluation of Amorphophallus albus. Chinese Agric Sci Bull. 2006;32:57–63.
Fan DM, Yue JP, Nie ZL, Li ZM, Comes HP, Sun H. Phylogeography of Sophora davidii (Leguminosae) across the ‘Tanaka-Kaiyong line’, an important phytogeographic boundary in Southwest China. Mol Ecol. 2013;22:4270–88.
Ying LX, Liu Y, Chen ST. Simulation of the potential range of Pistacia weinmannifolia in Southwest China with climate change based on the maximum-entropy (Maxent) model. Biodivers Sci. 2016;24:453–61.
Wright S. The interpretation of population structure by F-statistics with special regard to systems of mating. Evol. 1965;19:395–420.
Wang H, Pei D, Gu RS, Wang BQ. Genetic diversity and structure of walnut populations in central and southwestern China revealed by microsatellite markers. J Am Soc Hortic Sci. 2008;133:197–203.
Jin ZZ. Study on the floristic composition of seed plants in the dry-warm valley of Yunnan and Sichuan. Guihaia. 1998;18:313–21.
Zhang Y, Ye Q. Breeding system of the endangered species Psilopeganum sinense and its pollination process in a botanical garden. Plant Sci J. 2011;29:599–606.
Gibernau M. Pollinators and visitors of aroid inflorescences: an addendum. Aroideana. 2011;34:70–83.
Yang J, Li XD, Li XW, Shi QF, Li JQ. Analysis of AFLP variation of the endemic and rare species Psilopeganum sinense in Central China. J Wuhan Bot Res. 2007;25:226–34.
Keller JK, Waller DM. Inbreeding effects in wild populations. Trends Ecol Evol. 2002;17:230–41.
Stoeckel S, Grange J, Fernández JF, Bilger I, Frascaria-Lacoste N, Mariette S. Heterozygote excess in a self-incompatible and partially clonal forest tree species —Prunus avium L. Mol Ecol. 2006;15:2109–18.
Rasmussen KK, Kollmann J. Low genetic diversity in small peripheral populations of a rare European tree (Sorbus torminalis) dominated by clonal reproduction. Conserv Genet. 2008;9:1533–9.
Doyle JJ, Doyle JL. A rapid DNA isolation procedure from small quantities of fresh leaf tissues. Phytochemistry. 1987;19:11–5.
Kalinowski ST, Taper ML, Marshall TC. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol Ecol. 2007;16:1099–106.
Tamura K, Nei M, Kumar S. MEGA 4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–9.
Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinf Online. 2005;1:1–47.
Mantel N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967;27:209–20.
Rohlf FJ. NTSYS-PC: numerical taxonomy and multivariate analyses systems. Version 2.10m. New York: Exeter Publishing; 2000.
Pritchard JK, Stephans MJ, Donnelly PJ. Inference of population structure using multilocus genetype data. Enetics. 2000;155:945–59.
Acknowledgments
We are grateful to Jun Lu and Bao-Guo Huang for their helps in materials collection. We thank Professor Heng Li for valuable discussion and we thank local resident for their help in conservation initiatives. We also thank Molecular Biology Experiment Center, Germplasm Bank of Wild Species in Southwest China for technical support in designing and developing microsatellite primers of A. albus.
Funding
Support for this study was provided through grants from the NSFC-Yunnan joint fund to support key projects (grants to U1602264), the Yunnan Ten Thousand Talents Plan Young & Elite Talents Project (YNWR-QNBJ-2018-017), the Yunnan Science and Technology Innovation Team Program for PSESP (Plant Species with Extremely Small Populations) Conservation and Utilization (Grant No. 2019HC015), and the Young Academic and Technical Leader Raising Foundation of Yunnan Province (2015HB091). The role of the funding bodies is limited to direct funding of the project activities in the field and laboratory that results in this manuscript.
Author information
Authors and Affiliations
Contributions
GC and WBS designed the experiments. RT and EXL collected the materials. RT conducted the experiments and wrote the manuscript. YZZ analysed the data. JS, GC together with WBS completed modification of the content and language in this manuscript. GC and WBS contributed equally to this work. All of the authors read and agreed to the manuscript and this submission.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1
. Paired Nei’s genetic distance (below diagonal) and genetic identity (above diagonal) of 24 populations of Amorphophallus albus
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Tang, R., Liu, E., Zhang, Y. et al. Genetic diversity and population structure of Amorphophallus albus, a plant species with extremely small populations (PSESP) endemic to dry-hot valley of Jinsha River. BMC Genet 21, 102 (2020). https://doi.org/10.1186/s12863-020-00910-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-020-00910-x