
Abstract
Background
Fibrotic idiopathic interstitial pneumonias (fIIP) are a group of fatal lung diseases with largely unknown etiology and without definitive treatment other than lung transplant to prolong life. There is strong evidence for the importance of both rare and common genetic risk alleles in familial and sporadic disease. We have previously used genome-wide single nucleotide polymorphism data to identify 10 risk loci for fIIP. Here we extend that work to imputed genome-wide genotypes and conduct new RNA sequencing studies of lung tissue to identify and characterize new fIIP risk loci.
Results
We performed genome-wide genotype imputation association analyses in 1616 non-Hispanic white (NHW) cases and 4683 NHW controls followed by validation and replication (878 cases, 2017 controls) genotyping and targeted gene expression in lung tissue. Following meta-analysis of the discovery and replication populations, we identified a novel fIIP locus in the HLA region of chromosome 6 (rs7887 P meta = 3.7 × 10−09). Imputation of classic HLA alleles identified two in high linkage disequilibrium that are associated with fIIP (DRB1*15:01 P = 1.3 × 10−7 and DQB1*06:02 P = 6.1 × 10−8). Targeted RNA-sequencing of the HLA locus identified 21 genes differentially expressed between fibrotic and control lung tissue (Q < 0.001), many of which are involved in immune and inflammatory response regulation. In addition, the putative risk alleles, DRB1*15:01 and DQB1*06:02, are associated with expression of the DQB1 gene among fIIP cases (Q < 1 × 10−16).
Conclusions
We have identified a genome-wide significant association between the HLA region and fIIP. Two HLA alleles are associated with fIIP and affect expression of HLA genes in lung tissue, indicating that the potential genetic risk due to HLA alleles may involve gene regulation in addition to altered protein structure. These studies reveal the importance of the HLA region for risk of fIIP and a basis for the potential etiologic role of auto-immunity in fIIP.
Similar content being viewed by others


Background
The fibrotic idiopathic interstitial pneumonias (fIIPs) are a group of lung diseases characterized by progressive fibrosis of the alveolar interstitium that leads to significant morbidity and mortality; median survival of the most common form, idiopathic pulmonary fibrosis, is 2–3 years after diagnosis. Although pirfenidone [1] and nintedanib [2] have recently been shown to slow the progression of IPF, only lung transplantation has been proven to prolong survival. There is evidence for the importance of environmental factors [3–13] and both rare [14–21] and common genetic variation [22–29] in risk of fIIP. Cigarette smoking is the strongest known environmental risk factor for fIIP [30, 31], but up to one third of individuals with fIIP do not have a history of cigarette smoking.
Previously, we reported 10 genetic risk loci for fIIP based on a genome-wide association study; in aggregate, the genome-wide genotyped SNPs account for 31–33 % of the variation in risk of developing this disease [32]. The functions of the genes implicated to date in risk of fIIP indicate that host defense from inhaled insults, barrier function of the alveolar epithelium, and telomere maintenance are compromised in at least a subset of individuals with fIIP. While these studies have been revealing, the majority of risk for fIIP remains unexplained, suggesting that additional studies to identify genetic variation are warranted. In the current study, we attempt to identify additional fIIP genetic risk variants via genome-wide imputation association analyses using data from 1616 cases and 4683 out-of-study controls from our genome-wide association study (GWAS) [32]. We identify the human leukocyte antigen (HLA) region as a genetic risk locus for fIIP, demonstrating a potential role of auto-immunity in fIIP.
Results and discussion
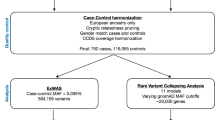
An overview of the study design is shown in Fig. 1.

Overview of Study Design. A discovery imputation GWAS among 1616 cases and 4683 controls was followed by validation and replication genotyping in 878 cases and 2017 controls. One novel locus was identified on Chromosome 6p21. Classical HLA alleles were imputed using genotyped SNPs among the GWAS cases and controls and were tested for association with fIIP. Lung tissue gene expression was compared between a subset of GWAS cases and non-GWAS controls and across genotypes for the most significant 6p21 SNP, rs7887. Gene expression was also compared across genotypes of the most significant HLA alleles (DQB1*06:02, DRB1*15:01) within the cases with lung tissue expression data
Imputation study
We imputed genotypes using the HapMap Phase 3 integrated panel [33] using IMPUTE2 [34, 35] and genotypes at 439,828 SNPs from the Illumina 660 Quad beadchip that met strict quality control criteria [32]. We tested for association at 998,072 imputed SNPs under an additive model for each SNP with imputation information as measured by .info > 0.5 using SNPTEST v2 [36]. We adjusted the p-values (divided each test statistic by 1.08, then recomputed p-values) for these imputed SNPs based on the inflation in the imputation test statistics (Additional file 1: Figure S1) observed among the 439,828 genotyped SNPs compared to the imputation test statistics for those SNPs obtained from an exact mixed model that accounts for subtle stratification and cryptic relatedness among the cases and controls [37]. After this adjustment and removal of the regions known to be associated with fIIP [32], neither the Q-Q plot of p-values (Additional file 1: Figure S2) nor the genomic control inflation factor (λ = 1.04) suggested systematic departures from the null hypothesis.
We identified 205 SNPs (Additional file 1: Table S1) associated with fIIP at a genome-wide level of significance (Additional file 1: Figure S3); 204 of these SNPs are in eight fIIP risk loci (3q26, 4q22, 5p15, 6p24, 7q22, 11p15, 15q14, and 17q21) we had previously reported. The remaining SNP, rs2169877 (P imputed = 2.8 × 10−08), is on chromosome 15q25 in an intron of the AKAP13 gene. We selected 15 of the genome-wide significant SNPs (including rs2169877) in addition to 152 SNPs among the 337 SNPs with 5 × 10−08 < P imputed <0.0001 for validation and replication genotyping (Additional file 1: Table S2); to avoid redundancy with known associated SNPs and among the SNPs selected for follow-up, we selected SNPs in weak LD (r 2 < 0.5) with previously-known genome-wide significant SNPs [32] and chose the most strongly-associated SNP among potential replication SNPs with pair-wise r 2 > 0.8. After genotype quality control and removal of putative duplicate samples between the GWAS and replication cases (see Methods), we successfully genotyped 1498 of the discovery GWAS cases for validation (we do not have access to GWAS out-of-study control DNA) and 878 cases and 2017 controls for replication at 148 of the SNPs.
After meta-analysis of the imputation and replication phases, 24 of the 148 SNPs were associated with fIIP at a genome-wide level of significance (P Meta ≤ 2.8 × 10−08; Additional file 1: Table S3). Among these, 6 SNPs (rs614549, rs7887, rs2844452, rs3020644, rs2280774, and rs3117116) represented 1 novel locus for fIIP in the HLA region at Chromosome 6p21; rs7887 in EHMT2 was the most strongly-associated SNP (P Meta = 3.7 × 10−09, Table 1; regional association shown in Fig. 2). All 6 SNPs had imputation.info scores >0.98. Rs2169877 at 15q25 was not genome-wide significant in the meta-analysis (P Meta =1.6 × 10−7).

Locus-specific plot for HLA region corresponding to discovery imputation GWAS results. The –log10 P values (y axis) of the SNPs are shown according to their chromosomal positions (x axis). The estimated recombination rates (cM/Mb) from the HapMap Project (NCBI Build 36) are shown as light blue lines, and the genomic locations of genes within the regions of interest in the NCBI Build 36 human assembly are shown as arrows. SNPs shown in red, orange, green, light blue and blue have r 2 ≥ 0.8, r 2 ≥ 0.6, r 2 ≥ 0.4, r 2 ≥ 0.2 and r 2 < 0.2 with the most highly-associated SNP, respectively. SNPs with no r 2 information with most-highly associated SNP shown in grey. Circles correspond to genotyped SNPs, squares correspond to imputed SNPs. P-values correspond to discovery cohort statistical evidence only; meta-analysis p-values can be found in Table 1 and Additional file 1: Tables S1-S3
Identification of independent associations within each locus
Among the genome-wide significant SNPs in regions previously known to be associated with fIIP, we compared the evidence for association between the newly-identified SNPs (N = 24) and those we reported previously (N = 46). Based on both the meta-analysis p-values and joint analysis of the combined GWAS and replication cases compared to replication controls, the top SNP remained the same as originally reported except for the 3q26 and 4q22 regions (Additional file 1: Table S3). At 3q26, rs12696304 near TERC (P Meta = 8.2 × 10−14) was more significant than rs6793295 (P Meta = 8.3 × 10−13), and at 4q22 in FAM13A, rs2609261 (P Meta = 2.4 × 10−12) was more significant than rs2609255 (P Meta = 2.2 × 10−11).
To assess the evidence for multiple independent association signals within each region accounting for our original GWAS (46 genome-wide significant SNPs) and the 24 SNPs identified in this study, we tested for association with each SNP in a given region after adjusting for the most significant SNP in that region based on the meta-analysis. We compared the combined case group (GWAS discovery + replication) to the replication control group. There was no strong evidence for independent signals within each region (Additional file 1: Table S4). In particular, in the HLA region, no other SNP was associated with fIIP after adjustment for rs7887 (all P >0.02; Table 1).
Classic HLA allele associations
To further interrogate the HLA region of association, we imputed classic HLA alleles for the discovery cohort using the HLA genotype imputation with attribute bagging (HIBAG) [38] pre-computed imputation probabilities for HLA-A, HLA-B, HLA-C, HLA-DRB1, HLA-DQ (A1, B1), and HLA-DPB1. The median posterior probabilities for the imputed alleles for each gene were >0.92 for all but the DRB1 gene (Additional file 1: Table S5). We tested for association under an additive (on the log-odds scale) model for each allele (Additional file 1: Table S6) and with each amino acid. Three alleles were associated with fIIP at a nominal p-value < 1 × 10−5, the level of association demonstrated by rs7887 in the GWAS discovery: DQA1*01:02 (P = 4.8 × 10−6), DQB1*06:02 (P = 6.1 × 10−8), and DRB1*15:01 (P = 1.3 × 10−7); DQB1*06:02 and DRB1*15:01 are very in high LD. While not genome-wide significant using the standard Bonferroni adjustment to achieve a study-wide α = .05 (5 × 10−8 corresponds to .05 divided by 1 million) based on the discovery set, both alleles are significant after false discovery rate adjustment for all the genome-wide SNP and HLA imputation statistical tests we conducted (FDR adjusted p-values = 0.015 and .029 for DQB1*06:02 and DRB1*15:01, respectively). After a step-wise analysis to delineate the conditionally independent HLA allele associations (see Methods), only DQB1*06:02 remained nominally significant. In a model with both the DQB1*06:02 allele and rs7887, both remained nominally significant (P adj = 4.8 × 10−6, and 1.0 × 10−3, respectively; Table 2), indicating the potential for more than one independent signal in the region (r 2 between rs7887 and DQB1*06:02/DRB1*15:01 = 0.22). Since the DRB1 alleles were imputed with less confidence than the DQB1 and DQA1 alleles (median posterior probability 0.89, 0.97 and 0.94, respectively), and each of the three HLA alleles are nominally significant after adjustment for rs7887 (Table 2), it is possible that the DQB1 alleles appear to be the only independent association due to less precision in allele definition for DRB1.
Lung tissue expression studies
To begin to explore the biologic relevance of the association evidence, we compared lung tissue gene expression via amplicon-based targeted RNA-seq between 87 cases of idiopathic pulmonary fibrosis (IPF, the most common form of fIIP) and 70 unaffected subjects for 66 genes in the 1.54 Mb of DNA defined by HLA region association (Additional file 1: Table S7); of the 86 genes annotated in the region, 75 could be designed (DQA1 and DQB1 not among them) and 9 did not pass QC thresholds (see Methods). We identified 34 genes that were differentially expressed in lung tissue from patients with IPF vs. controls at an FDR of 0.05 (Additional file 1: Table S8); 21 were differentially expressed at an FDR of <1 × 10−3 (Fig. 3; Additional file 1: Table S8). Eight of these 21 genes have a known immune or inflammatory function (C6orf25, LTA, LTB, AGER, HLA-DOB, C4A, C4B, and MICA). In addition, two of the genes (HSPA1L and HSPA1B) encode heat-shock proteins important for mediating protein folding, a mechanism thought to be involved in the pathogenesis of IPF [39].

Targeted RNA-Seq Expression Differences between IPF and Control Lung. The size of the dots corresponds to q-value; larger dots have smaller q-values. The color of the dots corresponds to the direction of expression changes; genes in blue have lower expression in case lung tissue compared to control lung tissue, genes in red have higher expression
To determine which of the gene expression changes are more likely important for development of disease as opposed to a result of the disease process, we explored whether either rs7887 or the HLA risk alleles are expression quantitative trait loci for genes in the region. We tested whether the alleles at rs7887 were associated with expression among the cases and controls separately. The alleles at rs7887 were not associated with expression of any gene after FDR correction in either group (Additional file 1: Table S9). Among cases, both the DRB1*15:01 and DQB1*06:02 alleles were associated with expression of the AIF1 gene (Q = 1.2 × 10−5 and Q = 1.2 × 10−5, respectively, Additional file 1: Tables S10 and S11); we were not able to infer HLA alleles for the controls in the RNA-seq study since they did not have genome-wide genotyping available. Several HLA genes, including the DQB1 gene, were not represented in our RNA-seq expression data, but were represented in existing Affymetrix gene array expression data for 66 IPF cases. The DRB1*15:01 and DQB1*06:02 alleles were associated with increased expression of the DRB5 (Q = 6.7 × 10−22 and Q = 1.06 × 10−21, respectively) and DQB1 (Q = 8.7 × 10−10 and Q = 1.93 × 10−17, respectively) genes, suggesting that the alleles may have etiologic effects through more than just the structure of their respective binding proteins.
Discussion
Similar to our other GWAS loci, the HLA fIIP risk locus will require follow-up resequencing in a large number of cases and controls to define the independent variant(s) contributing to fIIP risk. Several other small studies have implicated the HLA region in fIIP with little consensus on the alleles responsible [40–43]. Of particular interest given our findings, the DRB1*15:01 allele has been reported to be associated with IPF among a small sample from China [41]. In the time since we conducted the imputation analyses and conducted replication genotyping, more comprehensive imputation panels have been made available. We have conducted further imputation analyses based on the 1000 genomes panel; while there are no additional genome-wide signals in the absence of replication genotyping, there are additional SNPs that would be candidates for follow-up based on the screening approach we used in this study (Additional file 1: Table S12).
Our findings should be understood in the context of fibrotic interstitial lung disease, which is a relatively common manifestation of several immune-related disorders, including scleroderma, rheumatoid arthritis, and systemic lupus erythematosis. Interestingly, the DRB1:15:01 allele has been reported to be protective for development of systemic sclerosis among White and Hispanic subjects [44]. While we explicitly excluded cases with recognized connective tissue disease-associated or other known syndromic forms of fIIP, our findings highlight the concept that auto-immunity may drive the development of fIIP in a subset of patients with this disease. A recent case series indicated that 29 % of biopsy-confirmed IPF patients (that is, those without a characterized connective tissue disease or other known cause of their ILD) who underwent serology testing had a positive serology for auto-immunity [45]. In addition, the DRB1*15 and DQB1*06 alleles have been associated with interstitial lung disease in a sample of rheumatoid arthritis patients in Japan [43].
Conclusions
We have identified the HLA region as associated with fIIP. Two strongly-linked HLA alleles are associated with fIIP and affect expression of HLA genes in lung tissue, indicating that the potential genetic risk due to HLA alleles may involve gene regulation in addition to altered protein structure. In aggregate, the genome-wide genotyped and imputed SNPs are estimated to account for 35 % of the variability in risk of fIIP, a disease that was previously thought to be idiopathic. Importantly, the HLA locus and other risk loci will facilitate the identification of genes and processes that are involved in the etiology of fIIP that may lead to breakthroughs in disease pathogenesis and intervention. In particular for the HLA locus, linking the putative risk alleles to differential expression in tissue from the lung is one way to narrow the list of expression differences between cases and controls to those that are more likely to be drivers of disease risk as compared to consequences of the disease process. In addition, the association between the putative HLA risk alleles and transcript abundance of HLA genes provides alternative avenues of investigation into the functional role of the alleles.
Methods
Study populations, ethics, permission and consent
Case definition
We used standard criteria established by the American Thoracic Society/European Respiratory Society [46] to determine diagnostic classification of all patients in the discovery and replication phases. We excluded cases with known explanations for development of fibrotic FIIP including infections, systemic disorders, or relevant exposures (e.g. asbestos). To maximize power and minimize potential confounding by ancestry, we included only non-Hispanic white (NHW) participants that had been recruited as part of existing studies. All subjects gave written informed consent as part of IRB-approved protocols for their recruitment at each site (National Jewish Health, InterMune, Vanderbilt University, National Heart Lung and Blood Institute, Duke University, University of Pittsburgh, University of Texas Southwestern, Centre National de Génotypage, National Institute for Health Research Biomedical Research Unit, Royal Brompton Hospital, University of California San Francisco) and the GWAS study was approved by the National Jewish Health IRB and Colorado Combined Institutional Review Board (COMIRB).
GWAS discovery
We genotyped 1914 patients with fIIP from 7 cohorts (familial interstitial pneumonia [n = 566], National Jewish Health FIIP population [n = 238], InterMune IPF trials [n = 720], UCSF [n = 66], Vanderbilt University FIIP population [n = 105], and the National Heart Lung and Blood Institute Lung Tissue Research Consortium [n = 219]) and compared them to genotypes from 4683 out-of-study controls. After genotype quality control, we included 1616 cases in analyses.
A family with familial interstitial pneumonia (FIP) is defined by the presence of at least 2 cases of definite or probable IIP in individuals who are 3rd degree relatives or closer. Recruitment of families based at three major referral centers (Vanderbilt University, Duke University and National Jewish Health) has been ongoing since 1999. We included only 1 fIIP case among first degree relatives. The National Jewish Health fIIP cohort consists of patients with sporadic fIIP who were clinically evaluated and enrolled at National Jewish Health as part of ongoing natural history studies. Details of the recruitment criteria for the cases from the Intermune IPF γ-Interferon Intervention Trial have been described in detail [47]. Briefly, eligible patients had IPF, were 40 to 79 years old with clinical symptoms for at least 3 months and evidence of disease progression within the previous 12 months. We included all available cases regardless of treatment assignment. The National Heart Lung and Blood Institute Lung Tissue Research Consortium (NHLBI LTRC) was established to provide lung tissue and DNA for the research community. We included DNA from those subjects with a diagnosis of fIIP after receiving permission from the NHLBI LRTC.
We used de-identified control genotypes generated at Centre d’Étude du Polymorphisme Humain (CEPH) as part of other studies. Potential controls were those who self-reported NHW, had been genotyped on the same platform as our cases, and were appropriately approved for use as controls in other studies. We selected a subset of controls, corresponding to approximately 3 controls for 1 case, based on genetic similarity to the cases which passed our genotyping quality control thresholds as previously described [32].
Replication
We genotyped a total of 978 NHW fIIP cases and 2052 NHW controls for replication of the top SNPs from the GWAS. The replication controls were a subset (n = 1926) of the controls from the Chronic Obstructive Pulmonary Disease (COPD) Gene Study [48] and 126 controls from the University of Pittsburgh. We selected controls to be frequency matched to the replication cases based on age and gender. To exclude putative duplicate cases within the replication and between the GWAS and replication case groups, we calculated the kinship coefficient for each pair of individuals using the 329 SNPs constituting the combined replication genotyping from the original (181 SNPs) and imputation (148 SNPs) GWAS studies. There were no duplicate individuals within the GWAS; we removed 39 replication cases with kinship coefficient > .45 with either a GWAS case or another replication case. After duplicate removal and genotype quality control (see below), we included 878 cases and 2017 controls in any analyses that included replication samples.
Expression
We measured gene expression on a subset of Lung Tissue Research Consortium (n = 50) and National Jewish Health fIIP cases from the GWAS (n = 37) and National Jewish Health controls (n = 70). Whole-lung samples were obtained from International Institute for the Advancement of Medicine (Edison, NJ). Eligible cases and controls had sufficient RNA from lung tissue biopsy available for assay; cases with IPF were preferentially chosen over other fIIP diagnoses. Treatment status of IPF cases were not available for the 37 National Jewish Health cases; of the 50 LTRC IPF patients included in the RNA expression analysis, 33 (66 %) were on systemic steroid treatment, 14 (28 %) were not on systemic steroid treatment and 3 (6 %) did not have information on treatment status.
Genotyping
Genome-wide genotyping was carried out at CEPH using the Illumina 660 Quad beadchip (www.illumina.com). Details of the quality control and experiments have been reported in detail previously [32]. Replication genotyping was carried out at the Biomedical Genomics Center at the University of Minnesota. For validation and replication genotyping of imputation association results, we attempted to genotype 167 SNPs with adjusted imputation P-values less than 0.0001 (see Statistical Analyses) in 1027 independent cases and 2052 replication controls. In addition, to allow follow-up joint statistical tests (using raw genotypes from both GWAS cases and replication cases and controls) and validate the imputation genotypes, we also genotyped a large subset of GWAS cases (N = 1578). Details of the validation assays are described below. After genotyping quality control, we included 878 cases and 2017 controls in the replication, meta- and joint analyses and 1498 of the GWAS cases in the joint analyses.
Prior to genotyping, all samples were quality controlled by real-time Q-PCR quantitation (“QC1”) and uniplex genotyping using Taqman (“QC2”). Samples that failed QC1 or QC2, although carried forward through genotyping, were later removed from analysis.
Validation genotyping was accomplished with a combination of multiplexed (Sequenom iPLEX) and uniplex (Taqman) assays. First, assay design for multiplexed Sequenom iPLEX genotyping was performed on an input set of 167 SNPs (Additional file 1: Table S2), using a combination of web-based (AssayDesigner Suite, www.sequenom.com) and desktop (AssayDesigner) software tools (Sequenom, San Diego). Sequenom iPLEX genotyping is based on multiplexed locus-specific PCR amplification, multiplexed single-based extension (SBE) from locus-specific amplicons, and multiplexed resolution of SBE products base calling using matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOD) mass spectrometry.
Primers for the Sequenom assay were purchased from IDT (Coralville, Iowa), and all steps of the iPLEX procedure were carried out using reagents and methods from Sequenom (San Diego, CA) according to the manufacturer’s instructions. Reactions were carried out in 384-well plates and analyzed using the Sequenom MassARRAY Analyzer 4 system with iPLEX Gold reagents and SpectroCHIP arrays. Results were analyzed using a combination of commercial software (Typer 4, Sequenom) and custom tools for data management. Of 167 assays in 6 multiplexes, 148 were successful in generating usable genotyping data.
Gene expression
Total RNA was isolated from approximately 30 mg of snap-frozen or RNA-later preserved lung tissue using the Ambion mirVana kit (Life Technologies). RNA concentration was determined by Nanodrop ND-1000 (Thermo Scientific) and RNA integrity was determined using the 2100 Bioanalyzer (Agilent). cDNA single strand conversions were performed using the Superscript III.
Statistical analyses
Statistical analyses for the discovery GWAS based on observed genotypes, including selection of out-of-study controls, removal of genetic outliers, SNP quality control and tests for association have been described previously in detail [32].
Imputation
We imputed genotypes using the combined case and control discovery samples for all HapMap SNPs not on the Illumina 660 Quad beadchip across the entire genome. We used the multi-population reference panel data from HapMap3 for pre-phasing using Shapeit with appropriate default parameters [49, 50]. We performed imputation using IMPUTE2 [34, 35] and tested for association at only those SNPs with imputation information as measured by .info > 0.5 using SNPTEST (v2; [36]) with multiple Newton–Raphson iterations to estimate parameters. We adjusted for sex in all models. We computed the inflation factor across all the SNPs as the ratio of the median of the observed 1 degree of freedom chi-square test statistic obtained from the SNPTEST p-value to the theoretical median of 1 degree of freedom chi-square random variable. We adjusted the imputed p-values based on a comparison between the imputation test statistics and exact mixed model test statistics (from GEMMA [37]) for SNPs that were genotyped. For 439,827 SNPs that were genotyped and also imputed with .info > 0.5 (see Additional file 1: Figure S4 for distribution of .info scores), we computed the inflation factor as the ratio of the median of the observed 1 degree of freedom chi-square test statistics obtained from SNPTEST p-value from genotyped snps and the theoretical median of 1 degree of freedom chi-square random variable. We then computed adjusted imputed p-values across the genome by dividing the observed chi-square test statistic by the inflation factor. We compared the distribution of these adjusted p-values obtained under the additive model to that expected under the null hypothesis of no association across the genome and report the quantile-quantile (Q-Q plot) and genomic inflation factor (λ) to verify the absence of systematic biases. We selected a subset of SNPs with an adjusted imputation p-value <0.0001 for follow-up in the replication populations. Within novel loci, we selected the top SNP within a 500 kb window, and then only selected another SNP if that SNP had an estimated r 2 < 0.8 with the top SNP. We continued this process until all remaining SNPs had r 2 ≥ 0.8 with at least one selected SNP. Within previously-known loci, we used the top SNP from our original GWAS, and then only selected another SNP if that SNP had an estimated r 2 < .5 with the top SNP.
Replication association
We tested for association between each replication SNP and fIIP under an additive model (on the log-odds scale) in the replication cases and controls and estimated odds ratios and 95 % confidence intervals using PLINK. For SNPs with minor allele frequency < 5 %, we computed a permutation-based p-value with 100,000 permutations. The p-values were used in the meta-analysis of the GWAS and replication cohorts; based on the low level of imbalance in our study and the total count of minor alleles for the SNPs we tested, the meta-analysis should have appropriate maintenance of the type I error rate [51].
Meta-analysis
To obtain a joint measure of association between each of the 148 successfully genotyped SNPs in the replication set and fIIP, we performed a meta-analysis of the GWAS and replication results using the p-values from each analysis using METAL (http://www.sph.umich.edu/csg/abecasis/metal/). We used the weighted inverse normal method with weight equal to the square root of the total sample size in the i th study. SNPs with P Meta < 5 × 10−8 were considered genome-wide statistically significant. We created a locus-specific plot [52] of the discovery GWAS imputation results for the newly-identified chromosome 6 region that was genome-wide significant in the meta-analysis.
Multi-SNP models
To assess the independence of effects of the genome-wide significant SNPs from the meta-analysis, we used logistic regression models within each locus using the combined case group (GWAS and replication) and the replication controls. Specifically, within each locus with a genome-wide significant SNP, we tested for association between fIIP and each of the other validation panel SNPs within that locus after adjusting for the most significantly associated SNP in that locus (on chromosome 11p15, we adjusted for rs35705950).
HLA allele imputation and statistical analyses
Classical human leukocyte antigen (HLA) alleles at HLA-A,-B,-C,-DPB1,-DQA1,-DQB1, and -DRBI were imputed using the R-package HIBAG [38]. HIBAG uses an ensemble classifier and bagging techniques to arrive at an average posterior probability. The HIBAG-provided European-Ancestry reference panel was utilized for imputation. To account for imputation uncertainty, the allele dosage was utilized for all analyses. To filter-out extremely low frequency alleles, potentially yielding unstable estimates, a minimum best guess allele count of 10 was required in either the cases or controls for all alleles modeled. We tested for association between each of the alleles and fIIP using logistic regression, adjusting for the top three principal components of SNP variation, age and sex. We conducted a step-wise regression across all the imputed alleles, removing variables not independently associated with fIIP at the .01 level after adjustment for the other alleles in the model. Finally, we tested for association between pulmonary fibrosis and each of DRB1*15:01 and DQB1*06:02 adjusted for rs7887 and the first three principal components, age and sex, to determine the independence of effects.
RNA-seq expression analyses
After verifying that our amplicon-based targeted RNA-seq protocol achieved excellent enrichment for the small target regions, we used these regions (one per gene) to estimate gene expression. We used RSEM to assign reads to targeted regions using an EM algorithm. We removed one sample with fewer than 20,000 total reads and nine genes in the HLA region with fewer than 25 reads in more than 25 samples. We tested for differential gene expression in the lung between 87 cases and 70 controls with EdgeR36, using a negative binomial regression for the RNA-seq counts for each gene, adjusting for age and sex. A q-value < .05 was considered statistically significant. We used negative binomial regression, adjusting for age, sex, and total number of reads, to test for association between expression and rs7887 allele separately for cases and controls; we tested for association between alleles DRB1*15:01 and DQB1*06:02 and expression among cases only since these alleles could not be imputed for these controls without genome-wide SNP data.
Abbreviations
CEPH, Centre d’Étude du Polymorphisme Humain; FIIP, Fibrotic Idiopathic Interstitial Pneumonia; FIP, Familial Interstitial Pneumonia; GWAS. Genome-wide Association Study; HIBAG, HLA genotype Imputation with attribute BAGging; HLA, Human Leukocyte Antigen; NHLBI-LTRC, National Heart Lung and Blood Institute - Lung Tissue Research Consortium; NHW, Non-Hispanic White.
References
King Jr TE, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM, Kardatzke D, Lancaster L, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–92.
Richeldi L, Cottin V, Flaherty KR, Kolb M, Inoue Y, Raghu G, Taniguchi H, Hansell DM, Nicholson AG, Le Maulf F, et al. Design of the INPULSIS trials: Two phase 3 trials of nintedanib in patients with idiopathic pulmonary fibrosis. Respir Med. 2014;108(7):1023–30.
Baumgartner KB, Samet JM, Coultas DB, Stidley CA, Hunt WC, Colby TV, Waldron JA. Occupational and environmental risk factors for idiopathic pulmonary fibrosis: a multicenter case-control study. Collaborating Centers. Am J Epidemiol. 2000;152(4):307–15.
Hubbard R, Lewis S, Richards K, Johnston I, Britton J. Occupational exposure to metal or wood dust and aetiology of cryptogenic fibrosing alveolitis. Lancet. 1996;347:284–9.
Hubbard R, Cooper M, Antoniak M, Venn A, Khan S, Johnston I, Lewis S, Britton J. Risk of cryptogenic fibrosing alveolitis in metal workers. Lancet. 2000;355(9202):466–7.
Miyake Y, Sasaki S, Yokoyama T, Chida K, Azuma A, Suda T, Kudoh S, Sakamoto N, Okamoto K, Kobashi G, et al. Occupational and environmental factors and idiopathic pulmonary fibrosis in Japan. Ann Occup Hyg. 2005;49(3):259–65.
Tang YW, Johnson JE, Browning PJ, Cruz-Gervis RA, Davis A, Graham BS, Brigham KL, Oates JA, Jr, Loyd JE, Stecenko AA. Herpesvirus DNA is consistently detected in lungs of patients with idiopathic pulmonary fibrosis. J Clin Microbiol. 2003;41(6):2633–40.
Yonemaru M, Kasuga I, Kusumoto H, Kunisawa A, Kiyokawa H, Kuwabara S, Ichinose Y, Toyama K. Elevation of antibodies to cytomegalovirus and other herpes viruses in pulmonary fibrosis. Eur Respir J. 1997;10:2040–5.
Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng DS, Lane KB, Blackwell TR, Xu C, Markin C, Ware LB, et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol. 2008;294(6):L1119–26.
Stewart JP, Egan JJ, Ross AJ, Kelly BG, Lok SS, Hasleton PS, Woodcock AA. The detection of Epstein-Barr virus DNA in lung tissue from patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1999;159(4 Pt 1):1336–41.
Hubbard R, Venn A, Smith C, Cooper M, Johnston I, Britton J. Exposure to commonly prescribed drugs and the etiology of cryptogenic fibrosing alveolitis: a case–control study. Am J Respir Crit Care Med. 1998;157(3 Pt 1):743–7.
Musk AW, Pollard JA. Pindolol and pulmonary fibrosis. Br Med J. 1979;2(6190):581–2.
Erwteman TM, Braat MC, van Aken WG. Interstitial pulmonary fibrosis: a new side effect of practolol. Br Med J. 1977;2(6082):297–8.
Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, Lawson WE, Xie M, Vulto I, Phillips JA, 3rd, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356(13):1317–26.
Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC, Rosenblatt RL, Shay JW, Garcia CK. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci U S A. 2007;104(18):7552–7.
Thomas AQ, Lane K, Phillips 3rd J, Prince M, Markin C, Speer M, Schwartz DA, Gaddipati R, Marney A, Johnson J, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med. 2002;165(9):1322–8.
Lawson WE, Grant SW, Ambrosini V, Womble KE, Dawson EP, Lane KB, Markin C, Renzoni E, Lympany P, Thomas AQ, et al. Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax. 2004;59(11):977–80.
Wang Y, Kuan PJ, Xing C, Cronkhite JT, Torres F, Rosenblatt RL, DiMaio JM, Kinch LN, Grishin NV, Garcia CK. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet. 2009;84(1):52–9.
van Moorsel CH, van Oosterhout MF, Barlo NP, de Jong PA, van der Vis JJ, Ruven HJ, van Es HW, van den Bosch JM, Grutters JC. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am J Respir Crit Care Med. 2010;182(11):1419–25.
Cogan JD, Kropski JA, Zhao M, Mitchell DB, Rives L, Markin C, Garnett ET, Montgomery KH, Mason WR, McKean DF, et al. Rare Variants in RTEL1 Are Associated with Familial Interstitial Pneumonia. Am J Respir Crit Care Med. 2015;191(6):646–55.
Stuart BD, Choi J, Zaidi S, Xing C, Holohan B, Chen R, Choi M, Dharwadkar P, Torres F, Girod CE, et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat Genet. 2015.
Mushiroda T, Wattanapokayakit S, Takahashi A, Nukiwa T, Kudoh S, Ogura T, Taniguchi H, Kubo M, Kamatani N, Nakamura Y. A genome-wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J Med Genet. 2008;45(10):654–6.
Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, Fingerlin TE, Zhang W, Gudmundsson G, Groshong SD, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364(16):1503–12.
Zhang Y, Noth I, Garcia GN, Kaminski N. A variant in the promoter of MUC5B and idiopathic pulmonary fibrosis. N Engl J Med. 2011;364(16):1576–7.
Stock CJ, Sato H, Fonseca C, Banya WA, Molyneaux PL, Adamali H, Russell AM, Denton CP, Abraham DJ, Hansell DM, et al. Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis. Thorax. 2013.
Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y, Broderick SM, Wade MS, Hysi PG, Scuirba J, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med. 2013;1(4):309–17.
Borie R, Crestani B, Dieude P, Nunes H, Allanore Y, Kannengiesser C, Airo P, Matucci-Cerinic M, Wallaert B, Israel-Biet D, et al. The MUC5B Variant Is Associated with Idiopathic Pulmonary Fibrosis but Not with Systemic Sclerosis Interstitial Lung Disease in the European Caucasian Population. PLoS One. 2013;8(8), e70621.
Wei R, Li C, Zhang M, Jones-Hall YL, Myers JL, Noth I, Liu W. Association between MUC5B and TERT polymorphisms and different interstitial lung disease phenotypes. Transl Res. 2014;163(5):494–502.
Horimasu Y, Ohshimo S, Bonella F, Tanaka S, Ishikawa N, Hattori N, Kohno N, Guzman J, Costabel U. MUC5B promoter polymorphism in Japanese patients with idiopathic pulmonary fibrosis. Respirology. 2015;20(3):439–44.
Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: A risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1997;155:242–8.
Steele MP, Speer MC, Loyd JE, Brown KK, Herron A, Slifer SH, Burch LH, Wahidi MM, Phillips III JA, Sporn TA, et al. The Clinical and Pathologic Features of Familial Interstitial Pneumonia (FIP). Am J Respir Crit Care Med. 2005;172(9):1146–52.
Fingerlin TE, Murphy E, Zhang W, Peljto AL, Brown KK, Steele MP, Loyd JE, Cosgrove GP, Lynch D, Groshong S, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013;45(6):613–20.
International HapMap C, Altshuler DM, Gibbs RA, Peltonen L, Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, Yu F, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467(7311):52–8.
Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5(6), e1000529.
Howie B, Marchini J, Stephens M. Genotype imputation with thousands of genomes. G3 (Bethesda). 2011;1(6):457–70.
Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nat Rev Genet. 2010;11(7):499–511.
Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44(7):821–4.
Zheng X, Shen J, Cox C, Wakefield JC, Ehm MG, Nelson MR, Weir BS. HIBAG--HLA genotype imputation with attribute bagging. Pharmacogenomics J. 2014;14(2):192–200.
Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M, Lang G, Fink L, Bohle RM, Seeger W, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178(8):838–46.
Falfan-Valencia R, Camarena A, Juarez A, Becerril C, Montano M, Cisneros J, Mendoza F, Granados J, Pardo A, Selman M. Major histocompatibility complex and alveolar epithelial apoptosis in idiopathic pulmonary fibrosis. Hum Genet. 2005;118(2):235–44.
Xue J, Gochuico BR, Alawad AS, Feghali-Bostwick CA, Noth I, Nathan SD, Rosen GD, Rosas IO, Dacic S, Ocak I, et al. The HLA class II Allele DRB1*1501 is over-represented in patients with idiopathic pulmonary fibrosis. PLoS One. 2011;6(2), e14715.
Aquino-Galvez A, Perez-Rodriguez M, Camarena A, Falfan-Valencia R, Ruiz V, Montano M, Barrera L, Sada-Ovalle I, Ramirez R, Granados J, et al. MICA polymorphisms and decreased expression of the MICA receptor NKG2D contribute to idiopathic pulmonary fibrosis susceptibility. Hum Genet. 2009;125(5–6):639–48.
Furukawa H, Oka S, Shimada K, Sugii S, Ohashi J, Matsui T, Ikenaka T, Nakayama H, Hashimoto A, Takaoka H, et al. Association of human leukocyte antigen with interstitial lung disease in rheumatoid arthritis: a protective role for shared epitope. PLoS One. 2012;7(5), e33133.
Arnett FC, Gourh P, Shete S, Ahn CW, Honey RE, Agarwal SK, Tan FK, McNearney T, Fischbach M, Fritzler MJ, Mayes MD, Reveille JD. Major histocompatibility complex (MHC) class II alleles, haplotypes and epitopes which confer susceptibility or protection in systemic sclerosis: analyses in 1300 Caucasian, African-American and Hispanic cases and 1000 controls. Ann Rheum Dis. 2010;69(5):822–7.
Moua T, Maldonado F, Decker PA, Daniels CE, Ryu JH. Frequency and implication of autoimmune serologies in idiopathic pulmonary fibrosis. Mayo Clin Proc. 2014;89(3):319–26.
American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med 2002, 165(2):277–304.
King Jr TE, Albera C, Bradford WZ, Costabel U, Hormel P, Lancaster L, Noble PW, Sahn SA, Szwarcberg J, Thomeer M, et al. Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): a multicentre, randomised, placebo-controlled trial. Lancet. 2009;374(9685):222–8.
Regan EA, Hokanson JE, Murphy JR, Make B, Lynch DA, Beaty TH, Curran-Everett D, Silverman EK, Crapo JD. Genetic epidemiology of COPD (COPDGene) study design. COPD. 2010;7(1):32–43.
Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat Methods. 2012;9(2):179–81.
Delaneau O, Zagury JF, Marchini J. Improved whole chromosome phasing for disease and population genetic studies. Nat Methods. 2013;10(1):5–6.
Ma C, Blackwell T, Boehnke M, Scott LJ, Go TDI. Recommended joint and meta-analysis strategies for case–control association testing of single low-count variants. Genet Epidemiol. 2013;37(6):539–50.
Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26(18):2336–7.
Acknowledgements
We gratefully acknowledge the individuals who participated in the studies that contributed to this work.
Funding
This research was supported by the National Heart, Lung and Blood Institute (R01-HL095393, R01-HL097163, P01-HL092870, RC2-HL101715, U01-HL089897, U01-HL089856, U01-HL108642, and P50-HL0894932); NHLBI did not have a role in the design, collection, analysis or interpretation of the data, writing the manuscript nor the decision to submit the manuscript for publication.
Availability of data and materials
Genotype data from the discovery GWAS cases are deposited in dbGAP (phs000751.v1.p1). Microarray data on lung tissue is deposited in the Gene Expression Omnibus (GEO) repository as GSE31962. RNA sequencing count data will be deposited in GEO and is available from the authors.
Authors’ contributions
TEF and DAS designed the study; KKB, MPS, JEL, GPC, DL, SG, HRC, PLW, RMD, CKG, MSD, GG, HJI, NK, YZ, KFG, LHL, WRM, TMM, PLM, AUW, JDC, BJM, EAR, and MIS performed clinical, radiological, and pathological phenotyping of study subjects; WXB, KK, and SDS provided data and samples from the InterMune subjects; JT, RNK, CRM, JP coordinated the clinical evaluations; IVY and EM supervised and coordinated the laboratory work; EM, JDC, DSW, JJD, DZ, KS performed the laboratory work; DM organized the database; KBB supervised the replication genotyping; ML supervised the genome wide genotyping; MFM, MS, AP, DSK, MIS, and DAS provided advice on the design and relevance to pulmonary fibrosis; TEF, WZ, IVY, ALP, BSP, CDL, HA, PR, RZB, YK and BF analyzed the data; TEF, ML, and DAS developed the conceptual approaches to data analysis; TEF and DAS wrote the manuscript. All authors have read and approved the final version of the manuscript.
Competing interests
The authors have no competing interests to declare.
Consent to Publish
Not applicable.
Ethics
All subjects gave written informed consent as part of IRB-approved protocols for their recruitment at each site and the GWAS study was approved by the National Jewish Health IRB and Colorado Combined Institutional Review Boards (COMIRB).
Author information
Authors and Affiliations
Corresponding authors
Additional file
Additional file 1: Tables S1.
205 SNPs Associated with IIP at 5 × 10−8 in Imputation Analysis. Table S2: SNPs with 5×10−8 < Pimputed-adjusted < .0001. Table S3: GWAS-Significant SNPs after Meta-Analysis in All Regions (Most Significant in Region in Bold Type). Table S4: Table of 70 SNPs (46 original, 24 from imputation) Adjusted for Top SNP in Region if More than One SNP in Region. Top SNP Defined in Bold Type in Table S3a. All Cases (Discovery and GWAS) Compared to Replication Controls. Table S5: HLA Allele Imputation Accuracy Summary. Table S6: HLA Allele Association with IIP. Table S7: Chromosome 6p21 genes studied via RNA-seq. Table S8: Differential Expression by Case–control Status. Table S9: Differential Expression by Genotype at rs7887 Among Controls. Table S10: Differential Expression by Imputed Number of Copies of DRB1*15:01 Among Cases (RNA-seq). Table S11: Differential Expression by Imputed Number of Copies of DQB1*06*02 Among Cases (RNA-seq). Table S12: 214 Novel SNPs Meeting Carry-forward Threshold at P < 1 × 10−4 in Imputation Analysis using 1000 Genomes Reference. Figure S1: Q-Q plots of P-values from SNPTest using Imputed Dosage for a) Genotyped SNPs and b) Imputed SNPs Prior to Genomic Control Adjustment Based on Genotyped SNP P-values from GEMMA analysis (see manuscript statistical methods for details). Figure S2: Q-Q plot of imputed (adjusted) p-values (inflation factor = 1.04) after genomic control correction. Figure S3: Imputation GWAS Results Fig. 1: Imputation GWAS results with 1616 cases and 4683 controls under additive model. SNPs above red line were genome-wide significant at P < 5 × 10-8. A subset of these SNPs and SNPs between red and blue lines, corresponding to 5 × 10−8 < P-value < .0001, were selected for follow-up and genotyped in 878 cases and 2017 controls. Figure S4: Histogram of. INFO Scores from SNPTest. Only SNPs with .INFO >0.5 Included. Figure S5: Comparison of SNPTest p-values after genomic control (see manuscript statistical methods) and GEMMA dosage-values. GEMMA dosage p-values computed after initial study complete. (DOCX 8917 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fingerlin, T.E., Zhang, W., Yang, I.V. et al. Genome-wide imputation study identifies novel HLA locus for pulmonary fibrosis and potential role for auto-immunity in fibrotic idiopathic interstitial pneumonia. BMC Genet 17, 74 (2016). https://doi.org/10.1186/s12863-016-0377-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-016-0377-2