
Abstract
Genetic factors play a role in the risk of idiopathic pulmonary fibrosis (IPF). Specifically, MUC5B rs35705950 non-risk alleles and immunologic aberrations were associated with the IPF’s progression. However, rare genetic variants have not been systematically investigated in Chinese IPF patients. In this study, we aimed to improve understanding of the genetic architecture of IPF in the Chinese population and to assess whether rare protein-coding variants in the immunity pathway genes are enriched in the IPF patients with non-risk alleles at rs35705950. A case–control exome-wide study including 110 IPF patients and 60 matched healthy controls was conducted. rs35705950 was genotyped by Sanger sequencing. To identify genes enriched in IPF, gene-based association analyses were performed. Identified genes were included for further pathway analyses using gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG). Associations between rs35705950 and genes enriched in the immunity pathway were also tested. 226 genes that were enriched with deleterious variants were identified in IPF patients. Out of them, 36 genes were significantly enriched in GO and KEGG pathways in the IPF. Pathway analyses implicated that these genes were involved in the immune response and cell adhesion. Rare protein-altering variants in genes related to the immunity pathway did not significantly differ between patients with a MUC5B risk allele and individuals without risk allele. We drafted a comprehensive mutational landscape of rare protein-coding variants in the Chinese IPF and identified genes related to immune response and cell adhesion. These results partially explain changes in gene expression involved in the immunity/inflammatory pathways in IPF patients.
Similar content being viewed by others


Introduction
Idiopathic pulmonary fibrosis (IPF), one of the common types of idiopathic interstitial pneumonia (IIP), is characterized by progressive fibrosis of unknown aetiology1. IPF occurs primarily in men aged from 50 to 70, with a median survival of 2–3 years2. Two approved therapies, pirfenidone and nintedanib, appear to decelerate disease progression and have a mortality benefit3,4. Its pathogenic mechanisms have not been elucidated and no medical treatment to date has been found to cure IPF except lung transplantation5.
Genetic variants, rare and common, contribute to the susceptibility of IPF in both sporadic and familial cases6,7,8. Previous studies in patients with familial pulmonary fibrosis (FPF) or sporadic IPF have identified common variants (minor allele frequency (MAF) > 5%). A single nucleotide polymorphism (SNP) rs35705950 in the promoter region of the MUC5B gene demonstrated to be strongly associated with the IPF and familial interstitial pneumonia (FIP) in one genome-wide linkage analysis9 and the association was validated in multiple different cohorts10,11,12,13,14,15,16. Additionally, other common variants in several genes were also found to be associated with the disease, by genome-wide approaches, such as DNA repair-related genes (TERT17, TERC, OBFC1), host defense-related genes (ATP11A, TOLLIP), cell–cell adhesion-related genes (DSP, DPP9), profibrotic signaling pathway-related genes (AKAP13), FAM13A and SPPL2C10,11,18,19,20. In addition to the common variants, rare variants were also reported to be involved in two main pathways in IPF, including telomere maintenance and surfactant metabolism. Rare variants in multiple different telomerase related genes (TERT, TERC, RTEL1 and PARN) have been associated with both familial21,22,23,24,25 and sporadic IPF10,26,27,28. Two genes involved in encoding surfactant proteins A and C (SFTPA2 and SFTPC) have also been related to the IPF in sporadic studies29,30. However, these risk variants generally explain a relatively small proportion of IPF’s heritability; except for rs35705950.
Whole-exome sequencing (WES) has become an increasingly popular approach to identify rare alleles with direct functional consequences on protein products. However, WES has not been extended to the studies on sporadic IPF in the Chinese population. Besides, previous research on the IPF subjects indicated that MUC5B risk allele was associated with longer survival31 and aberrant immunity was related to IPF progression10,11,32,33,34,35,36,37. In this study, we performed WES to identify genes carrying excessive rare deleterious variants, investigated their aggregate effects by pathway analyses and assessed if candidate genes involved in the immunity pathway were enriched in IPF patients with non-risk alleles at rs35705950.
Results
Subjects’ characteristics and sequencing data
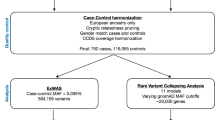
A total of 110 IPF patients and 60 matched controls were included in the study. Baseline characteristics of 170 participants were summarized in Table 1. Cases and controls were similar in age, gender proportion and smoking status due to matching. No significant difference was found in BMI between the two groups (P = 0.701).
Average sequencing depth of targeted exome regions in all tested samples was 110 × . The mean proportion of targets that were covered with at least 10 × was 99%.
Rare deleterious variants in IPF and candidate genes enriched with deleterious variants
After multi-step filtering (Supplementary Fig. S1), a total of 10,333 deleterious variants with MAF < 0.01 were identified in the IPF group, including a large proportion of nonsynonymous variants (n = 7093) and a small number of frameshift indels (n = 2136), stop-gain (n = 1058) and stop-loss (n = 46). After gene-based association analysis for all the above-mentioned variants by SKAT, we identified 226 genes that were enriched with rare, deleterious variants in the Chinese IPF samples (all P-FDR < 0.05) (Supplementary Table S1). Given the complexity of HLA, the association of HLA genes selected from SKAT with IPF was further determined by another collapsing analysis. The results showed that except HLA-G, other HLA genes were significantly associated with IPF (P-FDR < 0.05), and P value of HLA-G was equal to 0.05, which was at the critical level. The above HLA genes were included in the subsequent analysis (GO and KEGG), because the objective of this study was to find genes and pathways that might be related to IPF in a more conservative way, and to avoid missing genes potentially associated with the IPF. Finally, a total of 226 candidate genes were included in subsequent analyses.
Enrichment and pathway discovery
GO and KEGG pathway enrichment analyses were performed with the input consisting of 226 genes. Figure 1 shows that, for cellular components, mutated genes were mainly involved in vesicle, membrane, extracellular matrix and MHC protein complex (all P.adjusted < 0.05). In terms of biological process, ‘Interferon-gamma-mediated signaling pathway’ (P.adjusted = 7.852 × 10–5) and ‘Antigen processing and presentation of peptide antigen’ (P.adjusted = 2.190 × 10–3) showed the most significance, which can be observed in Table 2. As for the molecular function, candidate genes were significantly enriched in ‘peptide antigen binding’ (P.adjusted = 5.958 × 10–7) and ‘amide binding’ (P.adjusted = 3.599 × 10–3), similarly seen in Table 2. Additionally, KEGG pathways of the mutated genes included mainly ‘antigen processing and presentation’ (P.adjusted = 5.805 × 10–8), ‘phagosome’ (P.adjusted = 6.283 × 10–5), ‘cell adhesion molecules (CAMs)’ (P.adjusted = 3.585 × 10–4), ‘natural killer cell mediated cytotoxicity’ (P.adjusted = 5.858 × 10–3) and ‘Th1 and Th2 cell differentiation’ (P.adjusted = 0.042), seen in Table 3.

Cellular components terms of gene ontology analysis of candidate genes. The X-axis represents the ratio of genes enriched in the pathway to the total enriched genes, and the Y-axis represents the terms of enriched cellular components. The dot size represents the number of genes enriched in the pathway, and the color represents the P value. P value was adjusted by Benjamini–Hochberg false discovery rate.
Mutational landscape of genes enriched in GO and KEGG pathways in IPF
A total of 36 genes were significantly enriched in GO and KEGG pathways in IPF samples, seen in Fig. 2. All IPF samples (100%) had rare deleterious variants in HLA-DQB1, HLA-B, HLA-A, GCC2, HRNR, MUC17, COPZ2, SYN2, PCSK6, MST1, MUC4, SEC22B and LAMA5.

Mutational landscape of 36 candidate genes enriched in GO and KEGG pathways in the IPF samples. Mutation burdens are displayed on the X-axis panel (top), and cohort mutation percentage by a gene is displayed on the Y-axis panel (left). P value from SKAT of each gene is displayed on the Y-axis panel (right). Displayed genes are significantly enriched in the IPF samples. Each column represents an individual sample. Coloring of patient-gene intersection grid indicates mutation type (top legend).
Associations between MUC5B risk allele and genes enriched in the immunity pathway
Table 4 presents the associations between MUC5B rs35705950 and immunity-related genes with rare deleterious variations. No genes were found to be significantly associated with MUC5B risk allele (all P > 0.05).
Discussion
In this study of 110 IPF subjects and 60 matched controls, using gene-level association analyses, 226 genes with rare deleterious variants were significantly enriched in the Chinese IPF samples. A large proportion of these genes were first identified in this study. Among the 226 analyzed genes, only HLA-DRB1 and HLA-DQB1 genes and their relationships with pulmonary fibrosis were previously analyzed. One genome-wide imputation study reported that two risk alleles, DRB1*15:01 and DQB1*06:02, were associated with fibrotic idiopathic interstitial pneumonias38. The association between DRB1*15:01 and IPF was also confirmed in a previous study39. Additionally, these two risk alleles were related to the expression of the DQB1 gene38. In addition to HLA-DRB1 and HLA-DQB1, other HLA genes with rare deleterious variants were also significantly associated with IPF in our study, including HLA-DRB5, HLA-A, HLA-B, HLA-H, HLA-G, HLA-DQA1 and HLA-L. These findings reveal the importance of HLA region for the susceptibility of pulmonary fibrosis40.
We also attempted to identify GO category and biological pathways with the input of 226 candidate genes. A total of 36 genes were significantly enriched in the identified GO and KEGG pathways. GO-cellular components showed that these mutated genes were mainly related to vesicle, membrane, extracellular matrix and MHC protein complex. Accordingly, five pathways were highlighted in the KEGG enrichment analyses: ‘antigen processing and presentation’, ‘phagosome’ and ‘cell adhesion molecules (CAMs)’, ‘natural killer cell-mediated cytotoxicity’ and ‘Th1 and Th2 cell differentiation’. Similarly, several previous genome-wide association studies (GWAS) identified common variants in several genes that were relevant to host defense (ATP11A, TOLLIP11) in patients with IPF and cell–cell adhesion (DSP, DPP9) in patients with fibrotic IIP10. Another study by Aquino-Galvez et al.41 also indicated that MHC class I chain-related gene A (MICA) polymorphisms and abnormal expression of the MICA receptor NKG2D were related to the susceptibility of IPF. Besides, Th1/ Th2 cytokine gene polymorphisms were also involved in the etiology and pathogenesis of IPF42,43,44. Although this previous research concentrated on the common variants and our study focused on the rare deleterious variants, these findings suggested that mutated genes involved in the immunity pathway or the cell adhesion pathway might play a role in the risk or susceptibility of IPF.
As for the role of the immunity pathway in IPF, further evidence was obtained from the transcriptomic profiling of cultured lung fibroblasts in IPF patients, showing that 115 downregulated transcripts were enriched in the inflammation and immunity pathways such as defense response to virus, tumor necrosis factor (TNF) mediated signaling pathway, interferon-inducible absent in melanoma2 (AIM2) inflammasome as well as apoptosis45. Similarly, another transcriptomic analysis of nasal epithelium in IPF patients indicated that upregulated genes in IPF patients were related to immune response and inflammatory signaling46. In turn, the mutated genes enriched in the immunity pathway in our study might partially explain the changes of immunity-related gene expression in these previous studies. However, due to the different directions of immune-related gene expression changes were reported in these two transcriptomic studies, more research is needed to verify our findings and evaluate the function of these genes with rare deleterious variants.
Interestingly, two previous studies (one GWAS27 and one WES28) also focused on rare protein-altering variants on IPF patients. They found that four genes (TERT, TERC, PARN, RTEL1) significantly enriched with candidate rare variants were related to telomere maintenance. And the immune-related and cell adhesion pathways identified in our study may provide new ideas for us to understand the role of rare deleterious protein-altering variants in Chinese IPF.
Immunologic aberrations including immune cells32,35, genetic polymorphisms36 or gene expression changes34,37 were linked to the progression of IPF. Since IPF patients who do not carry the MUC5B risk allele have shorter survival from the time of diagnosis than those carrying the risk allele31, we also assessed whether immune-related genes with rare deleterious variants were enriched in MUC5B non-carriers. However, no significant association was found between them. This might suggest that multiple genetic factors and mechanisms might play roles in IPF progression27.
In summary, we evaluate the strengths and limitations of our study. This study was the first to explore rare protein-altering variants using WES in the Chinese IPF population. We aimed to identify genes and biological pathways enriched with rare deleterious variants, which would guide the future genetic and functional studies to elucidate the role of rare variants in the pathogenesis of IPF. Additionally, we included age and gender-matched healthy controls in this study, which was not achieved in the previous related studies27,28. However, there are still several limitations to be noted. First, our sample size was small, but we tried to improve our statistical power by using gene-level association analyses instead of single variant association analyses. Second, identified susceptibility genes and pathways have not been replicated and validated in an independent, larger case–control study, therefore, more studies with bigger sample size are needed for future validation analysis.
Conclusion
In this study, using WES, we identified 226 genes with rare deleterious variants enriched in Chinese IPF patients and drafted a comprehensive mutational landscape of rare protein-coding variants in 36 candidate genes enriched in GO and KEGG pathways. These candidate genes were mainly related to cell adhesion and immune response, which might partially explain changes of gene expression involved in immune-related pathways in IPF. Further validation studies with larger statistical power are needed to verify these findings and identify the underlying functional mechanisms.
Materials and methods
Study design and participants
A case–control study consisting of 110 IPF cases and 60 matched controls was conducted at the Peking Union Medical College Hospital (PUMCH) of the Chinese Academy of Medical Sciences and Peking Union Medical College (CAMS & PUMC), Dongcheng District, China. All the subjects were Han Chinese and were enrolled consecutively. All IPF cases had no family history of interstitial lung disease and diagnostic criteria of IPF were based on the American Thoracic Society/European Respiratory Society/Japanese Respiratory Society/Latin American Thoracic Association (ATS/ERS/JRS/ALTA) consensus statement in 201847. Two experienced pulmonologists and one radiologist independently reviewed the clinical and biopsy characteristics and HRCT scans of each patient. Criteria for controls selection included: (1) gender and age-matching, (2) exclusion of pulmonary fibrosis or a family history of interstitial lung disease. Ethical approval of this study was obtained from the Regional Ethics Committee of PUMCH (JS-1127/2016) and procedure of this research conforms to relevant regulations. All participants provided written informed consents. Additionally, demographic information, medical history, family history and other baseline information were collected from each participant.
Whole-exome sequencing-based approaches for mining of rare deleterious variants in IPF
The process of candidate variant selection and subsequent analysis was shown in Supplementary Fig. S1.
-
1)
Genomic DNA extraction
DNA was extracted from peripheral blood leukocytes using the QIAamp Genomic DNA mini kit (QIAGEN, CA, USA).
-
2)
Library construction and sequencing
A minimum of 1 μg of DNA per sample was used for the DNA library generation using Agilent SureSelect Human All Exon V6 kit (Agilent Technologies, CA, USA) according to the manufacturer’s protocol. First, genomic DNA samples were randomly fragmented by sonication (Covaris, Inc., Woburn, MA, USA) to an average size of 180 ~ 250 bp, followed by end-polishing and A-tailing and ligation of sequencing adaptors. Second, the libraries with special index were hybridized to biotinylated capture probes and they were captured for exome enrichment using magnetic beads with streptomycin, followed by PCR amplification. Third, the concentration of each captured library was accurately determined by quantitative PCR (qPCR) according to the manufacturer's protocol (Agilent Bioanalyzer 2100, Agilent, Santa Clara, CA, USA). Lastly, the qualified DNA libraries were sequenced on Illumina HiSeq X platform (Illumina Inc., San Diego, CA, USA) using 150 bp paired-end reads (PE150).
-
3)
Quality control
For each sample, the following filters were used to select clean reads with high quality: (1) remove the adaptor sequence in reads, (2) remove paired reads if more than 50% of bases were of poor quality (Phred quality ≤ 19) in either one read, (3) remove paired reads if the proportion of uncertain bases was over 10% in either one read.
-
4)
Detection and filtering of genomic alterations
Whole-exome valid sequencing reads were mapped to the reference human genome (GRCh37/hg19), using Burrows-Wheeler Aligner (BWA) software to generate original BAM files. Then, these files were sorted and realigned by SAMtools to compute the sequence coverage and depth48. Duplicate reads were marked and removed using the Picard suite. Finally, single-nucleotide variants (SNVs) and insertions and deletions (indels) were called with GATK49 and annotated by ANNOVAR50. Following filters were set to identify candidate variants: (1) keep mutations with coverage ≥ 10 × , unless the variant had high impact (e.g.: stop gain, stop loss, frameshift), which required coverage ≥ 5 × ; (2) remove variants with mutant allele frequency (MAF) ≥ 0.01 in East Asian (EAS) population in the Exome Aggregation Consortium (ExAC) database, 1000 Genomes or Genome Aggregation Database (GnomAD); (3) variations in the exonic or splicing region (10 bp upstream and downstream of splicing sites); (4) keep variants if the functional predictions by SIFT (dbNSFP version 3.0, D: Deleterious), PolyPhen-2 (dbNSFP version 3.0, D: Probably damaging), MutationTaster (dbNSFP version 3.0, A: Disease causing automatic or D: Disease causing) and CADD (dbNSFP version 3.0, PHRED-like score > 20) all indicated the SNV was not benign or if it had high impact (e.g.: stop gain, stop loss, frameshift).
Population structure analysis
Principal component analysis (PCA) for all samples was performed using software GCTA51 (version 1.93.2beta, http://cnsgenomics.com/software/gcta), including all candidate variants after multi-step filtering. Significant PCs were inferred using Tracy-Widom statistics (P value < 0.05)52,53. The results showed that no significant PC was found. There was no obvious difference between the case group and the control group (Supplementary Fig. S2). This confirmed that all the samples were Han Chinese and there was no population stratification.
Gene-based collapsing analysis
Single-variant tests are less capable of identifying rare variants than common variants54. To make them more capable of detecting association of rare variants that cluster in the individual genes, gene-level collapsing analysis was performed using SKAT55,56. Only 10,333 candidate variants were included for gene-based association analysis. Candidate genes enriched with rare deleterious variants were identified if P value (corrected by Benjamini–Hochberg false discovery rate (FDR)) from SKAT was less than 0.05. Considering the complexity of HLA region, we tried to use another common collapsing analysis28,57,58 to further determine the relationship between HLA genes and IPF. The detailed steps were as follows: for each of the candidate HLA genes (HLA-A, HLA-B, HLA-DQA1, HLA-DQB1, HLA-DRB1, HLA-DRB5, HLA-G, HLA-H, HLA-L) selected from SKAT, we assigned an indicator variable (1/0 states) to each individual based on the presence of at least one candidate variant in that gene (state 1) or no candidate variants in that gene (state 0). Then we used the two-tailed Fisher’s exact test for each gene to compare the rate of case subjects carrying a candidate variant compared with the rate of control subjects and P value was corrected by FDR.
Enrichment analysis and pathway analysis
To investigate the biological relevance of the candidate genes, gene ontology (GO) enrichment analysis was performed to categorize the function of these genes into three classes: ‘biological process’, ‘cellular components’, and ‘molecular function’. Additionally, the Kyoto Encyclopedia of Genes and Genomes (KEGG) database59 (http://www.genome.jp/kegg/pathway.html) was used to identify pathways that were enriched with candidate genes in IPF. P value was adjusted by FDR.
MUC5B rs35705950 genotyping
MUC5B rs35705950 within the promoter region of the MUC5B gene (chr11:g.1241221G.T, NCBI Build 37) was genotyped using the Sanger sequencing after the PCR amplification. Sanger sequencing was performed using the Big Dye v.3.1 terminator cycle sequencing kit and an Applied Biosystems 3730xl capillary sequencer (Applied Biosystems, CA, USA). Primers were as follows, forward, TGGCCAGAATGAGGGACAGT; reverse, GACGTCAAGGCCACAGCTAT. The risk allele of MUC5B 35705950 was T while the non-risk allele was G.
Statistical analysis
Statistical analysis was performed using SPSS software version 24.0 for Windows (SPSS Inc., Chicago, IL, USA) and R statistical software (version 3.51). Two-tailed P (or P.adjusted) < 0.05 was considered statistically significant. Results of continuous variables were reported as mean ± standard deviation (SD), while categorical variables were reported as a number with a percentage. Comparison of basic characteristics between two groups was done by using the Student’s t-test for continuous variables which fulfilled homogeneity of variance and by using a chi-square test for categorical variables. Logistic regression was used to assess associations between rs35705950 SNP and genes enriched in the immunity pathway. Results of logistic regression are presented as odd ratio (OR) with 95% confidence intervals (CIs).
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Richeldi, L., Collard, H. R. & Jones, M. G. Idiopathic pulmonary fibrosis. Lancet 389, 1941–1952. https://doi.org/10.1016/S0140-6736(17)30866-8 (2017).
Olson, A. L. et al. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am. J. Respir. Crit. Care Med. 176, 277–284. https://doi.org/10.1164/rccm.200701-044OC (2007).
Raghu, G. et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: Treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am. J. Respir. Crit. Care. Med. 192, e3–e19. https://doi.org/10.1164/rccm.201506-1063ST (2015).
Canestaro, W. J., Forrester, S. H., Raghu, G., Ho, L. & Devine, B. E. Drug treatment of idiopathic pulmonary fibrosis: Systematic review and network meta-analysis. Chest 149, 756–766. https://doi.org/10.1016/j.chest.2015.11.013 (2016).
Raghu, G. et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 183, 788–824. https://doi.org/10.1164/rccm.2009-040GL (2011).
Mathai, S. K., Newton, C. A., Schwartz, D. A. & Garcia, C. K. Pulmonary fibrosis in the era of stratified medicine. Thorax 71, 1154–1160. https://doi.org/10.1136/thoraxjnl-2016-209172 (2016).
Kropski, J. A., Blackwell, T. S. & Loyd, J. E. The genetic basis of idiopathic pulmonary fibrosis. Eur. Respir. J. 45, 1717–1727. https://doi.org/10.1183/09031936.00163814 (2015).
Fernandez, B. A. et al. A Newfoundland cohort of familial and sporadic idiopathic pulmonary fibrosis patients: Clinical and genetic features. Respir. Res. 13, 64. https://doi.org/10.1186/1465-9921-13-64 (2012).
Seibold, M. A. et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N. Engl. J. Med. 364, 1503–1512. https://doi.org/10.1056/NEJMoa1013660 (2011).
Fingerlin, T. E. et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 45, 613–620. https://doi.org/10.1038/ng.2609 (2013).
Noth, I. et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: A genome-wide association study. Lancet Respir. Med. 1, 309–317. https://doi.org/10.1016/S2213-2600(13)70045-6 (2013).
Zhang, Y., Noth, I., Garcia, J. G. & Kaminski, N. A variant in the promoter of MUC5B and idiopathic pulmonary fibrosis. N. Engl. J. Med. 364, 1576–1577. https://doi.org/10.1056/NEJMc1013504 (2011).
Stock, C. J. et al. Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis. Thorax 68, 436–441. https://doi.org/10.1136/thoraxjnl-2012-201786 (2013).
Horimasu, Y. et al. MUC5B promoter polymorphism in Japanese patients with idiopathic pulmonary fibrosis. Respirology 20, 439–444. https://doi.org/10.1111/resp.12466 (2015).
Wei, R. et al. Association between MUC5B and TERT polymorphisms and different interstitial lung disease phenotypes. Transl. Res. 163, 494–502. https://doi.org/10.1016/j.trsl.2013.12.006 (2014).
Jiang, H. et al. Association between MUC5B polymorphism and susceptibility and severity of idiopathic pulmonary fibrosis. Int. J. Clin. Exp. Pathol. 8, 14953–14958 (2015).
Mushiroda, T. et al. A genome-wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J. Med. Genet. 45, 654–656. https://doi.org/10.1136/jmg.2008.057356 (2008).
Mathai, S. K., Schwartz, D. A. & Warg, L. A. Genetic susceptibility and pulmonary fibrosis. Curr. Opin. Pulm. Med. 20, 429–435. https://doi.org/10.1097/MCP.0000000000000074 (2014).
Mathai, S. K., Yang, I. V., Schwarz, M. I. & Schwartz, D. A. Incorporating genetics into the identification and treatment of idiopathic pulmonary fibrosis. BMC Med. 13, 191. https://doi.org/10.1186/s12916-015-0434-0 (2015).
Allen, R. J. et al. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: A genome-wide association study. Lancet Respir. Med. 5, 869–880. https://doi.org/10.1016/S2213-2600(17)30387-9 (2017).
Tsakiri, K. D. et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. U. S. A. 104, 7552–7557. https://doi.org/10.1073/pnas.0701009104 (2007).
Armanios, M. Y. et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 356, 1317–1326. https://doi.org/10.1056/NEJMoa066157 (2007).
Stuart, B. D. et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat. Genet. 47, 512–517. https://doi.org/10.1038/ng.3278 (2015).
Cogan, J. D. et al. Rare variants in RTEL1 are associated with familial interstitial pneumonia. Am. J. Respir. Crit. Care Med. 191, 646–655. https://doi.org/10.1164/rccm.201408-1510OC (2015).
Kannengiesser, C. et al. Heterozygous RTEL1 mutations are associated with familial pulmonary fibrosis. Eur. Respir. J. 46, 474–485. https://doi.org/10.1183/09031936.00040115 (2015).
Cronkhite, J. T. et al. Telomere shortening in familial and sporadic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 178, 729–737. https://doi.org/10.1164/rccm.200804-550OC (2008).
Dressen, A. et al. Analysis of protein-altering variants in telomerase genes and their association with MUC5B common variant status in patients with idiopathic pulmonary fibrosis: A candidate gene sequencing study. Lancet Respir. Med. 6, 603–614. https://doi.org/10.1016/S2213-2600(18)30135-8 (2018).
Petrovski, S. et al. An exome sequencing study to assess the role of rare genetic variation in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 196, 82–93. https://doi.org/10.1164/rccm.201610-2088OC (2017).
Lawson, W. E. et al. Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax 59, 977–980. https://doi.org/10.1136/thx.2004.026336 (2004).
Campo, I. et al. A large kindred of pulmonary fibrosis associated with a novel ABCA3 gene variant. Respir. Res. 15, 43. https://doi.org/10.1186/1465-9921-15-43 (2014).
Peljto, A. L. et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA 309, 2232–2239. https://doi.org/10.1001/jama.2013.5827 (2013).
Gilani, S. R. et al. CD28 down-regulation on circulating CD4 T-cells is associated with poor prognoses of patients with idiopathic pulmonary fibrosis. PLoS ONE 5, e8959. https://doi.org/10.1371/journal.pone.0008959 (2010).
O’Dwyer, D. N., Ashley, S. L. & Moore, B. B. Influences of innate immunity, autophagy, and fibroblast activation in the pathogenesis of lung fibrosis. Am. J. Physiol. Lung. Cell. Mol. Physiol. 311, L590-601. https://doi.org/10.1152/ajplung.00221.2016 (2016).
Herazo-Maya, J. D. et al. Peripheral blood mononuclear cell gene expression profiles predict poor outcome in idiopathic pulmonary fibrosis. Sci Transl Med 5, 205ra136. https://doi.org/10.1126/scitranslmed.3005964 (2013).
Moore, B. B. et al. Inflammatory leukocyte phenotypes correlate with disease progression in idiopathic pulmonary fibrosis. Front Med. https://doi.org/10.3389/fmed.2014.00056 (2014).
O’Dwyer, D. N. et al. The Toll-like receptor 3 L412F polymorphism and disease progression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 188, 1442–1450. https://doi.org/10.1164/rccm.201304-0760OC (2013).
Trujillo, G. et al. TLR9 differentiates rapidly from slowly progressing forms of idiopathic pulmonary fibrosis. Sci Transl Med 2, 57ra82. https://doi.org/10.1126/scitranslmed.3001510 (2010).
Fingerlin, T. E. et al. Genome-wide imputation study identifies novel HLA locus for pulmonary fibrosis and potential role for auto-immunity in fibrotic idiopathic interstitial pneumonia. BMC Genet. 17, 74. https://doi.org/10.1186/s12863-016-0377-2 (2016).
Xue, J. et al. The HLA class II Allele DRB1*1501 is over-represented in patients with idiopathic pulmonary fibrosis. PLoS ONE 6, e14715. https://doi.org/10.1371/journal.pone.0014715 (2011).
Falfan-Valencia, R. et al. Major histocompatibility complex and alveolar epithelial apoptosis in idiopathic pulmonary fibrosis. Hum. Genet. 118, 235–244. https://doi.org/10.1007/s00439-005-0035-7 (2005).
Aquino-Galvez, A. et al. MICA polymorphisms and decreased expression of the MICA receptor NKG2D contribute to idiopathic pulmonary fibrosis susceptibility. Hum. Genet. 125, 639–648. https://doi.org/10.1007/s00439-009-0666-1 (2009).
Vasakova, M. et al. Th1/Th2 cytokine gene polymorphisms in patients with idiopathic pulmonary fibrosis. Tissue Antigens 67, 229–232. https://doi.org/10.1111/j.1399-0039.2006.00560.x (2006).
Whittington, H. A. et al. Analysis of an IL-10 polymorphism in idiopathic pulmonary fibrosis. Genes Immun. 4, 258–264. https://doi.org/10.1038/sj.gene.6363959 (2003).
Ahn, M. H. et al. A promoter SNP rs4073T>A in the common allele of the interleukin 8 gene is associated with the development of idiopathic pulmonary fibrosis via the IL-8 protein enhancing mode. Respir. Res. 12, 73. https://doi.org/10.1186/1465-9921-12-73 (2011).
Plantier, L., Renaud, H., Respaud, R., Marchand-Adam, S. & Crestani, B. Transcriptome of cultured lung fibroblasts in idiopathic pulmonary fibrosis: Meta-analysis of publically available microarray datasets reveals repression of inflammation and immunity pathways. Int. J. Mol. Sci. https://doi.org/10.3390/ijms17122091 (2016).
Sala, M. A. et al. Inflammatory pathways are upregulated in the nasal epithelium in patients with idiopathic pulmonary fibrosis. Respir. Res. 19, 233. https://doi.org/10.1186/s12931-018-0932-7 (2018).
Raghu, G. et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am. J. Respir. Crit. Care Med. 198, e44–e68. https://doi.org/10.1164/rccm.201807-1255ST (2018).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. https://doi.org/10.1093/bioinformatics/btp352 (2009).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. https://doi.org/10.1038/ng.806 (2011).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164. https://doi.org/10.1093/nar/gkq603 (2010).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82. https://doi.org/10.1016/j.ajhg.2010.11.011 (2011).
Patterson, N., Price, A. L. & Reich, D. Population structure and eigenanalysis. PLoS Genet. 2, e190. https://doi.org/10.1371/journal.pgen.0020190 (2006).
Belkadi, A. et al. Whole-exome sequencing to analyze population structure, parental inbreeding, and familial linkage. Proc. Natl. Acad. Sci. U. S. A. 113, 6713–6718. https://doi.org/10.1073/pnas.1606460113 (2016).
Asimit, J. & Zeggini, E. Rare variant association analysis methods for complex traits. Annu. Rev. Genet. 44, 293–308. https://doi.org/10.1146/annurev-genet-102209-163421 (2010).
Wu, M. C. et al. Powerful SNP-set analysis for case-control genome-wide association studies. Am. J. Hum. Genet. 86, 929–942. https://doi.org/10.1016/j.ajhg.2010.05.002 (2010).
Wu, M. C. et al. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 89, 82–93. https://doi.org/10.1016/j.ajhg.2011.05.029 (2011).
Guo, M. H., Plummer, L., Chan, Y. M., Hirschhorn, J. N. & Lippincott, M. F. Burden testing of rare variants identified through exome sequencing via publicly available control data. Am. J. Hum. Genet. 103, 522–534. https://doi.org/10.1016/j.ajhg.2018.08.016 (2018).
Wolock, C. J. et al. A case-control collapsing analysis identifies retinal dystrophy genes associated with ophthalmic disease in patients with no pathogenic ABCA4 variants. Genet. Med. 21, 2336–2344. https://doi.org/10.1038/s41436-019-0495-0 (2019).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361. https://doi.org/10.1093/nar/gkw1092 (2017).
Funding
The research leading to these results received funding from the National Key R & D Program of China (No. 2016YFC0905700) and the National Natural Science Foundation of China (No. 81670061). However, the funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
C.F., H.H. and Z.X. constructed the database and designed the study. C.F., Q.Z., N.W., X.J., J.G. and M.F. performed experiments and collected data. C.F., H.H., Y.F., M.F. and Z.X. analyzed the data. C.F. wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fang, C., Huang, H., Feng, Y. et al. Whole-exome sequencing identifies susceptibility genes and pathways for idiopathic pulmonary fibrosis in the Chinese population. Sci Rep 11, 1443 (2021). https://doi.org/10.1038/s41598-020-80944-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-80944-4
- Springer Nature Limited