
Abstract
Background
Evolutionary processes can cause strong spatial genetic signatures, such as local loss of genetic diversity, or conflicting histories from mitochondrial versus nuclear markers. Investigating these genetic patterns is important, as they may reveal obscured processes and players. The maternally inherited bacterium Wolbachia is among the most widespread symbionts in insects. Wolbachia typically spreads within host species by conferring direct fitness benefits, and/or by manipulating its host reproduction to favour infected over uninfected females. Under sufficient selective advantage, the mitochondrial haplotype associated with the favoured maternally-inherited symbiotic strains will spread (i.e. hitchhike), resulting in low mitochondrial genetic variation across the host species range.
Method
The common bluetail damselfly (Ischnura elegans: van der Linden, 1820) has recently emerged as a model organism for genetics and genomic signatures of range expansion during climate change. Although there is accumulating data on the consequences of such expansion on the genetics of I. elegans, no study has screened for Wolbachia in the damselfly genus Ischnura. Here, we present the biogeographic variation in Wolbachia prevalence and penetrance across Europe and Japan (including samples from 17 populations), and from close relatives in the Mediterranean area (i.e. I. genei: Rambur, 1842; and I. saharensis: Aguesse, 1958).
Results
Our data reveal (a) multiple Wolbachia-strains, (b) potential transfer of the symbiont through hybridization, (c) higher infection rates at higher latitudes, and (d) reduced mitochondrial diversity in the north-west populations, indicative of hitchhiking associated with the selective sweep of the most common strain. We found low mitochondrial haplotype diversity in the Wolbachia-infected north-western European populations (Sweden, Scotland, the Netherlands, Belgium, France and Italy) of I. elegans, and, conversely, higher mitochondrial diversity in populations with low penetrance of Wolbachia (Ukraine, Greece, Montenegro and Cyprus). The timing of the selective sweep associated with infected lineages was estimated between 20,000 and 44,000 years before present, which is consistent with the end of the last glacial period about 20,000 years.
Conclusions
Our findings provide an example of how endosymbiont infections can shape spatial variation in their host evolutionary genetics during postglacial expansion. These results also challenge population genetic studies that do not consider the prevalence of symbionts in many insects, which we show can impact geographic patterns of mitochondrial genetic diversity.
Similar content being viewed by others


Background
Range expansion studies have uncovered waves of demographic expansion in many species by comparing the genetic diversity of the initial source to that of the edge populations. Often, but not always (e.g. [1]), range expansions lead to reduced genetic diversity and stronger genetic differentiation at a species range limits compared to the source populations. These patterns are often due to the action of drift during rapid demographic spatial expansion and colonization [2, 3]. Under these conditions, certain alleles and genotypes have been shown to spread in the newly colonized regions due to allele surfing [4], or due to selection for local adaptation in novel environments at the range limits [1, 5,6,7,8]. Hidden processes and players may however confound these patterns, and challenge our full understanding of the evolutionary histories and genetic diversity of source and edge populations. Infections with maternally inherited symbionts, for example, can cause loss of genetic diversity across entire populations because of differential selection pressures on the infected versus uninfected host lineages, a process that may masquerade as selection or drift. The selective sweep of maternally inherited symbionts can lead to the hitchhiking of certain host haplotypes, which may not themselves be the major targets of selection [9,10,11,12].
The maternally inherited symbiotic bacteria Wolbachia can be found in up to half of all arthropod species [13, 14]. These bacteria are selfish passengers known for manipulating their host reproductive system via, for example, inducing the killing or feminization of the male progeny [15, 16], and the overproduction of daughters from unfertilized eggs (thelytoky [17]), or causing incompatibility between males and females of different infection status [18]. Some Wolbachia strains have also been shown to provide nutrients essential to the survival of their hosts [19], to protect against natural enemies [20,21,22], or to influence their host behaviours in ways that enhance fitness (e.g. mating rate [23], lekking [24], host choice [25], and more—see review [26]). These phenotypes have been selected as they improve the fitness of infected hosts over their uninfected counterparts, and therefore provide efficient means for the spread of the symbiont through generations, which can affect the inheritance pattern of the host DNA [27]. Rapid spread of maternally transmitted Wolbachia across populations and within species can lead to hitchhiking of the co-inherited mitochondrial haplotypes, increasing their frequencies in the host population. As a result, the overall mitochondrial diversity has been shown to decrease within infected populations [28].
In addition to potentially biasing population genetic signals of selection and drift, hidden Wolbachia infections could mislead, or challenge, species identification [29] and genetic and phylogenetic inferences based on the mitochondrion, as infection with such symbiont can lead to mito-nuclear discordance or affect diversification processes [30]. Integrating data about the infection-status of species as part of the routine protocol of genetic and phylogenetic studies, could for example inform on the obscure mitochondrial history of closely related species and hybrids [31].
The insect order Odonata (dragonflies and damselflies) includes approximately 6400 species belonging to 32 families [32]. Within these, the damselfly genus Ischnura (bluetails/forktails; Zygoptera: Coenagrionidae) is broadly distributed in both the Old and the New Worlds. Members of this damselfly genus and other odonates have been shown to readily undergo range shifts and expansion in response to climate change [33]. In the clade containing the common bluetail damselfly (Ischnura elegans), hybridization and introgression have been reported between I. elegans and the island bluetail (I. genei), the Sahara bluetail (I. saharensis) [34], and the Iberian bluetail (I. graellsii) [35], under secondary sympatry following recent range shifts [34,35,36]. Other studies have shown significant adaptive allele frequency changes along the northward range expansion gradient in I. elegans, consistent with selection caused by diverse novel environmental conditions, such as temperature, precipitation and wind speed [1]. Several Ischnura species, including I. elegans, often have heritable, female-limited colour polymorphisms that include a heritable male-mimic (androchrome; blue females) and one to two other morphs (red or green) [37,38,39,40]. Recent studies have shown that these morphs vary in their resistance and tolerance to parasitism by water mites [41], and in various aspects of thermal performance at the northward edge of their range [42, 43].
Although patterns and consequences of Wolbachia host interactions have been studied extensively in other insect groups (e.g. Hymenoptera [44]; Lepidoptera [10]; Diptera [45]), the recent studies by Thipaksorn et al. [46], Salunkhe et al. [47] and Lorenzo-Carballa et al. [48] possibly represent the only three systematic studies on Wolbachia infection in Odonata, and no previously published study has focused on Wolbachia in the genus Ischnura. Here, we investigated Wolbachia strain diversity in three Ischnura species (I. elegans, I. genei, and I. saharensis) and provide a first low-coverage assembly of the most-commonly found strain infecting I. elegans in Europe. By sequencing four mitochondrial markers (2375 bp) and one nuclear marker (512 bp), we quantified host genetic diversity, and (I) tested whether any Wolbachia-induced selective sweep that might have reduced genetic diversity across the I. elegans species range, and (II) looked for evidence of horizontal transfer of the symbiont between the three Ischnura species. Finally, by comparing the Wolbachia infection status of the three female colour morphs and the males of I. elegans, we tested whether there was any indication that the morphs differ in their infection status, just as they do in terms of ectoparasitic water mite infection [41]. This study thus reveals previously hidden players in the ecology and evolution of the range expanding species I. elegans, and three of its relatives.
Methods
Samples collection
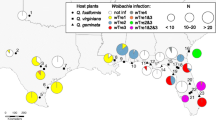
Ischnura damselflies were collected during the summer of 2015, 2016, 2019 or 2020 depending on their geographical origins. Individuals were caught in the field and stored in 95% ethanol in a − 20 °C freezer until further analysis. Specimens included 87 individuals from seven local populations in South Sweden (15 males and 72 females of all three morphs), and 105 other individuals from twelve other geographic regions, including Finland (Åland islands and mainland), Scotland, France, Cyprus, Greece, Ukraine, Belgium, the Netherlands, Italy, Montenegro and Japan (Table 1). We aimed for a minimum of three specimens per population but also included unique individuals from some regions. The seven Swedish local populations are all located within a few kilometres from each other; thus, samples were grouped under a unique ‘Southwest’ population for the rest of the study. Similarly, samples from Belgium and the Netherlands were grouped as one unique population, denoted as ‘Vinne-Walem’. In contrast, the samples from isolated parts of Italy were separated in three populations based on their relative geographical locations, and denoted either ‘Northern’, ‘Central’, or ‘Southern’ (Fig. 1, Table 1).

modified from maps freely available here: Europe (https://d-maps.com/carte.php?num_car=2232&lang=en), Japan (https://d-maps.com/carte.php?num_car=354&lang=en), Morocco (https://d-maps.com/carte.php?num_car=1132&lang=en)
Wolbachia strain diversity and penetrance from 17 populations across the geographical range of the damselfly Ischnura elegans. The top right window shows the data from the two populations in Japan. The sample in Morocco [AI] (bottom left) is from the species I. saharensis, while the samples from [CO], [SC] and [SD] are I. genei. Size of each pie chart is proportional to the number of individuals included in the study. ‘*’: Wolbachia infection rates in these populations are only based from the screening of one unique individual. Thus, for these populations we might be providing an over or under-estimations of the true local prevalence of the bacterium. Maps were
We also included 20 specimens belonging to three other Ischnura species in this study:
-
Thirteen specimens of I. pumilio, collected in 2019 from four Swedish populations. Ischnura pumilio co-occurs sympatrically with I. elegans in this region but falls in another major phylogenetic clade of the Ischnura tree, and is less closely related to I. elegans than are the following two species [49].
-
Three specimens of I. genei, an allopatric species to I. elegans endemic of the western Mediterranean region. Two samples of I. genei were collected from the two insular populations of Sardinia and Sicily (Italy), and the third sample is from Corsica (France) [50]
-
One unique specimen of I. saharensis collected from Morocco. The species is also an allopatric species to I. elegans and is distributed across North Africa.
Molecular work
Specimens were dissected in sterile conditions to avoid cross specimen contamination. We extracted the DNA from the abdomen of each damselfly, except for the Japanese samples, for which DNA was extracted from one leg, following the protocol of a Qiagen DNeasy Blood & Tissue Extraction Kit (Cat. #69506, Qiagen, USA). The quality of all DNA extracts was tested by PCR, through the amplification of the 5′-end region (~ 654 bp) of the cytochrome oxidase I (COI) mitochondrial gene using the primers LCO-1490/HCO-2198 designed by Folmer et al. [51]. Only samples that were positive for the COI amplification were included in the following analyses.
All sequences were deposited into the GenBank database (Accession #MZ463094-100; MZ501175-205; MZ508997-9001; MW509059-66; MZ893225-MZ893331). In total, we amplified and sequenced four mitochondrial regions (COI, COIb, COIIa and NDI) to test for a selective sweep, one nuclear marker (PRMT) to test for population bottlenecks, and two Wolbachia genes (ftsZ and wsp) to characterize strain diversity. Note: an extra Wolbachia gene (fbpa) was sequenced for few wEle1-infected specimens from Sweden (primers details in Additional file 1: Table S2). Purified PCR products from independent PCRs were sent to Macrogen (Macrogen Europe, Inc.) for single strand direct forward Sanger sequencing. All sequences were manually curated and aligned using Geneious Prime 2020.2.4 (https://www.geneious.com) and AliView 1.26 [52]. Double peaks in the chromatograms were treated as either evidence of contamination (for mtDNA), multiple infections (for Wolbachia DNA), polymorphism (for mtDNA and nuclear DNA), or sequencing noise (all). Wolbachia and mtDNA sequences showing such patterns were not included in the following analyses, while the analysis of those double peaks in the nuclear locus sequences allowed us to identify heterozygotic and homozygotic specimens at polymorphic sites (see below).
Ischnura elegans nuclear haplotype diversity
To identify diversity at the nuclear level, we isolated 400 bp of the successfully sequenced nuclear gene PRMT from 48 specimens (27 uninfected and 21 Wolbachia-infected) from 10 populations ([AB], [RK], [IK], [SW], [VC], [MI], [NI], [SI], [CY] and [GR]), with 2–12 specimens per population. However, Italy ([MI], [NI] & [SI]) and Japan ([RK] & [IK]) carry Wolbachia strains that are divergent from wEle1, and which may have altered the genetics of those populations in ways that are impossible to fully test due to our small sample size for these populations. Therefore, we did not include these sequences in further analyses. The final sample size was 31 sequences, including 23 Wolbachia-uninfected specimens and 8 Wolbachia-infected specimens from five populations (Table 1). To test whether the nuclear gene from Wolbachia-infected and uninfected damselflies has evolved under neutrality, we performed neutrality tests in DnaSP v6.0 [53] by calculating Tajima’s D (Tajima, 1989) and Fu and Li’s F [54] metrics, and by estimating nucleotide diversity (π) and haplotype diversity (Hd) (Table 2). We also estimated the observed heterozygosity levels at two nuclear polymorphic sites for both the Wolbachia-infected and uninfected specimens.
Wolbachia and mitochondrial haplotype diversity, phylogenies and haplotype networks
All Wolbachia Sanger-sequences were BLAST-ed against the Wolbachia PubMLST database (https://pubmlst.org/wolbachia/) [55] to find the corresponding or closest alleles at each locus. Additionally, all five MLST genes (ftsz, gatB, hcpA, coxA, and fbpA) and the wsp gene of the strain wEle1 were also extracted from the whole genome project of a Swedish I. elegans [56]. All Wolbachia reads were identified and isolated from the raw read data of the I. elegans genome project [56]. The wEle1 assembly was built by mapping reads to two previously sequenced Wolbachia genomes (wPip [57] and wMel [58]) using bwa mem version 0.7.8 [59]. The properly mapped pairs were extracted using samtools 1.8. The isolated Wolbachia paired reads were assembled into a draft genome using spades version 3.9.0 at kmers 21, 33, 55, 77 and 99. The wEle1 draft genome assembly (Nscaffold = 893; N50 = 5523 bp; longest scaffold = 53,331 bp, genome size = 1.4 MB) is available as Additional file 2, and the raw reads are available from NCBI under the Bioproject PRJNA575663.
We concatenated sequences of the ftsZ and wsp genes from each Wolbachia strain characterized in this study, and from five additional strains (wMel, wRi, wClec, wBm, wPip; Isolate id number: 1, 11, 36, 37, 1808 in Wolbachia pubMLST database, respectively) previously assigned to the A-, A-, F-, D-, and B-Wolbachia supergroups, respectively. The phylogenetic analyses were conducted in IQ-Tree on XSEDE [60] implemented in CIPRES v.3.2 [61], using the genes separately (Additional file 1: Fig. S1) or concatenated (Fig. 2). ‘Model Selection’ [62] was selected to allow for the search of the best model in CIPRES. The partition type was set to allow the two partitions (one for each gene) to have different speeds [63]. The best fit substitution models were decided by running ‘-m TESTNEW’ in IQ-Tree. Bootstrapping was conducted using ‘Ultrafast’ and ‘SH-aLRT’ bootstrap methods (Hoang et al. 2018) in IQ-Tree with 1000 replicates. The ‘TN + F + I’ and ‘TPM3 + F + G4’ models were applied to ftsZ and wsp genes, respectively, as the best fit models with highest BIC (Bayesian information criterion) scores. All other setting options were left as default. The pairwise genetic distances between Wolbachia strains were calculated in MEGA-X [64]. The best phylogenetic trees were visualized in FigTree v.1.4.4. (http://tree.bio.ed.ac.uk/software/figtree/), and rooted using the wBm-D and wClec-F strains as outgroups (Fig. 2).

Maximum likelihood tree of (A) the Wolbachia strains characterized in the present study and based on the concatenated sequences of the ftsZ and wsp genes, and of (B) five Wolbachia strains from five Ischnura species, including wEle1, based on the fbpa gene. C Phylogenetic tree of the Ischnura genus phylogeny, for comparison, as provided by [70]. In (B): the strains marked with red come from Ischnura species: wSen [44], ‘Wolbachia_I. spp.’, wCar and wTai [45]. Five additional strains (wBm, wClec, wMel, wRi, wPip) were also included in the trees as references for the different Wolbachia-supergroups A, B, D and F. The two Wolbachia trees were rooted using the D and F-Wolbachia supergroups as outgroups. Bootstrapping was conducted using ‘Ultrafast’ bootstrap method in IQ-Tree with 1000 replicates. Links between the B and C trees show the lack of concordance between the symbiont and host trees
Additionally, we built two types of mitochondrial haplotype networks: (A) one based on the COI 5′-end region only (598 bp), and (B) a second based on all four mitochondrial regions (2375 bp). The networks were built using POPART [65] with the median joining method [66] (Fig. 3). To the mitochondrial sequences produced by the present study, we added mitochondrial sequences from the same markers from any species of the I. elegans clade (i.e. I. elegans, I. genei, I. saharensis, I. graellsii and I. fountaineae), publicly available in GenBank before July 2020 (Additional file 1: Table S3). Note: only sequences with a length equal or longer to 600 bp were included to ensure the performance of the analyses.

Mitochondrial haplotype networks of I. elegans and two closely related species, I. genei and I. saharensis, based on (a and c) the mitochondrial COI gene only; and (b and d) all four mitochondrial markers, organised per country (a and b) or per infection status (c and d). Each circle represents one unique haplotype, which might be biased by differences in our sampling effort between populations. The size of the circle is proportional to the number of specimens carrying the same haplotype. The small black nodes indicate unobserved haplotypes. All other nodes were coloured by populations. The number of black bars between two nodes represent nucleotide differences between two haplotypes. The mitotypes of ‘Unknown’ infection status were collected from Genbank and EMBL (Additional file 1: Table S3)
Wolbachia selective sweep
The estimation of the timing of the Wolbachia sweep in I. elegans was carried out following the method described by Rich et al. [67]. We first estimated the neutral mutation rates at the third position of fourfold and twofold synonymous codons of the four mitochondrial genes separately. The open reading frames of mitochondrial genes were found by blasting the nucleotide sequence against the mitochondrial proteome of I. elegans [68]. The mitochondrial sequences of I. elegans and I. pumilio were aligned in MEGA X [64] in order to calculate the number of nucleotide differences and the number of fourfold and twofold synonymous sites between the two species. Jukes–Cantor correction [69] was applied to correct for multiple substitutions. Lastly, the neutral mutation rates were calculated based on the divergence time between I. elegans and I. pumilio, estimated between 10.4 and 21.7 My before present [70] (calculations were repeated twice, using each extreme of that divergence time range). Consequently, the age of the infection can be estimated using the following equation:
where t is the estimated time since the infection; S is the number of observed neutral polymorphisms in a set of mitochondrial haplotypes; \({\mu }_{a}\) and \({\mu }_{b}\) are the neutral mutation rates at the third position of fourfold and twofold synonymous codons, respectively; \({n}_{i}\) is the number of sampled sequences at the ith locus; \({l}_{i}\) and \({m}_{i}\) are the number of fourfold and twofold synonymous sites at the ith locus. Our estimation was only based on mitochondrial haplotypes that were carried by the infected individuals. With this method, the corrected number of substitutions was estimated as 137.91 = 245(− 3/4)ln[1 − (4/3) × (97/245)] and 111.11 = 332(− 1/2)ln[1 − 2 × (81/332)] among fourfold and twofold degenerate codons, respectively. Assuming the maximum estimate divergence time at 21.7 Mya [70], we estimated that neutral mutation rates on fourfold and twofold synonymous sites would be expected to be 1.30% and 0.77% per site per million years, respectively. If the minimum estimate of 10.4 Mya was assumed, the neutral mutation rates are estimated to be 2.71% and 1.61%, respectively. These estimations are biologically reasonable if we assume the general divergence rate of mtDNA in arthropods at 1.1–1.2% per site per million years (Brower 1994).
Finally, to test whether the mitochondrial genes from Wolbachia infected and uninfected damselflies have evolved under neutrality, we performed neutrality tests in DnaSP v6.0 [53] by calculating two population genetic statistics: Tajima’s D (Tajima, 1989) and Fu and Li’s F [54], and by estimating nucleotide diversity (π) and haplotype diversity (Hd).
Ischnura elegans colour polymorphism, sex differences, and Wolbachia infection in Sweden
All statistical analyses were performed in R version 3.6.1 [71]. We used Chi-square test to investigate the association between Wolbachia infection and sex, and between infection and colour morph in female I. elegans. As most populations had a limited and incomplete sampling per sex and colour morph (e.g. only one female from Åland and from Finland mainland; only two morphs present in the Japan, Cyprus and Scotland populations, see Zenodo open data submission https://doi.org/10.5281/zenodo.4445061), this test was only performed using the Swedish specimens. Fisher’s exact test was applied as an improvement of Chi-square test when the expected value of any cells of the contingency table is below five (Table 3).
Results
Wolbachia penetrance and prevalence in I. elegans and closely related species
There were geographical variations in Wolbachia penetrance across the I. elegans populations (Table 1; Fig. 1). In Europe, our data revealed that Wolbachia infection was more prevalent in the north-western than in the south-eastern regions (Fig. 1). The infection frequency was over 50% in ten populations, including Sweden [SW], Scotland [AB], the Netherlands and Belgium [VW], Finland mainland [FL], Northern Japan [IK], France [FR], Italy (NI, MI, SI), and Ukraine [VC] (Note: the sample size for the latter three countries being < 5, our data might represent under or over-estimates of the Wolbachia infection rates in these countries). In Sweden, where the majority of our specimens came from, the infection was nearly fixed, with a frequency of 97.7% (85 individuals infected out of 87 samples tested). In contrast, all specimens from Montenegro [BJ], Greece [GR], Cyprus [CY], Finland Åland [ÅL], Ukraine (DR, PC) and Central Japan [RK] were Wolbachia-free. Note that the infection screening of the Japanese samples was done using leg tissues, which do not always host Wolbachia cells. Although all samples from [IK] were found infected, the Wolbachia-free status of [RK] could still be an underestimation of the true infection rate in this population. Additionally, all 13 I. pumilio specimens from Sweden were infected. The I. genei specimen from Corsica Island [CO] was infected, while the other two from Sicily [SI] and Sardinia [SD] were not. The single I. saharensis specimen from Morocco was also uninfected.
Wolbachia strain diversity in Ischnura elegans, I. genei and I. saharensis
In total, three Wolbachia strains from the B-supergroup Wolbachia commonly found in insects, were identified in I. elegans (Table 1). They were denoted wEle1, wEle2, and wEle3. The strain wEle1 was widespread and found in Sweden [SW], Scotland [AB], Belgium and the Netherlands [VW], France [FR], Italy (MI, SI) and Ukraine [VC] (Fig. 1). In contrast, the strain wEle2 was restricted to Italy (NI, MI), and wEle3 was found in mainland Finland [FL] and Japan [IK]. The I. genei specimen from Corsica was also infected with the strain wEle1; while I. pumilio was found to carry two divergent strains, denoted wPum1 and wPum2, with eight specimens (out of 13) potentially carrying the two strains simultaneously. Note that we characterized these strains using only two markers, which combination should allow the differentiation of most strains, but possibly not of all strain variants [72]. Raw reads from the I. elegans whole genome sequencing project [56] were assigned to Wolbachia, and were used to build a de novo Wolbachia assembly of strain wEle, consisting of under 900 scaffolds. This fragmented Wolbachia assembly is 1.4 Mb long, suggesting it represents the full genome of a Wolbachia strain [73]. Although the circular genome assembly could not be built, we are confident these scaffolds are from a single bacterial infection that are not inserted in the genome of the host, as each scaffold did not contain any host genomic material.
The Wolbachia phylogeny based on the ftsZ and wsp genes (Fig. 2a) showed that all strains characterized in this study were from the B-supergroups (genetic distance between wPip and all five strains ranged from 0.077 to 0.113). The three strains isolated from I. elegans formed a monophyletic group, divergent from the two strains from I. pumilio, wPum1 and wPum2. The pairwise genetic distance ranged from 1.06e−3 to 2.04e−2 among the three I. elegans strains, and from 3.24e−2 to 7.49e−2 between wEle and wPum strains (Additional file 1: Table S1). When compared to the previous strain records from the Wolbachia PubMLST database [55], almost all strains carried the ftsZ allele #7, except the strain wPum2, which carried the ftsZ allele #73 (only one nucleotide difference from allele #7). The wsp allele #61 was characterized from wPum2, while the four other wsp alleles were new to the Wolbachia PubMLST database, for a total of 75 polymorphic sites.
Genetic diversity and Wolbachia-induced selective sweep in I. elegans
To identify effects of wEle1 infection on host genetic diversity, we identified 25 polymorphic sites over the analysed 400 bp of the nuclear locus PMRT sequenced from five I. elegans populations. We found 1–10 genotype(s) per population, with individual genotypes showing no less than 98.1% similarity. The haplotype diversity (Hd) at PMRT was 0.88 for the Wolbachia-infected specimens but higher (Hd = 0.91) for the uninfected specimens (Table 2). Additionally, observed heterozygosity levels at two informative polymorphic sites within the PMRT gene (position n = 47 and 228) were similar between infected and uninfected specimens: 25% and 50% for infected vs 23.5% and 43% for uninfected specimens (two-tailed P = 0.86; and P = 0.61, respectively).
Based on the analysis of the I. elegans COI locus only, we detected a total of 24 mitochondrial haplotypes (Haplo1 to 24) from 169 specimens (Fig. 3a, c). The combined analysis of all four mitochondrial markers revealed additional mitotypes (17 mitotypes, Haplo_A to Q), despite all four markers being screened in 47 I. elegans specimens only (Fig. 3b, d). Both mitochondrial haplotype network analyses (based on COI and COI + COIb + COII + NDI, respectively), however, showed similar patterns, with three main distinct clades emerging (Clade 1: Japan; Clade 2: Cyprus; and Clade 3: all other populations dominated by one common mitotype, the Haplo1 or Haplo_A; Fig. 3). Note that three haplotypes, Haplo23 from Italy, Haplo24 from Japan and Haplo19 from Cyprus, characterized with the COI gene only, fell outside the major clades of the tree. A BLAST search for these three haplotypes in the Barcode of Life Data system (BOLD v.4, [74]) showed that Haplo19 grouped with an unidentified Ischnura species from Iraq (99.5% similarity to an ‘early-released’ BOLD sample—Data not provided), while Haplo24 and Haplo23 grouped with I. elegans specimens from Pakistan (99.85% & 99.63% similarity to BOLD accession #MAODO254-11 & #MAODO255-11, respectively). The divergence may indicate that these three mitotypes represent either polymorphism in the COI gene, or species misidentification. By precaution, we removed the specimens from the rest of the analyses.
Most wEle1-infected specimens shared a single mitochondrial haplotype (Haplo1 or Haplo_A). However, the analysis of the four mitochondrial genes revealed that few wEle1-infected specimens alternatively carry one of three closely related mitotypes (Haplo_B, Haplo_C or Haplo_F, Fig. 3b, d), all three of which show little divergence from Haplo_A. Across the geographical range of I. elegans, the low mitochondrial haplotype diversity among wEle1-infected individuals (Hd = 0.13) contrasts with the high haplotype diversity found among the uninfected individuals (Hd = 0.70; when only COI is considered, Table 2). The Wolbachia-free Cyprus [CY] population maintained the largest mitochondrial haplotype diversity in our samples, with 11 haplotypes for 20 samples sequenced (Haplo1, Haplo13 to 22, Ho = 0.55). Altogether, these results are indicative of a wEle1-induced selective sweep in I. elegans, which is also supported by significantly negative results from the neutrality tests (Tajima’s D, Fu and Li’s F; Table 2). The other two Wolbachia strains characterized from I. elegans (wEle2 and wEle3) were also associated with few mitochondrial haplotypes, but these haplotypes were divergent from Haplo1. The Wolbachia strain wEle2 was associated with Haplo7 (Haplo_D and E) in Italy, while wEle3 was associated to Haplo24 (Haplo_P and Q) in Japan or Haplo9 in mainland Finland (Fig. 3). However, the effects of the Wolbachia strains wEle2 and wEle3 on mitochondrial haplotype diversity remain unclear, due to our small sample size. The Haplo1 commonly found in wEle1-infected I. elegans, was also characterized in the wElel1-infected I. genei sample from Corsica, and the uninfected I. saharensis sample from Morocco. This observation could suggest hybridization between these species or, alternatively, shared infection pre-speciation of these taxa. In contrast, the wPum-infected Swedish I. pumilio specimens carried Haplo4, and the three uninfected I. genei specimens from Italy carried either Haplo9 or Haplo10 (Fig. 3).
Based on the analysis of the sequences of the four mitochondrial loci, there were nine amino acid differences between the mitochondrial sequences of I. pumilio and I. elegans. Among the unchanged amino acid sites, we found 97 and 81 synonymous nucleotide differences among the 245 fourfold and 332 twofold degenerate codons, respectively. To correct for multiple substitutions and other mechanisms that cause higher number of substitutions more than the observed, we used the Jukes-Cantor correction. Given that we found four observed polymorphic sites in wEle1-infected individuals, we estimated the selective sweep of wEle1 to have happened between 20,860 and 43,543 years before present. When the timing was evaluated based on the mitochondrial COI locus only (598 bp), which was sequenced in more samples, the sweep was estimated between 10,159 and 21,174 years before present.
Ischnura elegans colour polymorphism, sex differences, and Wolbachia infection in Sweden
Within Sweden only, the area where most of our samples come from, we found that the three female colour morphs show similar Wolbachia infection frequencies (Fisher’s exact test, p = 0.63, Table 3) (Infection frequency: (A: blue) 1 = 27/27; (I: green) 1 = 22/22; (O: red) 0.96 = 23/24). Similarly, females and males were equally likely to be infected (Fisher’s exact test, p = 0.31) (Females: 0.99% or 72/73; Males: 0.93% or 14/15).
Discussion
The spread of maternally inherited symbionts is predicted to primarily affect the mitochondrial haplotype diversity of its host [12, 75], while external ecological and demographic factors like population range expansions and bottlenecks would be expected to reduce genetic diversity at both mitochondrial and nuclear levels. We found five B-supergroup Wolbachia strains in the three damselfly species of the genus Ischnura that we investigated. The common strain wEle1 was characterized in I. elegans and I. genei, while two additional strains (wEle2 and 3) were found in I. elegans only, and two divergent strains were found in the sympatric species I. pumilio (wPum1 and 2), which is sympatric with I. elegans in Sweden. In accord with a selective sweep driven by wEle1 in I. elegans across Western Europe, we would particularly like to highlight (I) the reduced mitochondrial haplotype diversity and non-neutral evolution of the mitochondrial haplotypes associated to wEle1-infected specimens, (II) the conserved nuclear haplotype diversity and levels of heterozygosity between infected and uninfected specimens. We estimated this selective sweep of Wolbachia to have occurred between 10,159 and 43,543 years ago, which is recent in the history of the host species (I. elegans and I. graellsii diverged 0.14 Mya [70]), but ancient enough to allow for the emergence of some diversity in the host mitochondrial DNA (Fig. 3), and largely concordant with the timing of the last glacial maximum (20,000 ya) in Europe [76]. Like other species of insects, I. elegans is currently shifting its geographic range northward in response to climate change [1, 33, 77, 78]. The timing of the selective sweep could suggest that I. elegans acquired the wEle1 during its ongoing northward range expansion since the last glacial period. In such situation, one would expect that the mitochondrial diversity would be affected by both the sweep, bottlenecks and drift due to range shift, while the nuclear diversity would be only affected by bottlenecks due to range shift. Although our Swedish population show the lowest mitochondrial diversity of all populations, the nuclear diversity is high, consistent with previous data showing high genetic diversity and lack of bottlenecking during the range expansion of I. elegans [1]. Our sample size is however small, and more comprehensive investigations of this may inform about the consequences of the spread of Wolbachia for the genetic diversity, the long-term success and the dynamics of this range expanding damselfly species.
Interactions between hosts and facultative symbionts are driven by complex sets of conflicts of interests between the partners, with outcomes that also depend on the environment [79, 80]. These dynamic systems will then either result in the fixation of the symbiont in the populations [81, 82], its decline [83, 84], or stability at intermediate frequencies [85, 86]. The striking success of wEle1 across the North-Western European range of I. elegans might suggest some benefits to the infected over the uninfected damselflies. Ischnura elegans is affected by parasitic water mites in nature, and previous studies have shown that tolerance and/or resistance levels to this parasite differ between sexes and female colour morphs, with for example the infuscans-obsoleta female morph showing the highest mite load when parasitized [41]. As Wolbachia is known to affect its host fitness in the presence of parasites [21, 22, 87], we tested whether the wEle1 infection rates varied between female colour morphs and sexes, which could then be linked to fitness variation for example in response to parasites in this host species. We found no differences in the wEle1-infection frequencies between sex or morph in I. elegans, but this result does of course not rule out the possibility that the symbiont could still protect against parasitism in I. elegans. Wolbachia are also known to manipulate their hosts in various other ways that may similarly support its success and spread in the host populations [9, 17, 88, 89]. The strain wEle1 is unlikely to manipulate its host reproductive system via feminization or male killing, as both females and males were found infected and the population sex-ratios were not systematically female biased [38, 41]. The strain could however induce cytoplasmic incompatibility, a type of sperm-egg incompatibility between males and females of different infection status [90], which would not affect population sex-ratio. Future mesocosm studies and mating experiments in semi-natural conditions [40] would allow the investigation of these hypotheses, to reveal the costs and/or benefits of Wolbachia in the damselfly species.
Although we found wEle1 at almost fixed frequencies in the Western European populations of I. elegans, the strain was rare in Eastern Europe (i.e. Greece [GR], Ukraine (DR, PC), and Montenegro [BJ]), and absent from a few other populations (i.e. Cyprus [CY], Italy (NI, MI), Åland [ÅL]). We showed that the few uninfected individuals found in the Western European populations carry the same mitochondrial haplotypes as their wEle1-infected conspecifics (Haplo1 and Haplo3), contrasting with the uninfected specimens from Eastern Europe that show a wide diversity of divergent mitochondrial haplotypes. This may suggest that the maternal transmission of the strain is not perfect and that few offspring from infected mothers can hatch uninfected in these populations. Additionally, two hypotheses could explain differences between populations: (I) wEle1 has spread across the western populations but did not yet invade the remaining populations, or (II) wEle1 has spread across the whole Europe in the past, but the infection was consequently lost in Eastern populations. For the first hypothesis, Wolbachia-infected population would show reduced mitochondrial haplotype diversity, while uninfected populations would have high mitochondrial diversity [10, 91]. In contrast, the second hypothesis suggests reduced haplotype diversity at the mitochondrial level and shared mitochondrial haplotypes, or closely related haplotypes, in both infected and uninfected populations [12, 88]. There is some support for each of these hypotheses, as we discuss more below.
The two populations originating from the Cyprus island in the Mediterranean Sea and from the Åland islands in the Baltic Sea, were both Wolbachia-uninfected. The Cyprus population is located at the southern range limit of I. elegans in Europe. There, the strong divergence of the mitochondrial haplotypes to those associated to the infection in Western Europe suggests that the population has remained uninfected potentially due to its geographic isolation. The population differ phenotypically in several aspects from other continental populations, particularly in terms of smaller average body size and deviant colour morph frequencies [92]. In contrast, although also geographically isolated, the island population on the Åland archipelago carries one unique haplotype that only differs by one nucleotide from the most common haplotype associated with wEle1 in Sweden, and is identical to the haplotype associated with wEle3 in mainland Finland (although based on one unique mitochondrial gene). These results better support the second hypothesis described above, in which the Åland population may have lost its infection recently. However, the I. elegans specimens collected in Åland all came from a single collection site in the southern part of the main island (i.e. Nåtö). Thus, these specimens might not be representative of the true average infection status of the entire Åland populations, and more mitochondrial genetic data will be needed to infer whether I. elegans colonized the Åland islands (I) after the sweep of wEle1 in Sweden, or (II) after a sweep of wEle3 in Finland and subsequently lost the infection and diverged at the mitochondrial level. Similarly, the uninfected individuals from Greece and Ukraine carry similar, to identical, mitochondrial haplotypes to the wElel1-infected samples, which is also consistent with recent infection loss and divergence from an original population.
As a facultative symbiont, Wolbachia is frequently lost [83, 93], either due to drift following the colonisation of new habitats [94] or selective pressures on the symbiont. High temperatures can also negatively affect Wolbachia titers in some Drosophila species [95], and lead to the loss of the infection in mosquitoes [96] and mites [97] reared under laboratory conditions. Additionally, local population variations can also be explained by imperfect transmission of the symbiont through generations, or/and locally variable negative selective pressures on the infected individuals. This is the case for example in the Åland population of the parasitoid wasp Hyposoter horticola (Hymenoptera) [87], which show strong variation in Wolbachia penetrance across local populations (from 0 to 100% infection rate, [86]). Infection patterns across the range of I. elegans could thus reflect a dynamic population history with local variations in infection status and penetrance.
Our study suggests a history of short-time separation between the Italian populations, and of longer-time separation between the Japanese and European populations. In the Italian peninsula, I. elegans damselflies carry two closely related mitochondria (Haplo1 and Haplo6), found in association with two closely related Wolbachia strains (wEle1 and wEle2, respectively). Both mitotypes and Wolbachia strains may have diverged after isolation in geographically separated refugia on the Italian peninsula during the last glacial maximum [76, 98]. In contrast, wEle3, the third strain characterized in all I. elegans specimens collected from Japan, and from mainland Finland, is highly divergent from both wEle1 and wEle2. The strain wEle3 is found in association with two mitochondrial haplotypes (Haplo9 and Haplo24), suggesting either haplotype diversity across the large range of this particular infection, or yet undetected bacterial diversity across the large geographical distance separating Japan from Finland. Additional sampling, screening and genotyping efforts are likely to uncover a higher strain diversity in the genus Ischnura than already suggested here and by previous studies (in I. senegalensis: [47], in I. taitensis: [93]).
Although a maternally inherited symbiont, Wolbachia has been shown to also transfer horizontally between species. Examples include, but are not restricted to, the horizontal transfer of Wolbachia between damselfly species of the genera Nesobasis and Melanesobasis in Fiji Islands in the Pacific Ocean [48]. Wolbachia can be horizontally transmitted via different means, including hybridization between host species [48], shared resources (e.g. shared hostplant [99]), or shared parasitism (e.g. shared mites [100] or parasitoids [101]). Damselflies are well-known for carrying and sharing ectoparasitic water mites [41, 102], however the role of such parasites as vectors of Wolbachia among Ischnura species remains unknown. In contrast, evidences of frequent hybridization and introgression have been shown in some odonate species due to latitudinal range expansion and the increasing sympatric interactions between closely related species [103], including in the genus Ischnura [34,35,36, 104]. The fact that the same mitochondrial haplotype and the same Wolbachia strain were isolated from both I. elegans and I. genei could suggest that horizontal transfer of the strain wEle1 between the two species occurred through hybridization.
Conclusion
The present biogeographic study of Wolbachia in the damselfly genus Ischnura revealed a wide diversity of previously hidden inherited symbiotic Wolbachia strains in the three species investigated. Furthermore, we detected a recent selective sweep of the Wolbachia strain wEle1 across the Western European populations of I. elegans, and we discuss the potential horizontal transfer of the strain through hybridization. The biogeographical pattern of the infection and the estimated timing of the sweep suggested that wEle1 might have spread across I. elegans populations during its host’s northern expansion after the last glacial maximum. Consequently, the mitochondrial haplotype diversity in this range expanding species has been highly reduced but started to recover from the successful spread of the symbiont. We found the symbiont in specimens of all colour morphs and both sexes, and thus the costs and benefits from the infections remain to be investigated. We clearly highlight the loss of host genetic diversity resulting from symbiotic associations and call for greater consideration of symbiotic infections in future research on the population genetics and ability of range expanding species in the context of climate change.
Availability of data and materials
The dataset supporting the conclusions of this article is available in the Zenodo repository https://doi.org/10.5281/zenodo.4445061; and in GenBank database (Accession #MZ463094-100; MZ501175-205; MZ508997-9001; MW509059-66; MZ893225-MZ893331) as stated in the text. The Illumina raw reads are deposited at NCBI short read archive (SRA) under accession numbers Bioproject PRJNA575663 and Biosample SAMN12906381 and SAMN12920919–20.
Abbreviations
- AB:
-
Aberdeen population, Scotland
- AI:
-
Ain Isker population in Morocco
- ÅL:
-
Åland population in Finland
- BJ:
-
Bojana population in Montenegro
- CO:
-
Corsica population in France
- COI :
-
Cytochrome c oxidase subunit 1 Coding gene, mitochondrial gene
- CY:
-
Cyprus population
- DR:
-
Dniest River population in Ukraine
- FbpA :
-
Fructose-bisphosphate aldolase protein Coding gene in Wolbachia
- FL:
-
Finland population
- FR:
-
France population
- FtsZ:
-
Cell division protein Gene in Wolbachia
- GR:
-
Greece population
- Haplo:
-
Mitochondrial haplotype
- Hd:
-
Haplotype diversity, as the probability of two haplotypes to be different
- Ho:
-
Observed haplotype diversity (NH/Nt)
- IK:
-
Ikeda population in Japan
- MI:
-
Central Italy population
- MLST:
-
MultiLocus Sequence Typing
- Na:
-
Non applicable
- NH :
-
Number of haplotypes
- NI:
-
North Italy population
- Nt:
-
Number of samples
- PC:
-
Pelican City population in Ukraine
- π (Pi):
-
Nucleotide diversity
- PMRT :
-
arginine methyltransferase Nuclear gene
- RK:
-
Rokkasho population in Japan
- S :
-
Number of polymorphic sites
- SC:
-
Sicily population in Italy
- SD:
-
Sardinia population in Italy
- SI:
-
South Italy population
- SW:
-
Sweden population
- VC:
-
Vostochne population in Ukraine
- VW:
-
Vinne-Walem population in the Netherlands/Belgium
- wEle1, wEle2, and wEle3:
-
Wolbachia strains characterized from the damselfly species Ischnura elegans
- wPum1, and wPum2:
-
Wolbachia strains characterized from the damselfly species Ischnura pumilio
- WSP :
-
Wolbachia surface protein gene
- ya :
-
Years ago
- Mya:
-
Million years ago
References
Dudaniec RY, Yong CJ, Lancaster LT, Svensson EI, Hansson B. Signatures of local adaptation along environmental gradients in a range-expanding damselfly (Ischnura elegans). Mol Ecol. 2018;27(11):2576–93.
Swaegers J, Mergeay J, Therry L, Larmuseau MH, Bonte D, Stoks R. Rapid range expansion increases genetic differentiation while causing limited reduction in genetic diversity in a damselfly. Heredity (Edinb). 2013;111(5):422–9.
Swaegers J, Mergeay J, Therry L, Bonte D, Larmuseau MH, Stoks R. Unravelling the effects of contemporary and historical range expansion on the distribution of genetic diversity in the damselfly Coenagrion scitulum. J Evol Biol. 2014;27(4):748–59.
Braga RT, Rodrigues JFM, Diniz-Filho JAF, Rangel TF. Genetic population structure and allele surfing during range expansion in dynamic habitats. Ann Braz Acad Sci. 2019;91(2):e20180179.
Duplouy A, Wong SC, Corander J, Lehtonen R, Hanski I. Genetic effects on life-history traits in the Glanville fritillary butterfly. PeerJ. 2017;5:e3371.
Alves I, Arenas M, Currat M, Hanulova AS, Sousa VC, Ray N, Excoffier L. Long-distance dispersal shaped patterns of human genetic diversity in Eurasia. Mol Biol Evol. 2016;33:946–58.
Gamboa M, Watanabe K. Genome-wide signatures of local adaptation among seven stoneflies species along a nationwide latitudinal gradient in Japan. BMC Genomics. 2019. https://doi.org/10.1186/s12864-019-5453-3.
Swaegers J, Mergeay J, Van Geystelen A, Therry L, Larmuseau MH, Stoks R. Neutral and adaptive genomic signatures of rapid poleward range expansion. Mol Ecol. 2015;24(24):6163–76.
Duplouy A, Hurst GD, O’Neill SL, Charlat S. Rapid spread of male-killing Wolbachia in the butterfly Hypolimnas bolina. J Evol Biol. 2010;23(1):231–5.
Charlat S, Duplouy A, Hornett EA, Dyson EA, Davies N, Roderick GK, Wedell N, Hurst GD. The joint evolutionary histories of Wolbachia and mitochondria in Hypolimnas bolina. BMC Evol Biol. 2009;9:64.
Hinrich J, Schulenburg Gvd, Hurst GDD, Tetzlaff D, Booth GE, Zakharov IA, Majerus MEN. History of infection with different male-killing bacteria in the two-spot ladybird beetle Adalia bipunctata revealed through mitochondrial DNA sequence analysis. Genetics. 2002;160(3):1075–86.
Rokas A, Atkinson RJ, Brown GS, West SA, Stone GN. Understanding patterns of genetic diversity in the oak gallwasp Biorhiza pallida: demographic history or a Wolbachia selective sweep? Heredity (Edinb). 2001;87(Pt 3):294–304.
Weinert LA, Araujo-Jnr EV, Ahmed MZ, Welch JJ. The incidence of bacterial endosymbionts in terrestrial arthropods. Proc Biol Sci. 1807;2015(282):20150249.
Zug R, Hammerstein P. Still a host of hosts for Wolbachia: analysis of recent data suggests that 40% of terrestrial arthropod species are infected. PLoS ONE. 2012;7(6):e38544.
Dyson EA, Kamath MK, Hurst GDD. Wolbachia infection associated with all-female broods in Hypolimnas bolina (Lepidoptera: Nymphalidae): evidence for horizontal transmission of a butterfly male killer. Heredity. 2002;88:166–71.
Hiroki M, Kato Y, Kamito T, Miura K. Feminization of genetic males by a symbiotic bacterium in a butterfly, Eurema hecabe (Lepidoptera: Pieridae). Naturwissenschaften. 2002;89(4):167–70.
Weeks AR, Breeuwer JA. Wolbachia-induced parthenogenesis in a genus of phytophagous mites. Proc Biol Sci. 2001;268(1482):2245–51.
Werren JH, Baldo L, Clark ME. Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol. 2008;6:741–51.
Nikoh N, Hosokawa T, Moriyama M, Oshima K, Hattori M, Fukatsu T. Evolutionary origin of insect-Wolbachia nutritional mutualism. Proc Natl Acad Sci USA. 2014;111(28):10257–62.
Aliota MT, Peinado SA, Velez ID, Osorio JE. The wMel strain of Wolbachia reduces transmission of Zika virus by Aedes aegypti. Sci Rep. 2016;6:28792.
Hedges LM, Brownlie JC, O’Neill SL, Johnson KN. Wolbachia and virus protection in insects. Science. 2008;322:702–702.
Teixeira L, Ferreira A, Ashburner M. The bacterial symbiont Wolbachia induces resistance to RNA viral infections in Drosophila melanogaster. PLoS Biol. 2008;6(12):e2.
de Crespigny FE, Pitt TD, Wedell N. Increased male mating rate in Drosophila is associated with Wolbachia infection. J Evol Biol. 2006;19(6):1964–72.
Jiggins FM, Hurst GD, Majerus ME. Sex-ratio-distorting Wolbachia causes sex-role reversal in its butterfly host. Proc Biol Sci. 2000;267(1438):69–73.
Abroon P, Ashori A, Duplouy A, Kishani Farahani H. Wolbachia impairs post-eclosion host preference in a parasitoid wasp. Naturwissenschaften. 2021;108:12p.
Bi J, Wang YF. The effect of the endosymbiont Wolbachia on the behavior of insect hosts. Insect Sci. 2020;27(5):846–58.
Mathé-Hubert H, Kaech H, Hertaeg C, Jaenike J, Vorburger C. Nonrandom associations of maternally transmitted symbionts in insects: the roles of drift versus biased cotransmission and selection. Mol Ecol. 2019;28(24):5330–46.
Jiggins FM. Male-killing Wolbachia and mitochondrial DNA: selective sweeps, hybrid introgression and parasite population dynamics. Genetics. 2003;164:5–12.
Kodandaramaiah U, Simonsen TJ, Bromilow S, Wahlberg N, Sperling F. Deceptive single-locus taxonomy and phylogeography: Wolbachia-associated divergence in mitochondrial DNA is not reflected in morphology and nuclear markers in a butterfly species. Ecol Evol. 2013;3(16):5167–76.
Telschow A, Gadau J, Werren JH, Kobayashi Y. Genetic incompatibilities between mitochondria and nuclear genes: effect on gene flow and speciation. Front Genet. 2019. https://doi.org/10.3389/fgene.2019.00062.
Sucháčková Bartoňová A, Konvička M, Marešová J, Wiemers M, Ignatev N, Wahlberg N, Schmitt T, Faltýnek Fric Z. Wolbachia affects mitochondrial population structure in two systems of closely related Palaearctic blue butterflies. Sci Rep. 2021;11(1):3019.
Waller JT, Willink B, Tschol M, Svensson EI. The odonate phenotypic database, a new open data resource for comparative studies of an old insect order. Sci Data. 2019;6(1):316.
Hickling R, Roy DB, Hill JK, Thomas CD. A northward shift of range margins in British Odonata. Glob Change Biol. 2005;11(3):502–6.
Sánchez-Guillén RA, Muñoz J, Rodríguez-Tapia G, Feria Arroyo TP, Córdoba-Aguilar A. Climate-induced range shifts and possible hybridisation consequences in insects. PLoS ONE. 2013;8(11):e80531.
Wellenreuther M, Muñoz J, Chávez-Ríos JR, Hansson B, Cordero-Rivera A, Sánchez-Guillén RA. Molecular and ecological signatures of an expanding hybrid zone. Ecol Evol. 2018;8(10):4793–806.
Sánchez-Guillén RA, Wellenreuther M, Cordero-Rivera A, Hansson B. Introgression and rapid species turnover in sympatric damselflies. BMC Evol Biol. 2011;11:210.
Svensson EI, Abbott JK, Gosden TP, Coreau A. Female polymorphisms, sexual conflict and limits to speciation processes in animals. Evol Ecol. 2007;23(1):93–108.
Svensson EI, Abbott J, Hardling R. Female polymorphism, frequency dependence, and rapid evolutionary dynamics in natural populations. Am Nat. 2005;165(5):567–76.
Le Rouzic A, Hansen TF, Gosden TP, Svensson EI. Evolutionary time-series analysis reveals the signature of frequency-dependent selection on a female mating polymorphism. Am Nat. 2015;185(6):E182-196.
Takahashi Y, Kagawa K, Svensson EI, Kawata M. Evolution of increased phenotypic diversity enhances population performance by reducing sexual harassment in damselflies. Nat Commun. 2014;5:4468.
Willink B, Svensson EI. Intra-and intersexual differences in parasite resistance and female fitness tolerance in a polymorphic insect. Proc R Soc B. 1847;2017(284):20162407.
Svensson EI, Willink B, Duryea MC, Lancaster LT. Temperature drives pre-reproductive selection and shapes the biogeography of a female polymorphism. Ecol Lett. 2020;23(1):149–59.
Lancaster LT, Dudaniec RY, Hansson B, Svensson EI. Do group dynamics affect colour morph clines during a range shift? J Evol Biol. 2017;30(4):728–37.
Oliveira DC, Raychoudhury R, Lavrov DV, Werren JH. Rapidly evolving mitochondrial genome and directional selection in mitochondrial genes in the parasitic wasp Nasonia (Hymenoptera: pteromalidae). Mol Biol Evol. 2008;25(10):2167–80.
Shoemaker DD, Dyer KA, Ahrens M, McAbee K, Jaenike J. Decreased diversity but increased substitution rate in host mtDNA as a consequence of Wolbachia endosymbiont infection. Genetics. 2004;168(4):2049–58.
Thipaksorn A, Jamnongluk W, Kittayapong P. Molecular evidence of Wolbachia infection in natural populations of tropical odonates. Curr Microbiol. 2003;47(4):314–8.
Salunkhe RC, Dhotre DP, Salunke BK, Patil VS, Mahale V, Andrew RJ, Patole MS, Narkhede KP, Shouche YS. Distribution and molecular characterization of Wolbachia endosymbionts in Odonata (Insecta) from Central India by multigene approach. Curr Sci. 2015;108(5):971–8.
Lorenzo-Carballa MO, Torres-Cambas Y, Heaton K, Hurst GDD, Charlat S, Sherratt TN, Van Gossum H, Cordero-Rivera A, Beatty CD. Widespread Wolbachia infection in an insular radiation of damselflies (Odonata, Coenagrionidae). Sci Rep. 2019;9:11933.
Willink B, Duryea MC, Svensson EI. Macroevolutionary origin and adaptive function of a polymorphic female signal involved in sexual conflict. Am Nat. 2019;194:707–24.
Sanmartín-Villar I, Cordero-Rivera A. The inheritance of female colour polymorphism in Ischnura genei (Zygoptera: Coenagrionidae), with observations on melanism under laboratory conditions. PeerJ. 2016;4:e2380.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994;3:294–9.
Larsson A. AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics. 2014;30(22):3276–8.
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 2017;34(12):3299–302.
Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133(3):693–709.
Baldo L, Hotopp JC, Jolley KA, Bordenstein SR, Biber SA, Choudhury RR, Hayashi C, Maiden MC, Tettelin H, Werren JH. Multilocus sequence typing system for the endosymbiont Wolbachia pipientis. Appl Environ Microbiol. 2006;72:7098–110.
Chauhan P, Swaegers J, Sánchez-Guillén RA, Svensson EI, Wellenreuther M, Hansson B. Genome assembly, sex-biased gene expression and dosage compensation in the damselfly Ischnura elegans. Genomics. 2021;113(4):1828–37.
Klasson L, Walker T, Sebaihia M, Sanders MJ, Quail MA, Lord A, Sanders S, Earl J, O’Neill SL, Thomson N, et al. Genome evolution of Wolbachia strain wPip from the Culex pipiens group. Mol Biol Evol. 2008;25(9):1877–87.
Wu M, Sun LV, Vamathevan J, Riegler M, Deboy R, Brownlie JC, McGraw EA, Martin W, Esser C, Ahmadinejad N, et al. Phylogenomics of the reproductive parasite Wolbachia pipientis wMel: a streamlined genome overrun by mobile genetic elements. PLoS Biol. 2004;2(3):E69.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60.
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32(1):268–74.
Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In: 2010 gateway computing environments workshop (GCE), New Orleans, LA; 2010. p. 1–8.
Kalyaanamoorthy S, Minh BQ, Wong TK, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587.
Chernomor O, Von Haeseler A, Minh BQ. Terrace aware data structure for phylogenomic inference from supermatrices. Syst Biol. 2016;65:997–1008.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–9.
Leigh JW, Bryant D. POPART: full-feature software for haplotype network construction. Methods Ecol Evol. 2015;6(9):1110–6.
Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48.
Rich SM, Licht MC, Hudson RR, Ayala FJ. Malaria’s Eve: evidence of a recent population bottleneck throughout the world populations of Plasmodium falciparum. Proc Natl Acad Sci USA. 1998;95(8):4425–30.
Feindt W, Herzog R, Osigus HJ, Schierwater B, Hadrys H. Short read sequencing assembly revealed the complete mitochondrial genome of Ischnura elegans Vander Linden, 1820 (Odonata: Zygoptera). Mitochondrial DNA B. 2016;1:574–6.
Jukes TH, Cantor CR. Evolution of protein molecules, vol. 21–132. New York: Academic Press; 1969.
Blow R, Willink B, Svensson EI. A molecular phylogeny of forktail damselflies (genus Ischnura) reveals dynamic macroevolutionary history of female colour polymorphisms. Mol Phylogenet Evol. 2021;160:107134.
Team RC. R: a language and environment for statistical computing. In: R Foundation for Statistical Computing, Vienna, Austria. 2019.
Bleidorn C, Gerth M. A critical re-evaluation of multilocus sequence typing (MLST) efforts in Wolbachia. FEMS Microbiol Ecol. 2018. https://doi.org/10.1093/femsec/fix163.
Sun LV, Foster JM, Tzertzinis G, Ono M, Bandi C, Slatko BE, O’Neill SL. Determination of Wolbachia genome size by pulsed-field gel electrophoresis. J Bacteriol. 2001;183(7):2219–25.
Ratnasingham S, Hebert PD. Bold: the barcode of life data system (http://www.barcodinglife.org). Mol Ecol Notes. 2007;7(3):355–64.
Hurst GD, Jiggins FM. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proc Biol Sci. 2005;272(1572):1525–34.
Clark PU, Dyke AS, Shakun JD, Carlson AE, Clark J, Wohlfarth B, Mitrovica JX, Hostetler SW, McCabe AM. The last glacial maximum. Science. 2009;325:710–4.
Lancaster LT, Dudaniec RY, Hansson B, Svensson EI. Latitudinal shift in thermal niche breadth results from thermal release during a climate-mediated range expansion. J Biogeogr. 2015;42(10):1953–63.
Lancaster LT, Dudaniec RY, Chauhan P, Wellenreuther M, Svensson EI, Hansson B. Gene expression under thermal stress varies across a geographical range expansion front. Mol Ecol. 2016;25(5):1141–56.
Jiggins FM. The spread of Wolbachia through mosquito populations. PLoS Biol. 2017;15(6):e2002780.
Correa CC, Ballard JWO. Wolbachia associations with insects: winning or losing against a master manipulator. Front Ecol Evol. 2016;3:153.
Zug R, Hammerstein P. Evolution of reproductive parasites with direct fitness benefits. Heredity (Edinb). 2018;120(3):266–81.
Koehncke A, Telschow A, Werren JH, Hammerstein P. Life and death of an influential passenger: Wolbachia and the evolution of CI-modifiers by their hosts. PLoS ONE. 2009;4(2):e4425.
Hurst GD, Jiggins FM, Pomiankowski A. Which way to manipulate host reproduction? Wolbachia that cause cytoplasmic incompatibility are easily invaded by sex ratio-distorting mutants. Am Nat. 2002;160(3):360–73.
Jansen VA, Turelli M, Godfray HC. Stochastic spread of Wolbachia. Proc Biol Sci. 2008;275(1652):2769–76.
Dyer KA, Jaenike J. Evolutionarily stable infection by a male-killing endosymbiont in Drosophila innubila: molecular evidence from the host and parasite genomes. Genetics. 2004;168(3):1443–55.
Duplouy A, Couchoux C, Hanski I, van Nouhuys S. Wolbachia infection in a natural parasitoid wasp population. PLoS ONE. 2015;10(8):e0134843.
van Nouhuys S, Kohonen M, Duplouy A. Wolbachia increases the susceptibility of a parasitoid wasp to hyperparasitism. J Exp Biol. 2016;219(Pt 19):2984–90.
Narita S, Nomura M, Kato Y, Fukatsu T. Genetic structure of sibling butterfly species affected by Wolbachia infection sweep: evolutionary and biogeographical implications. Mol Ecol. 2006;15(4):1095–108.
Charlat S, Engelstädter J, Dyson EA, Hornett EA, Duplouy A, Tortosa P, Davies N, Roderick GK, Wedell N, Hurst GD. Competing selfish genetic elements in the butterfly Hypolimnas bolina. Curr Biol. 2006;16(24):2453–8.
Majerus MEN. Symbionts, bacterial. In: Resh VH, editor. Encyclopedia of insects (second edition). Cardé, RT: Academic Press; 2009. p. 983–7.
Graham RI, Wilson K. Male-killing Wolbachia and mitochondrial selective sweep in a migratory African insect. BMC Evol Biol. 2012;12:204.
Willink B, Blow R, Sparrow DJ, Sparrow R, Svensson EI. Population biology and phenology of the colour polymorphic damselfly Ischnura elegans at its southern range limit in Cyprus. Ecol Entomol. 2021;46(3):601–13.
Bailly-Bechet M, Martins-Simões P, Szöllősi GJ, Mialdea G, Sagot MF, Charlat S. How long does Wolbachia remain on board? Mol Biol Evol. 2017;34:1183–93.
Reuter M, Pedersen JS, Keller L. Loss of Wolbachia infection during colonisation in the invasive Argentine ant Linepithema humile. Heredity (Edinb). 2005;94(3):364–9.
Hurst GD, Johnson AP, Schulenburg JH, Fuyama Y. Male-killing Wolbachia in Drosophila: a temperature-sensitive trait with a threshold bacterial density. Genetics. 2000;156(2):699–709.
Ross PA, Ritchie SA, Axford JK, Hoffmann AA. Loss of cytoplasmic incompatibility in Wolbachia-infected Aedes aegypti under field conditions. PLoS Negl Trop Dis. 2019;13(4):e0007357.
van Opijnen T, Breeuwer JA. High temperatures eliminate Wolbachia, a cytoplasmic incompatibility inducing endosymbiont, from the two-spotted spider mite. Exp Appl Acarol. 1999;23(11):871–81.
Galimberti A, Assandri G, Maggioni D, Ramazzotti F, Baroni D, Bazzi G, Chiandetti I, Corso A, Ferri V, Galuppi M, et al. Italian odonates in the Pandora’s box: a comprehensive DNA barcoding inventory shows taxonomic warnings at the Holarctic scale. Mol Ecol Resour. 2021;21(1):183–200.
Li SJ, Ahmed MZ, Lv N, Shi PQ, Wang XM, Huang JL, Qiu BL. Plantmediated horizontal transmission of Wolbachia between whiteflies. ISME J. 2017;11(4):1019–28.
Brown AN, Lloyd VK. Evidence for horizontal transfer of Wolbachia by a Drosophila mite. Exp Appl Acarol. 2015;66:301–11.
Ke F, You S, Huang S, Chen W, Liu T, He W, Xie D, Li Q, Lin X, Vasseur L, et al. Herbivore range expansion triggers adaptation in a subsequently-associated third trophic level species and shared microbial symbionts. Sci Rep. 2019;9(1):10314.
Gómez-Llano M, Narasimhan A, Svensson EI. Parasites mediate condition-dependent sexual selection for local adaptation in a natural insect population. Am Nat. 2020;196(3):344–54. https://doi.org/10.1086/710039.
Bybee S, Córdoba-Aguilar A, Duryea MC, Futahashi R, Hansson B, Lorenzo-Carballa MO, Schilder R, Stoks R, Suvorov A, Svensson EI, et al. Odonata (dragonflies and damselflies) as a bridge between ecology and evolutionary genomics. Front Zool. 2016;13:46.
Okude G, Fukatsu T, Futahashi R. Interspecific crossing between blue-tailed damselflies Ischnura elegans and I. senegalensis in the laboratory. Entomol Sci. 2020;23(2):165–72.
Acknowledgements
Thanks to F.F. Pan and S.W. Deng for their support over the course of this study. Thanks to C. Duplouy and S. Mäkelä for their assistance with collecting samples from France and Åland, and to H. Naraoka for providing samples from Rokkasho Japan. Thanks to C. Martel, Prof. N. Wahlberg and the members of the Systematic Biology Group at Lund University, to Prof. E. Svensson’s lab members, Prof. S. Bensch, Dr. C. Cornwallis and Prof. J-Å. Nilsson for fruitful discussions on the study. Diversity, Equality and Inclusion statement (https://www.nature.com/articles/d41586-020-02429-8). The authors highly value equity, diversity and inclusion in science. We would like to acknowledge the international character of our team, which significantly contributed to the completion and quality of the present study. It includes researchers from different countries, backgrounds and career stages. The first author is from China, the last author is from France, other authors are from India, Italy, Japan, Sweden and USA. There are three female and seven male authors. We cite a large body of studies from many of our peers without checking whether the citations are equally distributed across groups. We thus acknowledge some shortcomings in our study and strive to address these issues in future work.
Funding
Open access funding provided by Lund University. JD was funded by the Lund University Master program in Evolutionary Biology. AD was funded by a Marie Curie Sklodowska Individual Fellowship to AD (#790531, HostSweetHome) and by the Academy of Finland to AD (#328944). BH was funded by the Swedish Research Council (consolidator grant #2016-689). EIS was funded by the Swedish Research Council (VR # 2016-03356), the Gyllenstiernska Krapperupstiftelsen (#KR2018-0038) and Lunds Djurskyddsfond.
Author information
Authors and Affiliations
Contributions
AD & ES conceived the study. JD, ES, GA, RF, AG, BH, LTL, YT & AD collected samples. JD, PC, BH & AD produced the data. JD & AD analyzed the data and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
No consent to participate was needed for this study. The Italian Ministry of the environment, Land and Sea released a national permit for the collection of species included in European and Italian conservation directives, or to collect samples in regional or national protected areas (Prot. #0031783.20-11-2019).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Pairwise genetic distance between strains using MEGA X [91]. Table S2. List of the primers used in this study, and associated details. Table S3. GenBank accession numbers from additional sequences of the mitochondrial COI gene from diverse Ischnura species from different countries. Figure S1. The phylogeny of (A) ftsz gene and (B) wsp gene, separately
Additional file 2.
The wEle1 draft genome assembly (Nscaffold = 893; N50 = 5523 bp; longest scaffold = 53,331 bp,genome size = 1.4 MB) as a fasta file.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Deng, J., Assandri, G., Chauhan, P. et al. Wolbachia-driven selective sweep in a range expanding insect species. BMC Ecol Evo 21, 181 (2021). https://doi.org/10.1186/s12862-021-01906-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-021-01906-6