
Abstract
Background
Numerous recent studies have shown that resident symbiotic microorganisms of insects play a fundamental role in host ecology and evolution. The lepidopteran pest, African armyworm (Spodoptera exempta), is a highly migratory and destructive species found throughout sub-Saharan Africa, that can experience eruptive outbreaks within the space of a single generation, making predicting population dynamics and pest control forecasting extremely difficult. Three strains of Wolbachia have recently been identified infecting this species in populations sampled from Tanzania. In this study, we examined the interaction between Wolbachia pipiensis infections and the co-inherited marker, mtDNA, within populations of armyworm, as a means to investigate the population biology and evolutionary history of Wolbachia and its host.
Results
A Wolbachia-infected isofemale line was established in the laboratory. Phenotypic studies confirmed the strain wExe1 as a male-killer. Partial sequencing of the mitochondrial COI gene from 164 individual field-collected armyworm of known infection status revealed 17 different haplotypes. There was a strong association between Wolbachia infection status and mtDNA haplotype, with a single dominant haplotype, haplo1 (90.2% prevalence), harbouring the endosymbiont. All three Wolbachia strains were associated with this haplotype. This indicates that Wolbachia may be driving a selective sweep on armyworm haplotype diversity. Despite very strong biological and molecular evidence that the samples represent a single species (including from nuclear 28S gene markers), the 17 haplotypes did not fall into a monophyletic clade within the Spodoptera genus; with six haplotypes (2 each from 3 geographically separate populations) differing by >11% in their nucleotide sequence to the other eleven.
Conclusions
This study suggests that three strains of Wolbachia may be driving a selective sweep on armyworm haplotype diversity, and that based on COI sequence data, S. exempta is not a monophyletic group within the Spodoptera genus. This has clear implications for the use of mtDNA as neutral genetic markers in insects, and also demonstrates the impact of Wolbachia infections on host evolutionary genetics.
Similar content being viewed by others

Background
Recent studies into resident heritable symbiotic microorganisms have highlighted the central role they play in their insect host’s ecology and evolution [1, 2]. These symbionts can be classified as either obligate or facultative for host survival. Facultative symbionts are not essential for host development or reproduction, but their presence can impact upon host dynamics by manipulating host reproduction [3–5], as well as increasing host survival or fecundity [6–10], whereas removal of obligate symbionts results in the death of its host [11, 12]. One successful group of such symbiotic microorganisms belongs to the genus Wolbachia. These are intracellular, maternally-inherited bacteria belonging to the alpha-Proteobacteria group Rickettsia. They are among the most successful genus of bacteria, found in filarial nematodes, crustaceans, arachnids, and are estimated to occur in 20 - 70% of all insect species [13, 14].
Wolbachia have the ability to induce a number of reproductive manipulations of their hosts, such as feminisation of genetic males, induction of sperm-egg incompatibilities, thelytokous parthenogenesis, and male-killing [1, 15]. Through these processes, Wolbachia provides infected host-females with a relative reproductive advantage over uninfected females [16]. Each of these mechanisms increases the symbiont-infection frequency by ensuring that the number of infected daughters produced by an infected female is greater than the average production of daughters per female. A single insect or population may be infected with more than one symbiotic microorganism and different populations may have different infection statuses [17, 18].
Both Wolbachia and host mitochondria are maternally transmitted and subsequently can be co-inherited by the offspring. The high mutation rate of mitochondrial DNA makes it a valuable evolutionary marker when compared to corresponding nuclear DNA [19]. With this in mind, it also has the advantage of very low recombination, resulting in the whole mtDNA genome having the same genealogical history [20]. It is well documented that symbionts such as Wolbachia can impact upon mtDNA diversity within host populations [21–25]. Indeed, the strong linkage between the two co-inherited markers makes them ideal candidates for the investigation of symbiont invasion-history and symbiont impact upon host genetics [21, 26, 27]. Previous studies have documented the role of Wolbachia in driving dramatic changes within host populations, due to the phenotypes induced by the symbionts [28]. It is now widely accepted that endosymbiont screening and analysis should take place before any attempt to explain mtDNA patterns in terms of host ecology and evolution [20, 28].
The larval stage of the African armyworm, Spodoptera exempta (Lepidoptera: Noctuidae), is one of the most devastating crop pests in Africa, feeding upon many of the staple food crops such as maize, wheat, sorghum, millet, rice and pasture grasses. Most outbreaks occur on the eastern half of sub-Saharan Africa, as far north as Sudan and as far south as South Africa [29]. The adult moths are highly migratory, often flying hundreds of kilometres over consecutive nights [30], with moth movements largely determined by the seasonal progression of the inter-tropical convergence zone (ITCZ). Therefore, it is believed that early-season armyworm outbreaks in central Tanzania essentially act as “source” populations for moths that will subsequently migrate to northern Tanzania and further northwards towards the horn of Africa [29].
A baculovirus, SpexNPV, is known to be present in field populations of armyworms causing larval mortality, which can have significant impact upon host population dynamics during sporadic natural epizootics. In a recent study, we found that armyworm harboured three strains of Wolbachia, designated wExe1, wExe2 and wExe3[31]. Based on MLST classification, one of the strains, wExe1, was 100% identical to ST-125, which is a male-killing phenotype found in the nymphalid butterfly, Hypolimnas bolina[32]. The other two strains of Wolbachia are new sequence-types, assigned ST-222 (wExe2 – clade B) and ST-223 (wExe3 – clade A). Interestingly, both laboratory and field data showed that infection with wExe1 strain increased host susceptibility to a baculovirus, which could have clear implications for the evolutionary history of the host and pathogen [31].
In the current study, our aim was to address the following questions: (i) How robust is the male-killing phenotype and efficiency of wExe1 vertical transmission? (ii) In the field, is global Wolbachia infection associated with particular host mitochondrial genotypes? (iii) Is there evidence of a selective sweep within the host population? To answer these questions, we established laboratory cultures of wExe1-infected armyworm to assess the infection phenotype. We obtained partial sequences of Wolbachia genes and the mitochondrial cytochrome oxidase I (COI) gene from field-collected S. exempta larvae and adults collected from pheromone traps. We examined the diversity of mitochondrial haplotypes, and analysed mtDNA variation to explore the potential association with different Wolbachia infection statuses.
Results
Phenotypic effects of wolbachiainfection
Two wExe1-infected and several uninfected isofemale lines were established from field-collected pupae, and maintained under laboratory conditions for >4 generations. The two wExe1-infected lines had female-biased mean sex ratios of 1:0 (females:males) for the first 3 generations, suggesting an efficient transmission of the infection. On average, only 48.4% (n = 7,525) of the eggs hatched in the infected lines, compared to 94.3% (n = 10,366) in uninfected lines, indicating that Wolbachia caused the male-killing phenotype at the embryonic life-stage. In generation 4, a single wExe1-infected breeding pair produced a sex ratio of 10:3 (Table 1), suggesting that inefficient transmission can occur within this host-endosymbiont complex. PCR analysis of the offspring from this pair indicated a Wolbachia infection rate of 86.7% (n = 30) in the females, and 0% (n = 9) in the males. Tetracycline treatment cured the insects of Wolbachia infection, and produced sex ratios and egg hatch rates comparable to naturally uninfected isofemale lines (Table 1), demonstrating that Wolbachia was the cause of the observed distortions.
Wolbachia and host mtDNA COIgene diversity
Larvae (n = 932) were screened for the presence of Wolbachia from 59 outbreaks distributed across Tanzania over a 4-year period (see [31]). Wolbachia infections were not detected in any of the male moths (n = 334) collected in the 16 pheromone traps (over an area exceeding 750 000 km2). Three strains of Wolbachia were isolated, with MLST analysis indicating two new strains, and one strain identical to ST-125 (Figure 1; [31]). Strains wExe1, wExe2 and wExe3 were observed in 23 (39%), 18 (31%) and 19 (32%) of the 59 outbreaks [31], respectively, with 3 outbreaks containing all three strains, 11 with two, 29 with just one, and 16 with no Wolbachia present (Figure 2; Additional file 1: Table S1). Overall Wolbachia prevalence was 11.9%, 12.0% and 9.3% in sampled larvae over the three outbreak seasons. Mitochondrial COI sequences were obtained from a subsample of larval and adult-males collected from populations throughout Tanzania over two seasons (n = 164). A total of 17 different haplotypes were found in the armyworm samples (Figure 3a) [Genbank: JQ315120 - JQ315136]. The host COI haplotype diversity estimate was found to be low (haplotype diversity, Hd: 0.1861; nucleotide diversity, π: 0.00798).

Maximum-Likelihood (GTR+G+I model) phylogenetic tree for the concatenated MLST genes of Wolbachia isolates. The solid diamonds indicate the 3 strains isolated from S. exempta in this study. Where known, the names of the host species are given. The scale bar represents a 2% estimated difference in nucleotide sequence. Numbers given at each node correspond to the percentage bootstrap values (for 1000 repetitions). Replicate numbers of <60% were not included in the figure. Cimex lectularius Wolbachia ST-8 from Supergroup F is used as an outgroup.

The spatial prevalence of Wolbachia infections within armyworm larval populations sampled throughout Tanzania over the course of three field seasons. The numbers correspond to the field sites in Additional file 1: Table S1, numbered sequentially through the season.

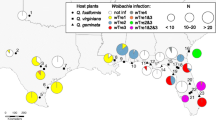
3a: Maximum-Likelihood (GTR+R model) phylogenetic tree of the mtDNA COI gene, indicating the paraphyletic nature of the Spodoptera genus. Haplo1, the most common of the S. exempta haplotypes, is indicated by the solid diamond. The scale bar represents a 2% estimated difference in nucleotide sequence. Numbers given at each node correspond to the percentage bootstrap values (for 1000 repetitions). Replicate numbers of <60% were not included in the figure. The nymphalid Hypolimnas bolina is used as an outgroup, 3b: A network analysis displaying the skew and selection for mtDNA haplotype haplo1 (H1) within the S. exempta populations. The filled grey segment in the H1 pie-chart indicates the total percentage of Wolbachia infections in the samples, all found within haplo1 haplotypes. The most divergent haplotypes are found furthest from the centre, and brackets indicate the difference in nucleotide substitutions with haplo1. Filled circles indicate “missing” haplotypes in the evolutionary chain.
According to neutral evolutionary theory, estimates of Tajima’s D and Fu & Li’s D* and F* statistics are expected to equal zero. Positive values are consistent with an excess of intermediate-frequency variants, whereas negative values indicate an excess of rare variants, as can result from a recent population bottleneck or a selective sweep. In the present study, estimates of D, D* and F* statistics were all negative for the COI gene (Tajima's D: -2.667, p < 0.001; Fu & Li's D*: -3.661, p < 0.02; Fu & Li's F*: -3.824, p < 0.02). Of the 164 sequences obtained, 148 (90.2%) belonged to the same haplotype, assigned haplo1 (Figure 3b). Significantly, all the Wolbachia infections detected in S. exempta were found associated with mtDNA haplo1, suggesting that recent selective sweeps associated with the invasion of Wolbachia have affected mtDNA diversity in the armyworm population, with a skewed prevalence of haplo1. Six of the haplotypes did differ by up to 11% in their nucleotide sequence to the other eleven (the largest difference observed was between haplo1 and haplo4; 73 nucleotide substitutions). Eight of the seventeen haplotypes displayed only a single nucleotide difference from haplo1, suggesting possible nucleotide substitution events occurring from haplo1. Apart from haplo1, all of the other haplotypes were very rare, each only detected in a single individual, making any inference on distribution-structuring or migratory behaviour difficult (Figure 4). This is most probably as a result of the highly migratory nature of this host species.

The spatial prevalence of COI hapolotypes within armyworm larval populations sampled throughout Tanzania over the course of two field seasons. The numbers correspond to the field sites in Additional file 1: Table S1, numbered sequentially through the season. A, B and C refer to the moth trap catches from those districts.
Host nuclear gene diversity
A skew of mtDNA can also result from demographic processes, such as an evolutionary bottleneck, although crucially the latter affects nuclear as well as mitochondrial loci. To investigate this further, we analysed approximately 150 bp of the 28S nuclear ribosomal protein gene from a geographically diverse sub-sample of the insects used to test the haplotypes (n = 20). There was high gene diversity with the vast majority of individuals differing by 1 – 4 nucleotide substitutions (Figure 5; gene diversity: 0.868; nucleotide diversity, π: 0.01400). Estimates of Tajima’s D and Fu & Li’s D* and F* statistics were non-significant, suggesting no selection for any particular nuclear genotype (D: -1.715, p > 0.05; D*: -1.976, p > 0.10; F*: -2.205, p > 0.05). This suggests that a bottleneck has not occurred, and that the COI skew is a result of a selective sweep, mostly likely driven by the invasion of Wolbachia infections.

Maximum-Likelihood (Kimura-2 parameter model) phylogenetic tree of the nuclear DNA 28S gene for armyworm collected at different sites. The solid diamonds indicate samples that were infected with Wolbachia. Samples had H1 haplotypes, unless otherwise indicated. The scale bar represents a 20% estimated difference in nucleotide sequence. Numbers given at each node correspond to the percentage bootstrap values (for 1000 repetitions). Replicate numbers of <60% were not included in the figure. The nymphalid Hypolimnas bolina is used as an outgroup.
Discussion
We explored the evolutionary history of Wolbachia pipientis infections in Tanzanian populations of the African armyworm, S. exempta. Three strains of Wolbachia have recently been detected at an infection prevalence of approximately 12% [31]. Based on previous MLST analysis (using the five genes ftsZ, coxA, fbpA, hcpA and gatB) [31], one of these strains, wExe1, is 100% identical to ST-125, which is a male-killing phenotype found in the Blue Moon Butterfly, Hypolimnas bolina[32] and, based on nucleotide identity for wsp and ftsZ genes, 100% identical to the Wolbachia isolate found in Tanzanian populations of the Common Acraea, Acraea encedon[33]. This indicates that an infection-transmission event may have occurred between host species at some point in evolutionary history, possibly via a shared generalist parasitoid [34–37]. Indeed, a recent study has shown that plant-mediated horizontal transmission may also be possible in some insect-endosymbiont systems [38]. The phenotypes of strains wExe2 and wExe3 are as yet undetermined. However, due to the absence of Wolbachia in male adult moths, it is possible that all three strains of Wolbachia display the male-killing phenotype. Male-killing Wolbachia have been previously identified in both Wolbachia Clade A [39] and Clade B e.g. [21, 32].
Male-killing is a widespread phenotype in lepidopteran-infecting Wolbachia and does appear to explain the sex ratio bias and egg-hatch rate in our infected lab lines. However, we cannot exclude the possibility of a more complicated scenario in the field, as Wolbachia is not the only cause of sex ratio distortion to be discovered in insects [40]. Studies on a range of species have revealed a number of insect endosymbionts that can cause similar phenotypic effects to Wolbachia[41–43], including the generation of complex fitness interactions between infection-agent and host sex chromosomes, leading to sex-biased mortality [44, 45]; DGGE analysis suggests that no secondary endosymbionts occur in the armyworm [31], but this remains an interesting avenue for more detailed future study.
The lack of geographical structuring (of both Wolbachia infection status and host mitochondrial genetics) throughout Tanzania is most likely due to the highly migratory nature of the armyworm, thereby preventing genetic differentiations from establishing. This failure to detect any spatial structuring in the armyworm population is consistent with early genetic analyses using iso-enzymes over a much wider East African geographical range [46]. The very large effective population size of this outbreaking species is also likely to be factor. The use of microsatellite markers is currently being examined as a technique allowing more sensitive exploration of the armyworm population structure [47].
Of particular interest was that all three strains of Wolbachia were found to be associated with the same host haplotype, haplo1. This haplotype was found in 90.2% of samples tested, suggestive of a selective sweep of this haplotype. No other host haplotype was observed to harbour Wolbachia, indicative of very low levels of horizontal symbiont transmission between individuals within a population. The high 28S nuclear gene variability, but selection for a single mtDNA haplotype, provides evidence that COI selection is being driven, and that this pattern is not due to demographic processes alone, such as a genetic bottleneck. We propose that this is due to the presence of multiple strains of Wolbachia (including the male-killer, wExe1). Two hypotheses have been previously proposed for why male-killers reduce mtDNA diversity, causing a sweep [48]. Firstly, the initial invasion of Wolbachia, and the subsequent selection for the beneficial parasite genes will result in selective sweeps of the co-inherited mtDNA molecules. Secondly, once the Wolbachia invasion has reached equilibrium within the host population, the effective population size of mtDNA will be reduced because mtDNA mutations in uninfected females will tend to be lost [48]. We propose that such a marked haplo1 sweep has occurred due to three separate Wolbachia-strain invasion events associated with mtDNA haplo1, each one enhancing the haplo1 sweep further. It is known that invasion events can cause selection for a particular haplotype, and therefore if three such events occurred in the same haplotype, a haplotype-skew towards 90% prevalence may not be unexpected. However, it must be stated that at this stage of our research, we can only speculate on this possibility.
So why is there such a large haplotype skew (90%) but only a moderate prevalence (12%) of Wolbachia? Several hypotheses have been proposed for maintaining Wolbachia prevalence equilibrium within host populations, including meta-population structure, mating preference, and environmentally-occurring antibiotics [39, 49]. All such spontaneous “cure” events would increase the ratio of uninfected individuals with the same haplotype to infected individuals. We propose two main hypotheses that may explain Wolbachia stability in our armyworm system. Firstly, inefficient transmission of male-killing Wolbachia. This occurs when an infected female produces offspring that are both infected and uninfected, resulting in individuals of both infection states having the same mtDNA profiles. Results from our laboratory cultures provide evidence for inefficient transmission in the S. exempta system, and this phenomenon would certainly help explain the skewed uninfected-haplo1 distribution. Wolbachia infection intensity within an individual may impact greatly upon the transmission efficiency between mother and offspring, whereby a reduction in infection intensity may lead to a decrease in vertical transmission efficiency and consequently loss of infection [50]. In contrast, excessive infection intensity may result in pathology, resulting in negative effects upon host fitness, as seen in the Drosophilla melanogaster Wolbachia strain wMelpop[51]. A number of previous studies have identified differences in endosymbiont intensity and infection-load within individual hosts [50, 52, 53]. We are currently developing quantitative assays to assess infection intensity and strain identification for the armyworm-Wolbachia system.
Secondly, Wolbachia-infected larvae may be more susceptible to an endemic armyworm baculovirus [31]. This would keep a check on Wolbachia prevalence in populations by causing greater mortality in Wolbachia-infected larvae than non-infected haplo1 larvae, thereby further exaggerating the haplo1 uninfected:infected ratio. We propose that a combination of these two mechanisms occurs in the field and contributes to maintaining stability of Wolbachia and preserving the mtDNA haplo1 skew within populations of armyworm. In addition, inter-strain Wolbachia stability may exist if the three strains are adapted to different 'environments'. Armyworm experience huge environmental variations over the course of a season e.g. host density, temperature, food-plant changes, disease challenge, etc. The three Wolbachia strains may be differentially adapted to hosts experiencing different conditions e.g. those that do better under low or high density conditions, or they interact differently with baculovirus strains. Indeed, in field populations there was no correlation between wExe3 prevalence and viral mortality, indicating that wExe3 may not increase virus susceptibility in armyworms (unlike wExe1 and wExe2) [31].
S. exempta appears to be paraphyletic at the mtDNA level within the genus Spodoptera, with >11% nucleotide divergence amongst haplotypes within what observational and nuclear-gene 28S molecular studies indicate is one species. As previously discussed, mtDNA is a popular method of identifying species, and assessing biodiversity via barcoding protocols [54, 55]. Historically these have found intra-specific mtDNA diversity to be very low (typically <1%), and inter-specific diversity to be higher (>2%). However, an increasing number of studies are finding high levels of mtDNA diversity within classical species [25, 56], leading to the hypothesis that cryptic species and races are present in greater numbers than previously thought [57, 58]. In addition to this, endosymbionts are capable of driving mtDNA introgression from other neighbouring species, thereby confounding the effect of paraphyly within a host species [20, 59, 60], such as that observed previously in Drosophila[61], Acraea butterflies [62] and Stomoxys flies [63]. This hypothesis may explain the paraphyly patterns observed in our S. exempta haplotype phylogeny.
Conclusion
All three strains of a Wolbachia infecting African armyworm S. exempta were associated with a single host haplotype, haplo1, which comprised 90.2% of the total samples tested. This study suggests that Wolbachia is driving a selective sweep for this particular haplotype, and that based on COI diversity, S. exempta is not a monophyletic group within the Spodoptera genus. This study supports previous research highlighting clear implications for the use of mtDNA as neutral genetic markers in insects.
Methods
Collection of armyworm samples and insect rearing
Larval outbreaks were sampled in Tanzania between 2007 and 2011 as previously reported [31]. Briefly, a minimum of thirty live larvae were collected in individual microtubes and stored in 100% ethanol for use in subsequent laboratory analysis. Adult males were caught throughout the season using a network of pheromone traps located across Tanzania. Trained trap operators collected specimens daily, and stored moths in 100% ethanol. The laboratory Spodoptera exempta culture was established from larvae collected in central Tanzania in January 2011, and high numbers were maintained at each generation to reduce inbreeding. All larval lines were maintained on a standard wheatgerm-based artificial diet [64] at a constant temperature of 25°C under a 12 hour light/dark cycle. Wolbachia-infected and Wolbachia-free insect lines were maintained in the same facility to ensure identical breeding conditions.
Wolbachiadetection and identification
DNA extractions were performed using the AllPrep DNA/RNA Mini Kit (Qiagen, Crawley, UK) according to the manufacturer's protocol. Larvae were screened for Wolbachia infection by PCR amplifying the Wolbachia-specific wsp and ftsZ genes, using the primers wsp-81F/wsp-691R [65] and ftsZfl/ftsZrl[66], respectively. Reaction mixtures (50 μl) contained PCR buffers (10 mM Tris–HCl pH 8.3 at 25°C; 50 mM KCl; 1.5 mM MgCl; 0.001% gelatin), 5 mM each of dATP, dTTP, dCTP and dGTP, 10 mM of the relevant primers, 1 unit Taq polymerase (Qiagen, Crawley, UK) and approximately 10 ng DNA template. PCR was carried out in a Techne TC-512 thermal cycler (Bibby Scientific Ltd., Stone, UK), under the following reaction conditions: (i) 94°C for 5 min, 1 cycle; (ii) 94°C for 30s, 52°C for 30s, 7°C for 30s, 40 cycles; and (iii) 72°C for 5 min, 1 cycle. Wolbachia MLST analysis was undertaken using PCR protocols for amplification of the five reported Wolbachia MLST genes (ftsZ, coxA, fbpA, hcpA and gatB) as described elsewhere [35]. PCR reaction-conditions were as above. All PCR products were run on a 1% agarose gel and visualized using ethidium bromide staining. PCR amplicons were purified (PCR Purification Kit, Qiagen), and sequenced (Source Bioscience, UK). The sequence data were analyzed against the Wolbachia MLST database (http://pubmlst.org/Wolbachia/).
Host mtDNA and 28S analysis
To study armyworm haplotype diversity and distribution, amplification of the S. exempta mitochondrial cytochrome oxidase I (COI) gene was undertaken, using universal primers LCO-1490 and HCO-1298 [67], which yielded approximately 640bp amplicons. A sample comprising at least four larvae per 2007–08 population (n = 121), and then a random number of larvae and adults from the field season 2009–10 (n = 43) was investigated. Polymerase chain reaction (PCR) mixtures (50 μL) contained PCR buffer (10 mm Tris–HCl pH 8.3 at 25°C, 50 mm KCl, 1.5 mm MgCl, 0.001% gelatin), 10 μm each of dATP, dTTP, dCTP and dGTP, 0.1 μm of each primer, 1 unit Taq polymerase (Qiagen, Crawley, UK) and approximately 10 ng genomic DNA template. PCR reactions were as above. All PCR products were run on a 1% agarose gel and visualized using ethidium bromide staining. Nuclear DNA diversity was studied by amplifying a partial segment of the 28S ribosomal gene, using primers 28SFor 5' AAA GAT CGA ATG GGG AGA TTC ATC 3' and 28SRev 5' CGT CCT ACT AGG GGA GAA GTG CAC 3' to yield an approximately 150 bp product. PCR reactions were as above. All PCR products were run on a 1% agarose gel and visualized using ethidium bromide staining.
Assessing wolbachiainduced phenotype traits
A proportion of all Wolbachia-infected armyworm lines used were treated with 0.03% tetracycline (10 mg/ml) to generate uninfected fly lines. This was achieved by feeding the armyworm larval stage, insuring ingestion of sufficient antibiotic to cure the Wolbachia infection, as previously described [68]. Following the tetracycline treatment, armyworms were maintained for a generation to recover before experimental use. All tetracycline-treated lines were tested with Wolbachia-specific PCR to test for Wolbachia infection. Only clean uninfected lines were used in subsequent experiments.
Statistical and phylogenetic analysis
PCR products were directly sequenced at Source BioScience UK Ltd. (Nottingham, UK). Sequences were viewed using BioEdit [69] and edited to remove universal primer regions. Preliminary identifications against previously published sequences were provided by BLAST [70]. Sequence alignment was performed with ClustalW 1.8 using default parameters [71]. Maximum-Likelihood phylogenetic analysis was performed having selected the most appropriate model based on lowest AIC score (MEGA 5.05 [72]). Sequences obtained in this study were deposited in GenBank [Genbank JQ315120-JQ315136]. Tajima’s D test, Fu and Li's D* and F* tests, haplotype diversity and nucleotide diversity were calculated using DnaSP vers. 5.10 [73]. Median-joining haplotype networks were drawn using Network (version 4.6.0.0; [74]).
References
Engelstaedter J, Hurst GDD: The ecology and evolution of microbes that manipulate host reproduction. Annu Rev Ecol Evol S. 2009, 40: 127-149. 10.1146/annurev.ecolsys.110308.120206.
Oliver KM, Degnan PH, Burke GR, Moran NA: Facultative symbionts in aphids and the horizontal transfer of ecologically important traits. Annu Rev Entomol. 2010, 55: 247-266. 10.1146/annurev-ento-112408-085305.
Zchori-Fein E, Perlman SJ, Kelly SE, Katzir N, MS H: Characterization of a ‘Bacteroidetes’ symbiont in Encarsia wasps (Hymenoptera: Aphelinidae): proposal of ‘Candidatus Cardinium hertigii’. Int J Syst Evol Micr. 2004, 54: 961-968. 10.1099/ijs.0.02957-0.
Zchori-Fein E, Gottlieb Y, Kelly SE, Brown JK, Wilson JM, Karr TL, Hunter MS: A newly discovered bacterium associated with parthenogenesis and a change in host selection behaviour in parasitoid wasps. P Natl Acad Sci USA. 2001, 98: 12555-12560. 10.1073/pnas.221467498.
Hunter M, Perlman S, Kelly S: A bacterial symbiont in the Bacteroidetes induces cytoplasmic incompatibility in the parasitoid wasp Encarsia pergandiella. Proc R Soc Lond B. 2003, 270: 2185-2190. 10.1098/rspb.2003.2475.
Scarborough CL, Ferrari J, Godfray HCJ: Aphid protected from pathogen by endosymbiont. Science. 2005, 310: 1781-10.1126/science.1120180.
Haine ER: Symbiont-mediated protection. Proc R Soc Lond B. 2008, 275 (1633): 353-361. 10.1098/rspb.2007.1211.
Vorburger C, Gehrer L, Rodriguez P: A strain of the bacterial symbiont Regiella insecticola protects aphids against parasitoids. Biol Lett. 2010, 6: 109-111. 10.1098/rsbl.2009.0642.
Jaenike J, Brekke TD: Defensive endosymbionts: a cryptic trophic level in community ecology. Ecol Lett. 2011, 14: 150-155. 10.1111/j.1461-0248.2010.01564.x.
Jaenike J: Population genetics of beneficial heritable symbionts. Trends Ecol Evol. 2012, 27: 226-232. 10.1016/j.tree.2011.10.005.
Dedeine F, Vavre F, Fleury F, Loppin B, Hochberg M, Bouletreau M: Removing symbiotic Wolbachia bacteria specifically inhibits oogenesis in a parasitic wasp. P Natl Acad Sci USA. 2001, 98: 6247-6252. 10.1073/pnas.101304298.
Fenn K, Blaxter M: Are filarial nematode Wolbachia obligate mutualist symbionts?. Trends Ecol Evol. 2004, 19: 163-166. 10.1016/j.tree.2004.01.002.
Werren JH, Windsor DM: Wolbachia infection frequencies in insects: evidence of a global equilibrium?. Proc R Soc Lond B. 2000, 267: 1277-1285. 10.1098/rspb.2000.1139.
Hilgenboecker K, Hammerstein P, Schlattmann P, Telschow A, Werren JH: How many species are infected with Wolbachia? - a statistical analysis of current data. FEMS Microbiol Lett. 2008, 281 (2): 215-220. 10.1111/j.1574-6968.2008.01110.x.
Werren JH, Baldo L, Clark ME: Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol. 2008, 6 (10): 741-751. 10.1038/nrmicro1969.
Vavre F, Charlat S: Making (good) use of Wolbachia: what the models say. Curr Opin Microbiol. 2012, 15: 263-268. 10.1016/j.mib.2012.03.005.
Perrot-Minnot M, Guo LR, Werren JH: Single and double infections with Wolbachia in the parasitic wasp Nasonia vitripennis: effects on compatibility. Genetics. 1996, 143: 961-972.
Werren J: Biology of Wolbachia. Ann Rev Entomol. 1997, 42: 587-609. 10.1146/annurev.ento.42.1.587.
Haag-Liautard C, Coffey N, Houle D, Lynch M, Charlesworth B, Keightley P: Direct Estimation of the Mitochondrial DNA Mutation Rate in Drosophila melanogaster. PLoS Biol. 2008, 6: 1706-1714.
Hurst GDD, Jiggins FM: Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proc R Soc Lond B. 2005, 272: 1525-1534. 10.1098/rspb.2005.3056.
Charlat S, Duplouy A, Hornett EA, Dyson EA, Davies N, Roderick GK, Wedell N, Hurst GDD: The joint evolutionary histories of Wolbachia and mitochondria in Hypolimnas bolina. BMC Evol Biol. 2009, 9 (64):
Rodriguero MS, Lanteri AA, Confalonieri VA: Mito-nuclear genetic comparison in a Wolbachia infected weevil: insights on reproductive mode, infection age and evolutionary forces shaping genetic variation. BMC Evol Biol. 2010, 4: 340-
Xiao JH, Wang NX, Li YW, Murphy RW, Wan DG, Niu LM, Hu HY, Fu YG, Huang DW: Molecular approaches to identify cryptic species and polymorphic species within a complex community of fig wasps. PLoS One. 2010, 29 (5(11)): e15067-
Thierry M, Becker N, Hajri A, Reynaud B, Lett JM, Delatte H: Symbiont diversity and non-random hybridization among indigenous (Ms) and invasive (B) biotypes of Bemisia tabaci. Mol Ecol. 2011, 20: 2172-2187. 10.1111/j.1365-294X.2011.05087.x.
Yu MZ, Zhang KJ, Xue XF, Hong XY: Effects of Wolbachia on mtDNA variation and evolution in natural populations of Tetranychus urticae Koch. Insect Mol Biol. 2011, 20: 311-321. 10.1111/j.1365-2583.2010.01066.x.
Dyer K, Jaenike J: Evolutionarily stable infection by a male-killing endosymbiont in Drosophila innubila: molecular evidence from the host and parasite genomes. Genetics. 2004, 168: 1443-1455. 10.1534/genetics.104.027854.
Delgado A, Cook J: Effects of a sex-ratio distorting endosymbiont on mtDNA variation in a global insect pest. BMC Evol Biol. 2009, 9: 49-10.1186/1471-2148-9-49.
Shoemaker DD, Keller G, Ross KG: Effects of Wolbachia on mtDNA variation in two fire ant species. Mol Ecol. 2003, 12: 1757-1771. 10.1046/j.1365-294X.2003.01864.x.
Rose DJW, Dewhurst CF, Page WW: The African Armyworm Handbook. 2000, NRI, Chatham, UK, 2
Rose DJW, Page WW, Dewhurst CF, Riley JR, Reynolds DT, Pedgley DE, Tucker MR: Down-wind migration of the African armyworm moth, Spodoptera exempta, studied by mark-and-capture and by radar. Ecol Entomol. 1985, 10: 299-313. 10.1111/j.1365-2311.1985.tb00727.x.
Graham RI, Grzywacz D, Mushobozi WL, Wilson K: Wolbachia in a major African crop pest increases susceptibility to viral disease rather than protects. Ecol Lett. 2012, 15: 993-1000. 10.1111/j.1461-0248.2012.01820.x.
Dyson EA, Kamath MK, Hurst GDD: Wolbachiainfection associated with all-female broods inHypolimnas bolina(Lepidoptera: Nymphalidae): evidence for horizontal transmission of a butterfly male killer. Heredity. 2002, 88: 166-171. 10.1038/sj.hdy.6800021.
Jiggins FM, Hurst GDD, Schulenburg JHG, Majerus MEN: Two male-killing Wolbachia strains coexist in a population of the butterfly Acraea encedon. Heredity. 2001, 86: 161-166.
Vavre F, Fleury F, Lepetit D, Fouillet P, Bouletreau M: Phylogenetic evidence for horizontal transmission of Wolbachia in host-parasitoid associations. Mol Biol Evol. 1999, 16: 1711-1723. 10.1093/oxfordjournals.molbev.a026084.
Baldo L, Hotopp JCD, Jolley KA, Bordenstein SR, Biber SA, Choudhury RR, Hayashi C, Maiden MCJ, Tettelin H, Werren JH: Multilocus sequence typing system for the endosymbiont Wolbachia pipientis. Appl Environ Microbiol. 2006, 72 (11): 7098-7110. 10.1128/AEM.00731-06.
Haine ER, Pickup NJ, Cook JM: Horizontal transmission of Wolbachia in a Drosophila community. Ecol Entomol. 2005, 30 (4): 464-472. 10.1111/j.0307-6946.2005.00715.x.
Gehrer L, Vorburger C: Parasitoids as vectors of facultative bacterial endosymbionts in aphids. Biol Lett. 2012, 8: 613-615. 10.1098/rsbl.2012.0144.
Caspi-Fluger A, Inbar M, Mozes-Daube N, Katzir N, Portney V, Belausov E, Hunter M, Zchori-Fein E: Horizontal transmission of the insect symbiont Rickettsia is plant-mediated. Proc R Soc Lond B. 2012, 279: 1791-1796. 10.1098/rspb.2011.2095.
Unckless RL, Jaenike J: Maintenance of a male-killing Wolbachia in Drosophilla innubila by male-killing dependent and male-killing independent mechanisms. Evolution. 2012, 66 (3): 678-689. 10.1111/j.1558-5646.2011.01485.x.
Weeks AR, Reynolds KT, Hoffmann AA: Wolbachia endosymbionts associated with cytoplasmic incompatibility dynamics and host effects: What has (and has not) been demonstrated?. Trends Ecol Evol. 2002, 17: 257-262. 10.1016/S0169-5347(02)02480-1.
Haselkorn T, Markow T, Moran N: Multiple introductions of the Spiroplasma bacterial endosymbiont into Drosophila. Mol Ecol. 2009, 18: 1294-1305. 10.1111/j.1365-294X.2009.04085.x.
Simon JC, Boutin S, Tsuchida T, Koga R, Le Gallic JF, Frantz A, Outreman Y, Fukatsu T: Facultative symbiont infections affect aphid reproduction. PLoS One. 2011, 6 (7): e21831-10.1371/journal.pone.0021831.
Hutchence KJ, Padé R, Swift HL, Bennet D, Hurst GDD: Phenotype and transmission efficiency of artificial and natural male-killing Spiroplasma infections in Drosophila melanogaster. J Invertebr Pathol. 2012, 109: 243-247. 10.1016/j.jip.2011.10.003.
Weeks AR, Velten R, Stouthamer R: Incidence of a new sex-ratiodistorting endosymbiotic bacterium among arthropods. Proc R Soc Lond B. 2003, 270: 1857-1865. 10.1098/rspb.2003.2425.
Kageyama D, Traut W: Opposite sex-specific effects of Wolbachia and interference with the sex determination of its host Ostrinia scapulalis. Proc R Soc Lond B. 2004, 271: 251-258. 10.1098/rspb.2003.2604.
den Boer M: Isoenzymes and migration in the African armyworm Spodoptera exempta (Lepidoptera, Noctuidae). J Zool. 1978, 185: 539-553.
Ibrahim KM, Yassin Y, Elguzouli A: Polymerase chain reaction primers for polymorphic microsatellite loci in the African armyworm, Spodoptera exempta (Lepidoptera: Noctuidae). Mol Ecol Notes. 2004, 4: 653-655. 10.1111/j.1471-8286.2004.00772.x.
Johnstone RA, Hurst GDD: Maternally inherited male-killing microorganisms may confound interpretation of mitochondrial DNA variability. Biol J Linn Soc. 1996, 58: 453-470. 10.1111/j.1095-8312.1996.tb01446.x.
Hurst GDD, Jiggins FM, Robinson SJW: What causes inefficient transmission of male-killing Wolbachia in Drosophila?. Heredity. 2001, 87: 220-226. 10.1046/j.1365-2540.2001.00917.x.
Kondo N, Shimada M, Fukatsu T: Infection density of Wolbachia endosymbiont affected by co-infection and host genotype. Biol Lett. 2005, 1: 488-491. 10.1098/rsbl.2005.0340.
Min KT, Benzer S: Wolbachia, normally a symbiont of Drosophila, can be virulent, causing degeneration and early death. P Natl Acad Sci USA. 1997, 94: 10792-10796. 10.1073/pnas.94.20.10792.
Ikeda T, Ishikawa H, Sasaki T: Regulation of Wolbachia density in the Mediterranean flour moth, Ephestia kuehniella, and the almond moth, Cadra cautella. Zool Sci. 2003, 20: 153-157. 10.2108/zsj.20.153.
Mouton L, Dedeine F, Henri H, Boulétreau M, Profizi N, Vavre F: Virulence, multiple infections and regulation of symbiotic population in the Wolbachia-Asobara tabida symbiosis. Genetics. 2004, 168: 181-189. 10.1534/genetics.104.026716.
Hebert P, Ratnasingham S, de Waard J: Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc R Soc Lond B. 2003, 270: S96-S99. 10.1098/rsbl.2003.0025.
Meusnier I, Singer GA, Landry JF, Hickey DA, Hebert PDN, Hajibabaei M: A universal DNA mini-barcode for biodiversity analysis. BMC Genomics. 2008, 12: 214-
Sun XJ, Xiao JH, Cook JM, Feng G, Huang DW: Comparisons of host mitochondrial, nuclear and endosymbiont bacterial genes reveal cryptic fig wasp species and the effects of Wolbachia on host mtDNA evolution and diversity. BMC Evol Biol. 2011, 11: 86-10.1186/1471-2148-11-86.
Funk W, Caminer M, Ron S: High levels of cryptic species diversity uncovered in Amazonian frogs. Proc R Soc Lond B. 2011, 279: 1806-1814. 10.1098/ rspb.2011.1653.
Nagoshi RN, Meagher RL, Hay-Roe M: Inferring the annual migration patterns of fall armyworm (Lepidoptera: Noctuidae) in the United States from mitochondrial haplotypes. Ecology and Evolution. 2012, 2: 1458-1467. 10.1002/ece3.268.
Whitworth TL, Dawson RD, Magalon H, Baudry E: DNA barcoding cannot reliably identify species of the blowfly genus Protocalliphora (Diptera: Calliphoridae). Proc R Soc Lond B. 2007, 274: 1731-1739. 10.1098/rspb.2007.0062.
Gompert Z, Forister M, Fordyce J, Nice C: Widespread mito-nuclear discordance with evidence for introgressive hybridization and selective sweeps in Lycaeides. Mol Ecol. 2008, 17: 5231-5244. 10.1111/j.1365-294X.2008.03988.x.
Solignac M, Monnerot M: Race formation, speciation, and introgression within Drosophila simulans, D. mauritiana, and D. sechellia inferred from mitochondrial DNA analysis. Evolution. 1986, 40: 531-539. 10.2307/2408575.
Jiggins FM: Male-killing Wolbachia and mitochondrial DNA. Selective sweeps, hybrid introgression and parasite population dynamics. Genetics. 2003, 164: 5-12.
Dsouli N, Delsuc F, Michaux J, De Stordeur E, Couloux A, Veuille M, Duvallet G: Phylogenetic analyses of mitochondrial and nuclear data in haematophagous flies support the paraphyly of the genus Stomoxys (Diptera: Muscidae). Infect Genet Evol. 2011, 11: 663-670. 10.1016/j.meegid.2011.02.004.
Cotter S, Wilson K: Heritability of immune function in the caterpillar Spodoptera littoralis. Heredity. 2002, 88: 229-234. 10.1038/sj.hdy.6800031.
Zhou WG, Rousset F, O'Neill S: Phylogeny and PCR-based classification of Wolbachia strains using wsp gene sequences. Proc R Soc Lond B. 1998, 265 (1395): 509-515. 10.1098/rspb.1998.0324.
Werren JH, Guo LR, Zhang W: Evolution and phylogeny of Wolbachia: reproductive parasites of Arthropods. Proc R Soc Lond B. 1995, 251: 55-63.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R: DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotech. 1994, 3: 294-299.
Kageyama D, Nishimura G, Hoshizaki S, Ishikawa Y: FeminizingWolbachiain an insect,Ostrinia furnacalis(Lepidoptera: Crambidae). Heredity. 2002, 88: 444-449. 10.1038/sj.hdy.6800077.
Hall T: BioEdit: a user friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser. 1999, 41: 95-98.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215: 403-410.
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22: 4673-4680. 10.1093/nar/22.22.4673.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum Parsimony methods. Mol Biol Evol. 2011, 28: 2731-2739. 10.1093/molbev/msr121.
Librado P, Rozas J: DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009, 25: 1451-1452. 10.1093/bioinformatics/btp187.
Bandelt H, Forster P, Röhl A: Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999, 16: 37-48. 10.1093/oxfordjournals.molbev.a026036.
Acknowledgements
This work was supported by a grant from the Sustainable Agriculture Research for International Development (SARID) programme, funded by the United Kingdom’s Biotechnology and Biological Sciences Research Council (BBSRC) and the Department for International Development (DfID). The views expressed are not necessarily those of DfID or BBSRC. Additional support was provided by Lancaster University, U.K. We are extremely grateful for the support and assistance of the Tanzanian Ministry of Agriculture, Food and Cooperatives and to Ms Yamini Tummala and Mr Phill Nott for technical support. We are especially grateful to Mr Wilfred Mushobozi for coordinating field activities in Tanzania and Mr David Grzywacz, Dr Alan Shirras, Professor Jenny Cory, Dr Glenn Rhodes and Dr Kamal Ibrahim for useful discussion.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
Both authors declare that they have no competing interests.
Authors’ contributions
RG designed the study, produced and analysed the data. KW instigated and supervised the study. Both authors co-wrote and approved the final manuscript.
An erratum to this article is available at http://dx.doi.org/10.1186/1471-2148-13-6.
Electronic supplementary material
12862_2012_2219_MOESM1_ESM.pdf
Additional file 1: Table S1. Wolbachia infection prevalence within the 59 sampled populations of African armyworm Spodoptera exempta. (PDF 37 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Graham, R.I., Wilson, K. Male-killing Wolbachia and mitochondrial selective sweep in a migratory African insect. BMC Evol Biol 12, 204 (2012). https://doi.org/10.1186/1471-2148-12-204
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-12-204