
Abstract
Background
Chinese grass shrimp (Palaemonetes sinensis) is an important species widely distributed throughout China, which is ecologically relevant and possesses ornamental and economic value. These organisms have experienced a sharp decline in population due to overfishing. Therefore interest in P. sinensis aquaculture has risen in an effort to alleviate fishing pressure on wild populations. Therefore, we investigated the genetic diversity and variation of P. sinensis to verify the accuracy of previous research results, as well as to assess the risk of diversity decline in wild populations and provide data for artificial breeding.
Methods
Palaemonetes sinensis specimens from seven locations were collected and their genetic variability was assessed based on mitochondrial COI gene segments. DNA sequence polymorphisms for each population were estimated using DNASP 6.12. The demographic history and genetic variation were evaluated using Arlequin 3.11. At last, the pairwise genetic distance (Ds) values and dendrograms were constructed with the MEGA 11 software package.
Results
Our study obtained sequences from 325 individuals, and 41 haplotypes were identified among the populations. The haplotype diversity (Hd) and nucleotide diversity (π) indices ranged from 0.244 ± 0.083 to 0.790 ± 0.048 and from 0.0004 ± 0.0001 to 0.0028 ± 0.0006, respectively. Haplotype network analyses identified haplotype Hap_1 as a potential maternal ancestral haplotype for the studied populations. AMOVA results indicated that genetic variations mainly occurred within populations (73.07%). Moreover, according to the maximum variation among groups (FCT), analysis of molecular variance using the optimal two-group scheme indicated that the maximum variation occurred among groups (53.36%). Neutrality and mismatch distribution tests suggested that P. sinensis underwent a recent population expansion. Consistent with the SAMOVA analysis and haplotype network analyses, the Ds and FST between the population pairs indicated that the JN population was distinctive from the others.
Conclusions
Our study conducted a comprehensive characterization of seven wild P. sinensis populations, and our findings elucidated highly significant differences within populations. The JN population was differentiated from the other six populations, as a result of long-term geographical separation. Overall, the present study provided a valuable basis for the management of genetic resources and a better understanding of the ecology and evolution of this species.
Similar content being viewed by others


Background
Chinese grass shrimp (Palaemonetes sinensis) is an important species belonging to the Palaemonidae family, which is widely distributed throughout China [20, 23]. These organisms are not only ecologically relevant but also possess ornamental and economic value [16, 36]. Additionally, P. sinensis is very popular in both domestic and foreign markets due to its pleasant flavor and high nutritional value [16, 37]. However, the wild populations of P. sinensis have gradually declined due to environmental pollution and overharvesting, and therefore interest in P. sinensis aquaculture has risen in an effort to potentially alleviate fishing pressure on wild populations by producing farmed shrimp to meet consumer demand. Therefore, our research group has conducted several studies on P. sinensis, including studies on morphology [38], physiology [3], immunology [9, 21, 22], genetic diversity [36], and phylogenetic relationships [37] among others. However, studies on the population genetic diversity and structure of this species remain scarce. It is well known that the study of population genetic diversity and genetic structure could provide guidance for the establishment of fishing quotas to prevent overharvesting [36]. At the same time, it is very necessary to understand the genetic structure and genetic diversity of wild P. sinensis populations, and to correctly evaluate the status of germplasm resources prior to artificial breeding, which can provide a basis for the selection of breeding populations [36]. Therefore, more research is needed to understand the genetics of this species.
COI gene sequence polymorphisms serve as “barcodes” to identify different species and assess cryptic diversity [15, 31] and have thus been increasingly used to investigate population genetics, taxonomy, molecular evolution, phylogeny relationship origins, and the diversity of Palaemonidae species such as Macrobrachium australiense [4], Macrobrachium olfersii [25], Palaemon longirostris and Palaemon garciacidi [5], and Palaemon capensis and Palaemon peringueyi [32]. In our previous study, we investigated the genetic diversity and structure of P. sinensis using transcriptome-derived microsatellite markers [37]. The results indicated that two populations, LD and SJ, had the lowest genetic diversity and were markedly different from the other populations. Given that microsatellites represent changes in the nuclear genome, the microsatellite markers obtained via transcriptome analysis represented sequences of coding regions in the genome, which are highly conserved and may underestimate population genetic diversity. Therefore, further studies are needed to establish the relationship and genetic variability among P. sinensis populations in China using different molecular markers. The COI gene polymorphisms represent changes in the mitochondrial DNA (mtDNA) genome. Thus, in this study, COI gene fragments were characterized to investigate the genetic diversity and population structure of seven wild P. sinensis populations in China. Through this study, we sought to verify the accuracy of previous research results, assess whether there is a risk of diversity decline in wild populations, and provide data for artificial breeding. Additionally, this genetic survey provides valuable information for the development of effective conservation and management strategies for this species and establishes a theoretical basis for the future study of the genetic biodiversity of P. sinensis.
Methods
Sampling, DNA extraction, and sequencing
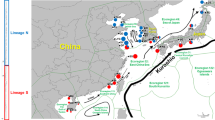
A total of 326 wild individuals were collected from seven locations in China (Table 1, Fig. 1), and stored in 75% alcohol directly. Genomic DNA was extracted from individual muscle samples using the TIAnamp Marine Animals DNA Kit (TIANGEN, China) according to the manufacturer’s instructions, and the quality of the extracted DNA was assessed via electrophoresis on a 1% agarose gel coupled with spectrophotometric analyses using a Thermo Scientific NanoDrop 2000 system.

Sampling collection sites of P. sinensis (see Table 1 for the full description of the populations). ArcMap 10.4 was used to produce a distribution map. The base map for the depiction was obtained freely from the URL: https://www.naturalearthdata.com/downloads/
The COI gene sequence was partially amplified via polymerase chain reaction (PCR) using the LCO1490 and HCO2198 primers [13]. Each PCR reaction (25 μL per reaction) contained 100 ng of template, forward and reverse primers (10 μmol/L; 1 μL each), 12.5 μL of 2× Taq PCR Master Mix (TIANGEN, China), and ddH2O [35]. Denaturation was conducted for 3 min at 94 ℃, followed by 35 cycles at 94 ℃ for 30 s, 53 ℃ for 40 s, and 72 ℃ for 40 s, with a final prolonged extension step at 72 ℃ for 10 min. The PCR products were then assessed via electrophoresis in 1% agarose gels [35], then purified and sequenced by Sangon Biotech (Shanghai) Co., Ltd.
Data analysis and phylogenetic relationship
All COI gene fragments were sequenced in both directions and the assembled sequences were manually inspected for quality assurance. Afterward, all sequences were aligned using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) [28]. DNA sequence polymorphisms including the highest number of haplotypes (h), number of segregating sites (S), average number of differences (K), haplotype diversity (Hd), and nucleotide diversity (π) values for each population were estimated using DNASP 6.12 [26]. Haplotypes for specific sample populations were also identified with DNASP 6.12 and haplotype networks were visualized with the PopART (Population Analysis with Reticulate Trees) network analysis software [19] using the TCS network inference method [8].
To estimate demographic history, Tajima’s D and Fu’s Fs tests were used to examine neutrality, and mismatch distribution analysis was also performed to assess the population expansion hypothesis using Arlequin 3.11 (10,000 permutations) [11]. Additionally, the relative population sizes before (θ0) and after (θ1) were also determined using Arlequin 3.11. The correlation between the observed and simulated distributions was tested using the SSD and HRI. The time of expansion was calculated with the formula
where μ = generation time × number of base pairs per sequence × mutation rate for the marker used, and τ was calculated in Arlequin 3.11. A mutation rate of 1.4% per million years [18] and a generation time of 1.5 years were assumed for all calculations. This generation time was established based on previous literature on other Palaemonidae shrimps species [32].
Genetic variation was evaluated via AMOVA [12] and pairwise FST were calculated using Arlequin 3.11 with 10,000 permutations [11]. SAMOVA 2.0 [10] was used to identify the defined drainage groups in the greatest variation among groups (FCT). The Ds values among populations based on the Kimura 2-parameter model were then calculated, after which the UPGMA and NJ dendrograms were constructed with the MEGA 11 software package [29].
Results
Sequence variation and genetic diversity
674 base pair sequences of the COI gene was obtained for downstream analyses. In 326 individuals of seven populations, 37 polymorphic sites were identified in this sequence, 21 of which were parsimony-informative. Moreover, the highest number of h, S, K, Hd, and π values were observed in the SL population (h = 15, S = 25, K = 1.885, Hd = 0.790 ± 0.048, and π = 0.0028 ± 0.0006), whereas the lowest values were observed in the DL population (h = 5, S = 4, K = 0.257, Hd = 0.244 ± 0.083, and π = 0.0004 ± 0.0001) (Table 2). Overall, most locations exhibited moderate to high haplotype diversity (0.244–0.790) due to large number of unique haplotypes. However, nucleotide diversity was relatively low, ranging from 0.0004 to 0.0028.
Haplotype network analysis
A total of 41 haplotypes were defined according to all variable positions in the COI gene from seven populations (Additional file 1: Table S1, GenBank Accession numbers: MT884019–MT884059). Among these haplotypes, 29 were singleton. For the shared haplotypes, 193 out of 326 (59.20%) individuals belong to one haplotype (Hap_1) in six populations (excluding JN population). There were 6 to 15 unique haplotypes within the populations but only eight haplotypes were shared between/among populations and no common haplotype was shared across all populations. A genealogy with one essential haplotype (Hap_1) was identified from the haplotype network based on the TCS inference method and all other haplotypes arose from it through one or several mutational steps (Fig. 2). Due to its central position in the network and exhibiting the highest frequency, haplotype Hap_1 might be the maternal ancestral haplotype for the P. sinensis populations in this study. Among other haplotypes, seven shared haplotypes were identified (Hap_6, Hap_8, Hap_9, Hap_15, Hap_18, Hap_20, and Hap_24) and 33 unique haplotypes were found to derive from the above-mentioned haplotypes within seven mutational steps. For the JN population, six singleton haplotypes were derived from its dominant haplotype (Hap_15) within one or two mutation steps.

TCS network of P. sinensis based on COI haplotypes. The sizes of the circles indicate the number of individuals with each given haplotype. The number in the bracket represents the mutation steps between haplotypes and the black circles on the line represent an additional mutational change
Population expansion
Both Tajima’s D and Fu’s Fs tests showed negative values for all populations (Table 3), most of which reached a significant level (P < 0.05). The results for four populations (DL, PJ, SL, and JN) were found to be extremely significant (P < 0.01) for both tests, indicating a departure from mutation-drift equilibrium and potential population demographic expansion.
The population sizes before expansion (θ0) were zero for all populations, whereas population size after expansion (θ1) in the SL, SY, and SH populations were noticeably larger than in the other populations (Table 3). Furthermore, mismatch distribution did not differ significantly from the sudden expansion model when using either the sum of squared deviations (SSD) or Harpending’s raggedness index (HRI) for goodness-of-fit. A pattern of population expansion for P. sinensis was supported by the unimodal mismatch analysis (Fig. 3). Further, SSD and HRI were not statistically different from the model-predicted frequency (SSD = 0.009, P > 0.05; HRI = 0.201, P > 0.05) (Table 3).

Mismatch distributions of the P. sinensis haplotypes in each population. The bars represent the observed values, whereas the curves represent the expected values
Coalescence analyses demonstrated that the age of expansion in units of mutational time (τ) was highly variable (0.469–3.000) among the populations. Using a mutation rate of 1.4% per million years [18], the expansion of the P. sinensis population was estimated to have occurred approximately 16,000 to 106,000 years ago. The value of τ for the entire dataset was 1.783, corresponding to a demographic expansion origin of approximately 63,000 years before the present time for P. sinensis (Table 3).
Genetic divergence and distance among populations
The analysis of molecular variance (AMOVA) test indicated that the genetic variances within and among populations were 26.93% and 73.07%, respectively (Table 4). Furthermore, separating the seven populations into two groups maximized the variation among groups (FCT) as suggested by spatial analysis of molecular variance (SAMOVA) analysis (Table 4). The two groups were PJ + SL + AS + DL + SY + SH and JN. Analysis of molecular variance using the optimal two-group scheme indicated that the highest variation (53.36%) occurred among groups, whereas the variance within populations was 45.11%.
The pairwise Wright’s fixation index (FST) values among the seven studied populations ranged from − 0.0029 to 0.7052, most of which were highly significant (P < 0.01) (Table 5). Additionally, based on the criteria for genetic differentiation proposed herein, JN exhibited very high genetic differentiation (FST > 0.25) relative to other populations, as demonstrated by its FST values ranging from 0.4555 to 0.7052, all of which were found to be extremely significant [33]. The pairwise genetic distance (Ds) values between the seven examined P. sinensis populations were calculated according to the Kimura 2-parameter model (Table 5, below diagonal). The values ranged from 0.0007 to 0.0034, further supporting the low genetic differentiation between the populations. Figure 4 illustrates the generated UPGMA and NJ trees based on the Kimura 2-parameter genetic distance. The JN population was separated from all six remaining populations.

UPGMA (A) and NJ (B) clustering of P. sinensis populations based on the mitochondrial COI gene
Discussion
Genetic diversity
Previous reports have explored the genetic diversity of different Palaemonidae populations based on the mtDNA COI gene. However, our study is the first to examine the population-wide genetic variability of P. sinensis using mtDNA. Compared with other Palaemonidae, the haplotype diversity (Hd = 0.627) of P. sinensis was similar to that of P. capensis (Hd = 0.607) and P. peringueyi (Hd = 0.795) in South Africa [32], but lower than several other species such as M. australiense (Hd = 0.924) [4], M. olfersii (Hd = 0.94) [25], and P. longirostris and P. garciacidi (Hd = 0.97) [5]. The lower genetic diversity observed herein was similar to our previous reports using microsatellite markers, which may be due to poor swimming ability, as well as the time or space constraints during P. sinensis sampling [37].
Overall, the P. sinensis populations showed high haplotype diversity (Hd = 0.627) but low nucleotide diversity (π = 0.0004) in this study. The remarkably lower nucleotide diversity (π < 0.005) may be due to the commercial overexploitation of wild stocks, as P. sinensis is a prevalent specialty food and is also sought-after as a pet in this region. During sample collection, we found that some fishermen would engage in illegal fishing during the closed season, and this illegal fishing would lead to a sharp decline in the population size, resulting in a decrease in genetic diversity. Overharvesting can drive the decline of genetic diversity [1, 24, 27, 34], which is closely related to the long-term adaptability and survivability of populations. Low genetic diversity can affect the fitness [6] and the ability of individuals to survive and adapt to future environments [27, 34]. Therefore, the unnecessary cross-basin introduction should be avoided to preserve the local genetic resources. Moreover, in addition to fishing restrictions, the population size of low genetic diversity populations can be recovered by releasing shrimp from adjacent populations.
Genetic variation
Based on the outcome of AMOVA, the genetic differentiation within populations of P. sinensis, which accounted for 73.07% of the total genetic variation, was much higher than that among populations. Moreover, according to the maximum variation among groups (FCT) determined by SAMOVA, analysis of molecular variance using the optimal two-group scheme (JN and the other populations) indicated that the highest variations were found among groups (53.36%). Similarly, both the pairwise Ds and FST values revealed that the JN population was genetically different from the other populations. According to the criterion made by Thorp [30], the pairwise Ds values between pairs of P. sinensis populations ranged between 0.0007 and 0.0034, indicated that they were closely populations (Ds < 0.2). In contrast, the pairwise FST values among the other six populations were low or moderate [2]. The low or moderate differences between the pairwise FST values of the six populations indicated that they might share the same ancestors, and the haplotype analysis results also supported this conclusion. This was likely because the samples in the present study were collected from two geographical areas. Specifically, the JN population belonged to the Huaihe River Drainage in East China, whereas the other six populations inhabit the Liaohe River Basin and Related River Drainage in Northeast China. Therefore, the divergence between the JN population and the other six populations is due to the long-term geographical separation of their ancestors, which is consistent with our previous study using microsatellite markers [37]. Nevertheless, although no apparent geographic division was found based on genealogic reconstructions (Fig. 2) and the haplotypes of the JN population were distinct from those of the other populations, no distinct branches were observed in the Fig. 2. This may be because the estimated time of population expansion (16,000 to 106,000 years ago) was too short to form geographically unique clade [7].
Historical demographics
Neutrality tests (Tajima’D and Fu’s Fs) and mismatch distribution analysis were performed to understand the demographic dynamics of seven P. sinensis populations. Both mismatch distribution analysis and significant negative values of neutrality tests indicated a pattern of recent population expansion in most populations. The insignificant values of PSSD and PRag (P > 0.05) did not reject the hypothesis of the population growth in all seven populations. Moreover, the extremely significant negative values in the majority of Fu’s Fs statistics in populations DL, PJ, SL, SY, and JN also confirmed the expansions of these five populations despite the negative but non-significant Fu’s Fs value in population AS and SH.
The demographic history was reflected in the genetic indices of this species, which showed low nucleotide diversity (π < 0.005) and higher haplotype diversity (Hd > 0.5) (Table 2). As described by Grant et al. [14], high Hd and low π values can be attributed to rapid population expansion after a period of low effective population size, which enhances the retention of new mutations. This is consistent with the large number of unique and low-frequency haplotypes found in the present study (Additional file 1: Table S1). This is also consistent with the negative Tajima’s D value observed in our study, which was probably due to the population expansion caused by larger scale breeding after a sharp decline in population size [17]. Additionally, low diversity parameters (Hd < 0.5, π < 0.005) were identified in the DL, PJ, AS and JN populations, indicating that they may have recently experienced a bottleneck or founder effect produced by minority populations [14]. The genetic purity of these populations was not suitable for further selective breeding as a base population. Therefore, in situ conservation and the introduction of individuals from adjacent populations is critical in order to improve genetic diversity and reduce diversity decline as a result of inbreeding [14, 17].
Conclusions
The genetic variability of seven P. sinensis populations was assessed based on partial sequences of the COI gene. The results of AMOVA showed highly significant differences within populations. Forty-one haplotypes were detected among the populations, and the haplotype network indicated that haplotype Hap_1 might be the maternal ancestral haplotype for the studied P. sinensis populations. Neutrality and mismatch distribution tests suggested that P. sinensis underwent recent population expansion. The result of SAMOVA analysis, haplotype network analysis, Ds, and FST among pairs of populations suggested that the JN population was distinctive from the other six populations, which was due to long-term geographic separation. Overall, our results provide important insights for the development of genetic resource management strategies and enable a better understanding of the ecology and evolution of this species. Lastly, we propose several strategies for future P. sinensis genetic germplasm protection and aquaculture. First, illegal fishing should be strictly limited during the closed fishing season to avoid reductions in P. sinensis population size. Second, the DL, PJ, AS and JN populations were not suitable for further selective breeding as a base population, which highlights the need for in situ conservation efforts. Moreover, populations with low genetic diversity can be recovered by releasing shrimp from adjacent populations, and unnecessary cross-basin introduction should be avoided to preserve the local genetic resources.
Availability of data and materials
Sequence data from this article have been deposited with the GenBank Data Library under the Accession numbers: MT884019–MT884059.
Abbreviations
- COI:
-
Cytochrome c oxidase subunit I
- mtDNA:
-
Mitochondrial DNA
- DNA:
-
Deoxyribonucleic acid
- h :
-
Number of haplotypes
- S :
-
Number of segregating sites
- K :
-
Average number of differences
- H d :
-
Haplotype diversity
- π :
-
Nucleotide diversity
- PCR:
-
Polymerase chain reaction
- θ 0 :
-
Population sizes before expansion
- θ 1 :
-
Population size after expansion
- SSD:
-
Sum of squared deviations
- HRI:
-
Harpending’s raggedness index
- AMOVA:
-
Analysis of molecular variance
- SAMOVA:
-
Spatial analysis of molecular variance
- τ:
-
Age of expansion in units of mutational time
- F CT :
-
Variation among groups
- F ST :
-
Wright’s fixation index
- D s :
-
Genetic distance
- UPGMA:
-
Unweighted pair group method with arithmetic means
- NJ:
-
Neighbor-joining
References
Allendorf FW, England PR, Luikart G, Ritchie PA, Ryman N. Genetic effects of harvest on wild animal populations. Trends Ecol Evol. 2008;23:327–37.
Balloux F, Lugon-Moulin N. The estimation of population differentiation with microsatellite markers. Mol Ecol. 2002;11:155–65.
Bao J, Li XD, Yu H, Jiang HB. Respiratory metabolism responses of Chinese mitten crab, Eriocheir sinensis and Chinese grass shrimp, Palaemonetes sinensis, subjected to environmental hypoxia stress. Front Physiol. 2018;9:1559.
Carini G, Hughes JM. Population structure of Macrobrachium australiense (Decapoda: Palaemonidae) in Western Queensland, Australia: the role of contemporary and historical processes. Heredity. 2004;93:350–63.
Cartaxana A. Morphometric and molecular analyses for populations of Palaemon longirostris and Palaemon garciacidi (Crustacea, Palaemonidae): evidence for a single species. Estuar Coast Shelf S. 2015;154:194–204.
Chapman JR, Nakagawa S, Coltman DW, Slate J, Sheldon BC. A quantitative review of heterozygosity-fitness correlations in animal populations. Mol Ecol. 2009;18:2746–65.
Chen PC, Shih CH, Chu TJ, Wang D, Lee YC, Tzeng TD. Population structure and historical demography of the Oriental River Prawn (Macrobrachium nipponense) in Taiwan. PLoS ONE. 2015;10:e0145927.
Clement M, Posada D, Crandall KA. TCS: a computer program to estimate gene genealogies. Mol Ecol. 2000;9:1657–60.
Dong J, Zhao YY, Yu YH, Sun N, Li YD, Wei H, Yang ZQ, Li XD, Li L. Effect of stocking density on growth performance, digestive enzyme activities, and nonspecific immune parameters of Palaemonetes sinensis. Fish Shellfish Immun. 2018;73:37–41.
Dupanloup I, Schneider S, Excoffier L. A simulated annealing approach to define the genetic structure of populations. Mol Ecol. 2002;11:2571–81.
Excoffier L, Laval G, Schneider S. Arlequin ver. 3.11: an integrated software package for population genetics data analysis. Evol Bioinform Online. 2005;1:47–50.
Excoffier L, Smouse PE, Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–91.
Folmer O, Black M, Wr H, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994;3(5):294–99.
Grant WS, Bowen BW. Shallow population histories in deep evolutionary lineages of marine fishes: insights from sardines and anchovies and lessons for conservation. J Hered. 1998;89(5):415–26.
Hurzaid AT, Chan T, Nor SAM, Muchlisin ZA, Chen W. Molecular phylogeny and diversity of penaeid shrimps (Crustacea: Decapoda) from South-East Asian waters. Zool Scr. 2020;49(11):1–18.
Imai T, Oonuki T. Records of Chinese grass shrimp, Palaemonetes sinensis (Sollaud, 1911) from western Japan and simple differentiation method with native freshwater shrimp, Palaemon paucidens De Haan, 1844 using eye size and carapace color pattern. Bioinvasions Rec. 2014;3:163–8.
Jiang BJ, Fu JJ, Dong ZJ, Fang M, Zhu WB, Wang LM. Maternal ancestry analyses of red tilapia strains based on d-loop sequences of seven tilapia populations. Peer J. 2019;7:e7007.
Knowlton N, Weigt LA. New dates and new rates for divergence across the Isthmus of Panama. Proc R Soc Lond B. 1998;265:2257–63.
Leigh JW, Bryant D. PopART: full-feature software for haplotype network construction. Methods Ecol Evol. 2015;6(9):1110–6.
Li XZ, Liu RY, Liang XQ. The zoogeography of Chinese Palaemonoidea. Biodivers Sci. 2003;11:393–406.
Li YD, Li X, Xu WB, Han ZB, Zhao YY, Dong J, Wei H, Chen QJ. Comparative iTRAQ-based quantitative proteomic analysis of the Chinese grass shrimp (Palaemonetes sinensis) infected with the isopod parasite Tachaea chinensis. Parasite Vector. 2019;12:415.
Li YD, Xu WB, Li X, Jiang HB, She QX, Han ZB, Li XD, Chen QJ. Comparative transcriptome analysis of Chinese grass shrimp (Palaemonetes sinensis) infected with isopod parasite Tachaea chinensis. Fish Shellfish Immun. 2018;82:153–61.
Liu RY, Liang XZ, Yan SL. A study of the Palaemonidae (Crustacea: Decapoda) from China II. Palaemon, Exopalaemon, Palaemonetes and Leptocarpus. Stud Marina Sin. 1990;31:229–65.
Pinsky ML, Palumbi SR. Meta-analysis reveals lower genetic diversity in overfished populations. Mol Ecol. 2014;23:29–39.
Rossi N, Luis MF, Crandall KA. Molecular analysis of the freshwater prawn Macrobrachium olfersii (Decapoda, Palaemonidae) supports the existence of a single species throughout its distribution. PLoS ONE. 2013;8(1):e54698.
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–7.
Ryman N, Utter F, Laikre L. Protection of intraspecific biodiversity of exploited fishes. Rev Fish Biol Fisheries. 1995;5:417–46.
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539.
Tamura K, Stecher G, Kumar S. MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol. 2021;7:3022–7.
Thorp JP. The molecular clock hypothesis: biochemical evolution, genetic differentiation, and systematic. Annu Rev Ecol Syst. 1982;13:139–68.
Ward RD, Zemlak TS, Innes BH, Last PR, Hebert PD. DNA barcoding Australia’s fish species. Philos Trans R Soc B Biol Sci. 2005;360(1462):1847–57.
Wood LE, Sammy DG, Daniels SR, Tzen-Yuh C. Phylogeographic patterning among two codistributed shrimp species (Crustacea: Decapoda: Palaemonidae) reveals high levels of connectivity across biogeographic regions along the South African coast. PLoS ONE. 2017;12(3):e0173356.
Wright S. Evolution and genetics of populations. Chicago: University of Chicago; 1978.
Yorisue T, Iguchi A, Yasuda N, Yoshioka Y, Sato T, Fujita Y. Evaluating the effect of overharvesting on genetic diversity and genetic population structure of the coconut crab. Sci Rep. 2020;10:10026.
Zhao YY, Zheng XH, Kuang YY, Ma LM, He C, Zhu XC, Dong J, Wei H, Sun XW. Comparison of mitochondrial genomes of seven kinds of Carassius auratus auratus. J Shenyang Agric Univ. 2018;49(4):417–24.
Zhao YY, Zhu XC, Li YD, Han ZB, Xu WB, Dong J, Wei H, Li XD. Mitochondrial genome of Chinese grass shrimp, Palaemonetes sinensis, and comparison with other Palaemoninae species. Sci Rep. 2019;9:17301.
Zhao YY, Zhu XC, Li Z, Xu WB, Li XD. Genetic diversity and structure of Chinese grass shrimp, Palaemonetes sinensis, inferred from transcriptome-derived microsatellite markers. BMC Genet. 2019;20(1):75.
Zhao YY, Zhu XC, Xu WB, Jiang Y, Han ZB, Wang SY, Dong J, Wei H, Bai WL, Li XD. Morphological differences among four populations of Chinese grass shrimp (Palaemonetes sinensis). J Ecol. 2017;36(8):2252–6.
Acknowledgements
Not applicable.
Funding
This work was supported by the Cultivation Plan for Youth Agricultural Science and Technology Innovative Talents of the Liaoning Province (No.2015044), the Open Project of the National and Local Joint Engineering Laboratory for Freshwater Fish Breeding (HSY2020K1), the Talent Introduction Program of Shenyang Agricultural University (No. 880416005), and The Earmarked Fund for Modern Agro-industry Technology Research System (CARS-48). The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
YZ conceived and designed the experiments. YJ, ZL and XL performed the experiments. WX, HW and YL collected the samples. YZ and XZ analyzed the data and wrote the manuscript. YZ and XL provided reagents and materials and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Permissions were not necessary for collecting these samples. All efforts were made to minimize the suffering of the animals. All animal procedures in this study were conducted according to the animal husbandry guidelines of Shenyang Agriculture University. The studies in animals were reviewed and proved by the Ethical Committee and Experimental Animal Committee of Shenyang Agriculture University (Reference number: 201906019).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Distribution of the COI haplotypes of P. sinensis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhao, Y., Zhu, X., Jiang, Y. et al. Genetic diversity and variation of seven Chinese grass shrimp (Palaemonetes sinensis) populations based on the mitochondrial COI gene. BMC Ecol Evo 21, 167 (2021). https://doi.org/10.1186/s12862-021-01893-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-021-01893-8