
Abstract
Background
Although parasitic isopods can negatively affect the reproduction and ingestion of several commercially important crustaceans, little is known regarding the mechanisms that underlie these effects.
Methods
In the present study, the iTRAQ (isobaric tags for relative and absolute quantification) approach was applied to identify differentially expressed proteins in the Chinese grass shrimp Palaemonetes sinensis infected with the parasitic isopod Tachaea chinensis.
Results
On the basis of our analysis, we identified 1262 proteins from a total of 4292 peptides. There was a significant difference in the expression of 182 proteins between the control and infected groups, among which 69 were upregulated and 113 were downregulated after T. chinensis infection. The differentially expressed proteins revealed that parasitism may inhibit the immune response, thereby increasing host vulnerability to additional lethal infection. Furthermore, T. chinensis may secrete anticoagulants to inhibit hemolymph clotting. Moreover, the isopod parasite placed a heavy metabolic burden on the host, particularly with respect to glucose metabolism.
Conclusions
Our study is the first to use the iTRAQ-based proteomic approach to analyze the effects of an isopod parasite on its host. The results we obtained using this approach will make a valuable contribution to understanding the molecular mechanisms underlying isopod parasitism on crustaceans.
Similar content being viewed by others

Background
Parasitic isopods have a negative impact on a variety of commercially important fish and crustacean hosts [1]. The rapid development of aquaculture has led to increased international interest in parasites; however, unlike for parasites such as protozoans, trematodes, copepods and acanthocephalans, there have been relatively few studies on isopod parasites. Although many researchers have focused on the phylogenetic systematics [2,3,4], life history [5,6,7] and epidemiology [1, 8] of parasitic isopods, many aspects of the molecular mechanisms underlying the negative impacts on the host, as well as host responses, remain poorly understood.
Ectoparasitic isopods feed primarily on blood (hemolymph) that oozes from wounds of the host after the integument has been perforated by the isopod’s mandibles, which can cause host stress, tissue damage, secondary infection and mortality [1, 8,9,10]. Although trypsin inhibitors and anticoagulants have been found in isopod parasites, the mechanism by which it evades the immune response of the host during parasitism is still unclear; Manship et al. [11] proposed that the mechanism may include hemostasis, which limits blood loss by vasoconstriction and platelet aggregation. This is similar to the mechanism used by ticks [11]. Moreover, most studies have tended to focus on isopod–fish interactions, whereas comparatively little attention has been devoted to the interactions between isopods and crustaceans. To date, numerous approaches have been applied in studying the defense system response of crustaceans during pathogen infection, including high-throughput expressed sequence tag analysis, suppression subtractive hybridization and simple gene investigation [12,13,14]. In a previous study, we identified a number of critical genes related to the host response after isopod parasitism [12]; however, mRNA may not provide information regarding the presence of different protein isoforms or the posttranslational modifications of proteins. Therefore, an investigation of changes in the host proteome is important, since proteins, unlike transcripts, directly reflect the host’s response [15, 16].
Tachaea chinensis, one of the common ectoparasites of economic shrimps, is widely distributed in China and neighboring countries [17]. Tachaea chinensis is approximately 0.8 cm long, mainly attaches to the ventral thoracic region of shrimps and can readily be detected by the naked eye after parasitization (Additional file 1: Figure S1). In the present study, we performed a quantitative proteomic profiling using isobaric tags for relative and absolute quantification (iTRAQ) of the Chinese grass shrimp, Palaemonetes sinensis. The profiling was performed for shrimps infected with a parasite and for uninfected control shrimps to determine the integrated molecular mechanisms underlying T. chinensis parasitization and the resultant host responses. Our data fill a major knowledge gap in research on parasitic isopods, and thereby provide an important empirical basis for disease prevention and control efforts. Additionally, our results should support further research on the molecular biology of isopods.
Methods
Animals
The T. chinensis (1.24 ± 0.13 cm) used in this study were collected from a rice field in Panjin City, Liaoning Province, China, in April 2018, and transported to the aquaculture laboratory at Shenyang Agricultural University. Palaemonetes sinensis (3.48 ± 0.35 g) were caught using a net cage in a lake nearby the laboratory (no isopod parasitism had previously been reported in this area). These shrimps were acclimated in two 300-l square fiberglass recirculation tanks. Each tank was linked to a circular flow system. The water temperature was maintained at 22 ± 0.5 °C and the photoperiod was set at a light:dark cycle of 12:12 h. After 2 weeks of acclimation, 20 healthy P. sinensis individuals (3.16 ± 0.41 cm, n = 20) were transferred into individual plastic tanks (15.8 cm diameter and 32.1 cm height), each of which contained 5 l of water from the acclimation tanks, and environmental conditions were the same as those used for the acclimation period. Subsequently, 10 T. chinensis specimens were transferred into 10 tanks separately (one per host) and formed the infected group. The shrimps in the remaining 10 tanks were used as the controls. According to the results of our previous study, most parasitized shrimp began dying after 15 days (unpublished data); therefore, after 7 days in the present study, all 10 control shrimps and all 10 infected shrimps (without parasites) were placed separately in twenty 2-ml RNAse-free tubes that were immediately frozen in liquid nitrogen for storage until protein extraction.
Protein digestion and iTRAQ labeling
For each sample, 200 µg of protein was diluted in 200 µl of uric acid (UA) buffer (8 M urea, 150 mM Tris-HCl, pH 8.0) and transferred onto a 10-kDa ultrafiltration filter. The samples were centrifuged at 14,000× g for 15 min and washed with 200 µl of UA buffer. After incubation in 100 µl of 50 mM iodoacetamide in UA buffer for 30 min in the dark, the samples were centrifuged again at 14,000× g for 10 min. The filters were washed three times with 100 µl of UA buffer and then twice with 100 µl of dissolution buffer. Finally, 2 μg of trypsin (iTRAQ Reagents, SCIEX, Foster City, USA) was added to each filter and digested overnight at 37 °C.
iTRAQ labeling was performed using an iTRAQ Reagent-8 plex Multiplex Kit according to the manufacturer’s instructions (Applied Biosystems, Foster City, USA). The proteins in the control group were labeled with reagent 118, 119 and 120, whereas those in the infected group were labeled with reagent 121, 122 and 123.
SCX fractionation and LC-MS/MS analysis
The iTRAQ-labeled samples were purified by strong cation exchange (SCX) chromatography using a Polysulfoethyl 4.6 × 100 mm column (5 μm, 200 Å; PolyLC Inc, Columbia, MD, USA) in an AKTA Purifier 100 (GE Healthcare, Piscataway, USA). After SCX fractionation, 33 fractions were collected and combined into 10 pools and desalted on C18 cartridges (Sigma-Aldrich, Saint Louis, USA). Each fraction was segregated using a nano HPLC Easy nLC system (Thermo Finnigan, Hemel Hempstead, UK) incorporating two Thermo Fisher Scientific (Hemel Hempstead, UK) EASY columns (2 cm × 100 μm, 5 μm-C18 for sampling and 75 μm × 100 mm, 3 μm-C18 for analysis). The flow rate was set at 250 nl/min. The peptides were eluted with a gradient of mobile phase A (0.1% formic acid in water) for 5 min, and then in a gradient starting from 0 to 35% mobile phase B (0.1% formic acid in 84% acetonitrile) for 45 min, followed by an 8 min linear gradient to 100%. Finally, the samples were maintained in 100% mobile phase B for 2 min. Each sample was subjected to mass spectrometry (MS) survey using a Q-Exactive mass spectrometer (Thermo Finnigan). Briefly, a full MS survey scan was performed for a mass range of 300 to 1800 m/z with resolution of 70,000 at m/z 200. High energy collisional dissociation (HCD) fragmentation was used for MS/MS, and the 10 most intense signals in the survey scan were fragmented. Normalized collision energy was set at 30 eV with a 0.1% underfill ratio.
Data analysis
The raw files were analyzed by using Proteome Discoverer v.1.4 software (Thermo Fisher Scientific, Karlsruhe, BW, Germany). Identification of the proteins was conducted using the MASCOT v.2.2 search engine (Matrix Science Ltd., London, UK). The protein identification and quantitation parameters are shown in Table 1. Differential proteins were analyzed for significant down- or upregulation, which was calculated using Protein Pilot. The values of the intensities of the three reporter ions for each experimental group were averaged and then the difference was statistically assessed. The fold change was set to > 1.2 for protein upregulation and < 0.83 for protein downregulation.
These proteins were linked to the following databases for downstream analysis: Quick GO (Gene Ontology analysis), KOG (Clusters of orthologous groups for eukaryotic complete genomes) and KEGG (Kyoto Encyclopedia of Genes and Genomes).
Verification of the proteome data
The proteome data were confirmed at both the transcriptional and protein levels. At the transcriptional level, the significantly expressed proteins were compared with the expression of their coding genes in our previous transcriptome data. Moreover, 55 shrimps were used to validate the proteome by using the activity of cytochrome c oxidase (CCO) and hemocyanin content: 5 for the control and 50 for the experimental group. The T. chinensis (0.88 ± 0.15 cm) and P. sinensis (3.61 ± 0.29 g) were collected in November 2018. After acclimation for 2 weeks, each of the 50 shrimps in the experimental group were artificially infected with a single isopod and randomly divided into five groups. One shrimp was collected from each group at 0 (control), 24, 48, 72, 96 and 120 h after the artificial infection; from these, 200 μl of hemolymph was extracted and divided into two aliquots. Each 100-μl subsample was centrifuged for 10 min (900× g, 4 °C) and the resulting supernatants were immediately used for the determination of hemocyanin content and CCO activity. We detected no significant difference among groups with regard to shrimp weight.
The hemocyanin measurements were performed spectrophotometrically. The supernatant samples were diluted with 1.9 ml of buffer (10 mmol/l CaCl2, 50 mmol/l Tris-HCl, pH = 8.0) and readings were taken at 334 nm (O.D.334). The concentration of hemocyanin was calculated using the following equation: hemocyanin content (mg/ml) = 2.33 × O.D.334 × 100. CCO activity was measured using a CCO testing kit (A090-1-1, Nanjing Jiancheng Biological Product, Nanjing, China) according to the manufacturer’s guidelines.
Statistical analyses
Statistical analyses were performed using analysis of variance (ANOVA). Significant differences between means were determined by Duncan’s test at a significance level of P < 0.05. All statistical analyses were performed using SPSS 22.0 software (version 22.0; IBM, Armonk, NY, USA).
Results
Protein profiling
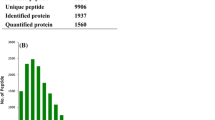
The iTRAQ analysis of the present study showed 17,131 queries in the 162,387 spectra. The protein database contained a total of 41,095 protein sequences, and the fragment and peptide mass tolerances were set to ± 20 ppm and ± 0.1 Da, respectively. Among these, a total of 1262 unique proteins were identified across the 4292 peptides (Fig. 1a). There were 225 proteins between 10 and 20 kDa, followed by 209, 132 and 149 proteins of 20–30, 30–40 and 40–50 kDa, respectively, whereas 165 proteins had a mass of over 100 kDa (Fig. 1b).

Basic information statistics. a Coverage of proteins by the identified peptides. b Distribution of identified proteins among different molecular weights
Functional annotation and classification
All 1262 unique proteins were subjected to GO, KOG and KEGG database analyses (Additional file 2: Table S1). According to the GO analysis, 824, 775 and 789 proteins were assigned to the categories “biological processes”, “molecular function” and “cellular components”, respectively (Fig. 2). The top five most frequent categories of biological processes in our study were “cellular processes” (83.96%), “single-organism processes” (82.31%), “metabolic processes” (78.93%), “multicellular organismal process” (49.15%) and “developmental process” (43.12%). To predict and classify their possible functions based on reference to orthologs from other species, all proteins were annotated using the KOG database. As shown in Fig. 3, a total of 1090 proteins were categorized into 25 groups, among which “General function prediction only” accounted for the largest group (677), followed by “signal transduction mechanisms” (541) and “posttranslational modification, protein turnover, chaperones” (452). In KEGG analysis, a total of 504 proteins were mapped against the KEGG pathway database. The most enriched pathways were as follows: carbon metabolism (45), biosynthesis of amino acids (26), glycolysis/gluconeogenesis (25), citrate cycle (18) and pyruvate metabolism (17) (Fig. 4).

GO annotation of the identified proteins from the proteome database of the infected and uninfected shrimps. Most proteins can be divided into the three major categories: a biological process; b cellular component; c molecular function

KOG annotation of the identified proteins from the proteome database of the infected and uninfected shrimps

KEGG annotation of the identified proteins from the proteome database of the infected and uninfected shrimps
iTRAQ analysis and identification of differentially expressed proteins
Using a 1.2-fold increase or decrease in protein expression as a benchmark for a physiologically significant change, 182 differentially expressed proteins (P < 0.05) were identified between the control and infected groups. Among these differentially expressed proteins, 69 were upregulated and 113 were downregulated after T. chinensis infection (Additional file 3: Table S2). According to the GO enrichment analysis, 1691, 243 and 441 proteins were enriched for biological process, cell component and molecular function, respectively; of these 349, 62, and 129 were statistically significant (Additional file 4: Figure S2). Moreover, 78 KEGG pathways were enriched in this dataset, of which seven were statistically significant. Among the 78 KEGG pathways, 8, 8, 6, 6 and 4 mapped to the top five enriched pathways “carbon metabolism”, “phagosome”, “glycolysis/gluconeogenesis”, “biosynthesis of amino acids” and “photo transduction-fly”, respectively (Additional file 5: Figure S3).
After eliminating the uncharacterized proteins among the 182 proteins differentially expressed between the control and infected groups, we obtained 30 upregulated proteins (Table 1) and 64 downregulated proteins (Table 2). As shown in Table 1, the differentially expressed proteins related to cytoskeletal function, including slow muscle myosin S1 heavy chain, paramyosin, bestrophin homolog, cuticular protein 34, troponin C, peritrophin and tubulin alpha chain were significantly upregulated after infection. Moreover, five cytochrome c oxidase proteins and two transient receptor potential cation channel subfamily M member proteins were also identified. The highest ratio was observed for hyperglycemic hormone (2.54). Among the 64 significantly downregulated differentially expressed proteins, 30 proteins were related to immunity, notably Toll-like receptor, cathepsin L, tachylectin and annexin. The remainder included four macroglobulin- and 20 hemocyanin-related proteins (Table 2). Moreover, actin-related proteins, including beta actin and cardiac muscle actin, were significantly downregulated.
Verification of the protein data
According to association analysis of the proteome and transcriptome data, 20 of the 150 differentially expressed proteins (13.33%) shared the same annotation with transcripts. Among these 20 proteins, 16 (80%) showed expression trends similar to the transcriptome expression trends in the control and infected group, including nine upregulated and seven downregulated proteins/genes (Fig. 5a).

Verification of the protein data. a Transcriptional and proteome expression patterns of identical proteins/genes. b The hemocyanin content (Hc) and cytochrome c oxidase (CCO) activity verification
The results of our analysis of hemocyanin content and CCO activity over a 120 h period after infection are shown in Fig. 5b. After parasitization by T. chinensis, the hemocyanin content of shrimps significantly decreased by 1.17 ± 0.31 (F(4, 20) = 21.21, P = 0.002); 1.57 ± 0.42 (F(4, 20) = 40.97, P < 0.001); 1.21 ± 0.27 (F(4, 20) = 20.54, P = 0.002); 1.87 ± 0.48 (F(4, 20) = 40.71, P < 0.001); and 2.01 ± 0.55 (F(4, 20) = 72.41, P < 0.001) mg/ml at 24, 48, 72, 96 and 120 h after isopod parasitism, respectively. Compared with the control group (0 h), the activity of CCO significantly increased by 7.57 ± 0.88% (F(4, 20) = 84.98, P < 0.001); 4.31 ± 0.46% (F(4, 20) = 16.99, P = 0.003); 16.45 ± 2.54% (F(4, 20) = 189.89, P < 0.001); and 20.17 ± 3.26% (F(4, 20) = 169.89, P < 0.001) in shrimps parasitized for 48, 72, 96 and 120 h, respectively.
Discussion
Investigation of the response to different pathogens represents an important research goal for elucidating immune and related components in the defense system of crustaceans [18,19,20]. In recent years, the isopod parasite T. chinensis has resulted in heavy economic losses of commercially farmed crustacea, particularly in Penaeus vannamei, Macrobrachium nipponense, Exopalaemon carinicauda and Palaemonetes sinensis [21,22,23]. Therefore, understanding the pathogenic mechanism of this parasitic disease has become crucial for achieving sustainable crustacean production. In the present study, using the iTRAQ-based quantitative proteomic approach, we aimed to gain a better understanding of the molecular mechanisms underlying the effects of this isopod parasite and shrimp proteome changes to the parasite.
Hemocyanin has been studied in several host-pathogen interactions, which have indicated that hemocyanin also plays an important role in the immune system of shrimps [14, 24,25,26] and has been found to be significantly upregulated during viral and bacterial infection [27,28,29,30]. However, patterns in the variation of hemocyanin in parasitized and healthy shrimps remain unclear. In the present study, we identified 20 hemocyanin-related proteins (31.25%) among differentially expressed proteins that were downregulated in response to parasitism. Moreover, we found that the hemocyanin content decreased concomitant with an increase in the duration of parasitism. These results are similar to those obtained for fish hosts, in which hemoglobin concentrations also decreased in response to isopod parasitization [31]. These findings indicate that T. chinensis could use its mandible to feed, suck the hemolymph and promote hematological changes in P. sinensis. Therefore, similar to other blood-feeding parasites, T. chinensis may also be implicated in the transmission of blood-dwelling diseases between shrimps, which we intend to examine in our future research.
Most invertebrates need to rapidly prevent the loss of blood or equivalent fluids through inflicted injuries [32]. In shrimps, the hemolymph clotting system, which comprises transglutaminase and clotting proteins, plays an important role in the innate immune response and prevention of blood loss during injury and wound healing [33]. However, in the present study, we found that a putative clotting protein, cytoplasmic type actin 3 and three macroglobulin proteins were significantly downregulated in parasitized shrimps and blood coagulation. Moreover, there was a 0.87-fold downregulation of transglutaminase, which catalyzes the gelation of the plasma (Additional file 2: Table S1). It is well known that blood-feeding parasites, such as ticks, mosquitoes, fleas and leeches need to evade the clotting system of their vertebrate hosts and maintain blood flow during feeding [34, 35]. Many researchers consider that isopod parasites may inject anticoagulants or other compounds directly into the blood to obtain their “blood meal” [11, 31]. In order to suck the hemolymph, T. chinensis may secrete various anticoagulants, which could work cooperatively and prevent blood clotting by downregulating the expression of clotting-related proteins at the site of injury.
Moreover, we found that five actin proteins and a Toll-like receptor protein were significantly downregulated. In contrast, these proteins have been found to be upregulated during viral and bacterial infection [36]. Crustaceans infected with viruses or bacteria exhibit a significant change in the signaling pathways related to the immune response, including MAPK, Toll-like receptor, PI3K-Akt and Jak-STAT pathways [34,35,36,37]; however, in our previous study, we detected no significant changes in these pathways during T. chinensis parasitization [12]. Parasite survival largely depends on circumventing the host’s immune system. Therefore, to ensure survival until transmission to the next host, parasites such as T. chinensis may have to balance between evading the host immune response, similar to acanthocephalans parasitizing gammarids [37, 38], and increasing host vulnerability to additional lethal infection.
According to the KEGG analysis performed in the present study, two pathways involved in glucose metabolism, namely carbon metabolism and glycolysis/gluconeogenesis, were among the top five statistically significant different pathways. Moreover, proteins associated with these two glycometabolism-related pathways, including crustacean hyperglycemic hormone (CHH), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and carbohydrate sulfotransferase (CST), were significantly upregulated. These results are similar to those obtained from our corresponding transcriptome analysis [12]. Carbohydrate dynamics and CHH concentrations were also found to be upregulated during infection with a dinoflagellate parasite in Nephrops norvegicus [39]. However, the upregulated levels of CHH, GAPDH, and CST were caused either directly by parasitic disruption of the shrimp’s endocrine system or indirectly by interfering with the positive and negative feedback loops.
Conclusions
To the best of our knowledge, this is the first study to investigate the response of shrimps to an isopod parasite using an iTRAQ-based proteome method. The differentially expressed proteins related to hemocyanin content and the hemolymph coagulation system were identified as downregulated after isopod infection. This may indicate that, similar to blood sucking parasites, T. chinensis could inhibit the hemolymph clotting system of P. sinensis during parasitization. Moreover, isopod parasites place a heavy metabolic burden on their hosts, particularly with regards to glucose metabolism. Overall, our study provides valuable empirical data that will support future molecular research on isopod parasitization of crustaceans.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD014542. All analyzed data are available from the corresponding author upon reasonable request.
Abbreviations
- iTRAQ:
-
isobaric tags for relative and absolute quantification
- GO:
-
gene ontology
- KOG:
-
clusters of orthologous groups for eukaryotic complete genomes
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- CHH:
-
hyperglycemic hormone
- GAPDH:
-
glyceraldehyde-3-phosphate dehydrogenase
- CST:
-
carbohydrate sulfotransferase
References
Williams JD, Boyko CB. The global diversity of parasitic isopods associated with crustacean hosts (Isopoda: Bopyroidea and Cryptoniscoidea). PLoS ONE. 2012;7:e35350.
Ashby B, King KC. Friendly foes: the evolution of host protection by a parasite. Evol Lett. 2017;1:211–21.
Hata H, Sogabe A, Tada S, Nishimoto R, Nakano R, Kohya N, et al. Molecular phylogeny of obligate fish parasites of the family Cymothoidae (Isopoda, Crustacea): evolution of the attachment mode to host fish and the habitat shift from saline water to freshwater. Mar Biol. 2017;164:105.
Nagler C, Hyžný M, Haug JT. 168 million years old “marine lice” and the evolution of parasitism within isopods. BMC Evol Biol. 2017;17:76.
Hispano C, Bultó P, Blanch AR. Life cycle of the fish parasite Gnathia maxillaris (Crustacea: Isopoda: Gnathiidae). Folia Parasitol. 2014;61:277–84.
Smi NJ, Basson L, Van As JG. Life cycle of the temporary fish parasite, Gnathia africana (Crustacea: Isopoda: Gnathiidae). Folia Parasitol. 2003;50:135–42.
Hadfield KA, Smit NJ, Avenantoldewage A. Life cycle of the temporary fish parasite, Gnathia pilosus (Crustacea: Isopoda: Gnathiidae) from the east coast of South Africa. J Mar Biol Assoc UK. 2009;89:1331–9.
Smit NJ, Bruce NL, Hadfield KA. Global diversity of fish parasitic isopod crustaceans of the family Cymothoidae. Int J Parasitol Parasites Wildl. 2014;3:188–97.
Wilson GDF. Global diversity of isopod crustaceans (Crustacea; Isopoda) in freshwater. Hydrobiologia. 2008;595:231–40.
Poore GC, Bruce NL. Global diversity of marine isopods (except Asellota and crustacean symbionts). PLoS ONE. 2012;7:e43529.
Manship BM, Walker AJ, Jones LA, Davies AJ. Blood feeding in juvenile Paragnathia formica (Isopoda: Gnathiidae): biochemical characterization of trypsin inhibitors, detection of anticoagulants, and molecular identification of fish hosts. Parasitology. 2012;139:744–54.
Li YD, Xu WB, Li X, Jiang HB, She QX, Han ZB, et al. Comparative transcriptome analysis of Chinese grass shrimp (Palaemonetes sinensis) infected with isopod parasite Tachaea chinensis. Fish Shellfish Immunol. 2018;82:153–61.
Bachère E, Mialhe E, Rodriguez J. Identification of defence effectors in the haemolymph of crustaceans with particular reference to the shrimp Penaeus japonicus (Bate): prospects and applications. Fish Shellfish Immunol. 1995;5:597–612.
Zhang Y, Gao G, Lin R, Aweya JJ, Tao M, Wang F. Transcriptome analyses reveal Litopenaeus vannamei hemocytes response to lipopolysaccharide. Fish Shellfish Immunol. 2018;76:187–95.
Wen L, Dong Q, Yan X. Proteomic analysis of differentially expressed proteins in hemolymph of Scylla serrata response to white spot syndrome virus infection. Aquaculture. 2011;314:53–7.
Marcos-López M, Rodger HD, O’Connor I, Braceland M, Burchmore RJS, Eckersall PD, et al. A proteomic approach to assess the host response in gills of farmed Atlantic salmon Salmo salar L. affected by amoebic gill disease. Aquaculture. 2017;470:1–10.
Hua CJ, Li WX, Zhang D, Zou H, Li M, Jakovlić I, et al. Basal position of two new complete mitochondrial genomes of parasitic Cymothoida (Crustacea: Isopoda) challenges the monophyly of the suborder and phylogeny of the entire order. Parasit Vectors. 2018;11:628.
Shields JD. The impact of pathogens on exploited populations of decapod crustaceans. J Invertebr Pathol. 2012;110:211–24.
Small HJ, Pagenkopp KM. Reservoirs and alternate hosts for pathogens of commercially important crustaceans: a review. J Invertebr Pathol. 2011;106:153–64.
Stentiford GD. Diseases in aquatic crustaceans: problems and solutions for global food security. J Invertebr Pathol. 2012;110:139.
Lu XD, Zou WM, Peng HL, Shu WU, Chen W, Xie JG, et al. New records of two parasitic isopods in Peal River. J Fish Sci China. 2007;14:1023–6.
Cheng RY, Niu Y, Guo YM. A new record of freshwater isopod crustacean from Henan (Crustacea: Isopoda: Corallanidae). J Henan Normal Univ. 2011;565:28–33.
Matsuei S. A new record of Tachaea chinensis Thielemann (Isopoda Corallanidae) from Okinawajima Island, Ryukyu Islands, Japan. Jpn J Ornithol. 2014;52:65–8.
Zhang YL, Yan F, Hu Z, Zhao XL, Min SY, Du ZH, et al. Hemocyanin from shrimp Litopenaeus vannamei shows hemolytic activity. Fish Shellfish Immunol. 2009;27:330–5.
Xu J, Ruan L, Li Z, Yu X, Li S, Shi H, et al. Characterization of four hemocyanin isoforms in Litopenaeus vannamei. Acta Oceanol Sin. 2015;34:36–44.
Coates CJ, Nairn J. diverse immune functions of hemocyanins. Dev Comp Immunol. 2014;45:43–55.
Zhao ZY, Yin ZX, Weng SP, Guan HJ, Li SD, Xing K, et al. Profiling of differentially expressed genes in hepatopancreas of white spot syndrome virus-resistant shrimp (Litopenaeus vannamei) by suppression subtractive hybridisation. Fish Shellfish Immunol. 2007;22:520–34.
Pan D, He N, Yang Z, Liu H, Xu X. Differential gene expression profile in hepatopancreas of WSSV-resistant shrimp (Penaeus japonicus) by suppression subtractive hybridization. Dev Comp Immunol. 2005;29:103–12.
Gesheva V, Idakieva K, Kerekov N, Nikolova K, Mihaylova N, Doumanova L, et al. Marine gastropod hemocyanins as adjuvants of non-conjugated bacterial and viral proteins. Fish Shellfish Immunol. 2011;30:135–42.
Boonchuen P, Jaree P, Tassanakajon A, Somboonwiwat K. Hemocyanin of Litopenaeus vannamei agglutinates Vibrio parahaemolyticus AHPND (VPAHPND) and neutralizes its toxin. Dev Comp Immunol. 2018;84:371–81.
Nair GA, Nair NB. Effect of infestation with the isopod, Alitropus typus M. Edwards (Crustacea: Flabellifera: Aegidae) on the haematological parameters of the host fish, Channa striatus (Bloch). Aquaculture. 1983;30:11–9.
Cerenius L, Söderhäll K. Coagulation in invertebrates. J Innate Immun. 2011;3:3–8.
Maningas MBB, Hidehiro K, Ikuo H. Molecular mechanisms of the shrimp clotting system. Fish Shellfish Immunol. 2013;34:968–72.
Shu QT, Glanfield A, Gobert GN, Jones MK. Heme and blood-feeding parasites: friends or foes? Parasit Vectors. 2010;3:108.
Santiago PB, Araújo CND, Motta FN, Praça YR, Charneau S, Bastos IMD, Santana JM. Proteases of haematophagous arthropod vectors are involved in blood-feeding, yolk formation and immunity—a review. Parasit Vectors. 2017;10:79.
Zhang X, Zhang X, Yuan J, Du J, Li F, Xiang J. Actin genes and their expression in pacific white shrimp, Litopenaeus vannamei. Mol Genet Genomics. 2018;293:479–93.
Cornet S, Franceschi N, Bauer A. Immune depression induced by acanthocephalan parasites in their intermediate crustacean host: consequences for the risk of super-infection and links with host behavioural manipulation. Int J Parasitol. 2009;39:221–9.
Franceschi N, Rigaud T, Moret Y, Hervant F, Bollache. Behavioural and physiological effects of the trophically transmitted cestode parasite, Cyathocephalus truncatus, on its intermediate host, Gammarus pulex. Parasitology. 2007;134:1839–47.
Stentiford GD, Chang ES, Chang SA, Neil DM. Carbohydrate dynamics and the crustacean hyperglycemic hormone (CHH): effects of parasitic infection in Norway lobsters (Nephrops norvegicus). Gen Comp Endocrinol. 2001;121:13–22.
Acknowledgements
We thank Panjin Guanghe Fisheries Co. Ltd for providing the shrimps.
Funding
This work was supported by the earmarked fund for Modern Agro-industry Technology Research System (no. CARS-48), the National Natural Science Foundation of China (no. 31502190) and the Talent Introduction Program of Shenyang Agricultural University (nos. 880416005 and 880417024).
Author information
Authors and Affiliations
Contributions
YL and QC conceived and designed the study. XL, WX and ZH collected the specimens and performed the experiments. YZ, JD, HW and QC analyzed the data. YL drafted the manuscripts. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Our study did not involve endangered or protected species. In China, catching grass shrimp from rice fields does not require specific permits. All efforts were made to minimize animal suffering and discomfort. The experimental protocol was approved by the Animal Ethics Committee of Shenyang Agriculture University, Shenyang, China.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Picture of T. chinensis attached to the ventral thoracic region of P. sinensis.
Additional file 2: Table S1.
Total proteins compared to GO, KOG, and KEGG databases.
Additional file 3: Table S2.
Lists of differentially expressed proteins.
Additional file 4: Figure S2.
GO enrichment analysis.
Additional file 5: Figure S3.
KEGG enrichment analysis.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
Cite this article
Li, Y., Li, X., Xu, W. et al. Comparative iTRAQ-based quantitative proteomic analysis of the Chinese grass shrimp (Palaemonetes sinensis) infected with the isopod parasite Tachaea chinensis. Parasites Vectors 12, 415 (2019). https://doi.org/10.1186/s13071-019-3675-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-019-3675-5