
Abstract
Background
Invasive species are increasingly driving biodiversity decline, and knowledge of colonization dynamics, including both drivers and dispersal modes, are important to prevent future invasions. The bee species Megachile sculpturalis (Hymenoptera: Megachilidae), native to East-Asia, was first recognized in Southeast-France in 2008, and has since spread throughout much of Europe. The spread is very fast, and colonization may result from multiple fronts.
Result
To track the history of this invasion, codominant markers were genotyped using Illumina sequencing and the invasion history and degree of connectivity between populations across the European invasion axis were investigated. Distinctive genetic clusters were detected with east–west differentiations in Middle-Europe.
Conclusion
We hypothesize that the observed cluster formation resulted from multiple, independent introductions of the species to the European continent. This study draws a first picture of an early invasion stage of this wild bee and forms a foundation for further investigations, including studies of the species in their native Asian range and in the invaded range in North America.
Similar content being viewed by others


Background
Invasion occurs when a species is accidentally or intentionally introduced to and establishes within such a new environment. But invasive pollinators, like invasive bee species, often deviate from the negative perspective of invaders, especially when it comes to pollinators of crops. Bees are generally considered beneficial for the pollination services they provide, but they may have surprising negative impacts on local ecosystems where they do not naturally occur [1,2,3,4]. Negative impacts can take different forms, including through disrupting plant-pollinator networks, competition with native species for nest sites or other resources, pollinating invasive plants, the introduction of novel pathogens, and economic losses [5,6,7,8]. Among bees, those which nest in cavities opportunistically are disproportionately invasive, i.e. Afranthidium (Immanthidium) repetitum, and, relatedly, wood-nesting bees make up larger proportions of the fauna in more remote islands due to higher dispersal abilities [9, 10]. In these ways, invasive bees can be promising study organisms for investigating range expansion and dispersal patterns [11].Given increasing reports of invasions that come with globalization of human society [12], it is crucial that we learn more about the dynamics of bee invasions to better protect native and managed ecosystems.
Megachile (Callomegachile) sculpturalis Smith, 1853, one of hundreds of species amongst Megachile [13], is the first translocated bee species to become established in Europe. Megachile sculpturalis is a protandric wild bee with an active season from June to mid-September in Europe [14,15,16,17]. Males body size ranges between 12 and 22 mm, and females between 20 and 28 mm. Both sexes are characterized by their narrow body, bright orange hairy thorax hair and dusky wings (Fig. 1) [18, 19]. Megachile sculpturalis is a cavity-nesting wild bee using preexisting holes in wood. The species naturally occurs in East-Asia (Japan, China, South-Korea and Taiwan), but was first established outside its native range in the eastern USA around 1994, and is now considered as invasive species [14, 20, 21]. Although it is classified as a pollen generalist, this polylectic species presents a strong preference for pollen of Styphnolobium japonicum and Ligustrum sp. in Europe. Both plants are native to Asia and often used as ornamental plants in human habitats [16, 17, 22]. The bee species shows aggressive behavior towards the local bee fauna from time to time, and events of direct evictions have been observed in the US and in Europe [23, 24]. A recent study detected a negative correlation between its presences in trap nests and the emergence of native bee species raising the question of the magnitude of the impact of M. sculpturalis on the local bee populations [25].

Sex dimorphism of Megachile sculpturalis: a males have a three-toothed mandible and shiny yellow hair on their supraclypeal plate, and lack a metasomal scopa. b females are bigger, with a largely black metasomal scopa except for the first two sternites where hairs are yellow. c As a protandric species, male larvae are positioned at the front in the nests and emerge earlier than females
In Europe, this species was first recognized in a small city near Marseille (Allauch, France) in 2008 [26]. There are relatively few examples of other invasive aculeate species in Europe, besides Vespa velutina first recognized in 2005 also in South-France, and Megachile disjunctiformis found in northern Italy in 2011 [27,28,29]. Megachile sculpturalis most likely found its way to Europe as a stowaway on ships while nesting in dead wood [26, 30]. Following its initial discovery in France, the species was reported from northern Italy in 2009 [16]. In the following years, M. sculpturalis colonized many European countries remarkably fast. Its range expansion from France and Italy continued to Spain, Switzerland, Germany, Austria, Hungary, Slovenia, Croatia, Serbia, Bosnia Herzegovina reaching now to the Crimean Peninsula [16, 22, 23, 30,31,32,33,34,35,36,37]. A recent compilation of records from our collections and a citizen science approach, show range extensions to eastern locations, including Vienna (2017) [38], Gyöngyös (2015) [33], and Belgrade (2017) [32]. This suggests that the current distribution pattern in Europe was attained by two distinctive mechanisms of dispersal. First, after its introduction in South-France, M. sculpturalis probably colonized most countries by stepwise dispersal. Additionally, accidental transport in wood could explain the large distances-over hundreds of kilometers-between isolated reports eastward [23].
The invasion of M. sculpturalis had been tracked relatively well, because of the conspicuous size of the species (for an European bee) and its typical association with anthropogenic habitats [23]. Thus, we are able to investigate the early spread of this species in real-time and generate baseline genetic diversity data to better understand the structure and colonization pathways, before widespread admixture within the invasive range occurs. On the basis of genetic diversity, the success of translocated populations might be linked not only to the number of founding individuals but also the number of different colonization events [39,40,41]. Population genetic analyses are therefore an important tool to study the patterns and intensity of dispersal over geographic ranges [42,43,44,45].
To conduct such analyses, allele frequencies from selected neutral marker loci are compared to assess gene flow, which is a crucial parameter studying population dynamics and invasion biology [39, 46, 47]. Great advances have been made in the study of genetic diversity in non-model organisms over the last decade. Codominant datasets from microsatellites or simple sequence repeats (SSRs) were the standard for assessing connectivity and genetic structure between populations for a long time [45, 48, 49]. The main advantage of the genotyping by amplicons sequencing (SSR-GBAS) approach is the direct accessibility of the sequence of an allele without the need to indirectly use size as a proxy. Here, we call the method SSR-GBAS, as in earlier studies SSR-GBS was used which led to confusion because of the frequent view of using GBS exclusively for RAD methods [50, 51]. Variants using similar allele call methods have also been termed SSR-Seq [52]. The single-base-pair resolution enables enhanced sensitivity in detecting alleles and hidden size-homoplasy with a greater potential for individual identification as well as the ability to resolve low genetic diversity and spatial patterns at very fine scales [53,54,55]. SSR-GBAS can also be applied for species with large genomes, where little or no genetic data are available. Another major advantage of parallel sequencing is the high number of samples that can be included in single sequencing runs, therefore circumventing some additional shortcomings of microsatellites aside from their exploring only length-polymorphisms [50, 56, 57]. The approach is especially promising for samples providing little or degraded DNA [55], which is often the case when working with small organisms such as wild bees. Overall, this type of marker is ideal for exploring the invasion dynamics of exotic species.
Here, we present and validate a set of SSR loci as markers suitable for a genotyping by amplicon sequencing approach. We use these markers for a first investigation of genetic variations of M. sculpturalis across Europe. The degree of connectivity between widespread populations is explored to detect signs of one or multiple colonizing events in Central Europe. The present study sets the baseline for further population genetic analyses, including specimens not only from Europe but also from its native range in Asia and North America. It should serve as a model for a deeper understanding of the interdependencies of successful colonization dynamics and mechanisms of dispersals, such that we may better predict and control the spread of invasive species in the future.
Results
Output statistics
To study the genetic variability of European M. sculpturalis populations, 48 SSR markers were designed and tested on 81 samples of European M. sculpturalis specimens. Out of all 48 markers, two (FB12 and FB47) were excluded due to an unusually high number of alleles as a possible consequence of duplication or scoring errors. The marker set included 20 pentanucleotide (two were excluded) and another 28 tetranucleotide motifs. Primer information is summarized in Table 1, including polymorphism information content, number of different alleles and unbiased, expected heterozygosity (uHe).
Genetic diversity of European populations
The partial Illumina MiSeq run resulted in 3,043,426 paired reads for the library analyzed with an average of around 41,000 reads per sample.
Of the 81 total specimens, seven samples were missing ten or more markers and were excluded from further analysis. In total 74 samples were included. The sample group FR represented the highest number of polymorphic loci with 95.65%, followed by the sample group CH 93.48% and SFR with 89.13% and finally, VIE with 67.39%, which is probably a result of the uneven sample sizes across regions. Specimens from Vienna had the highest number of private alleles (VIE = 0.383), in contrast to low levels in samples from the group CH = 0.191 and SFR = 0.021. The remaining sample group FR had 0.298. This pattern is also reflected by the level of genetic distance between the groups (GD, Table 3). Population diversity statistics were first applied including all 74 samples. Due to the haplo-diploid system, females are heterozygous, and males are supposed to be homozygote, which should decrease Ho. When females (n = 42) were separated from the males, we saw a similar picture as for all samples: the Ho was lower than He in all groups and for females overall, but not for the group VIE (Table 2).
Genetic differentiation (Pairwise F-statistics = Fst) was low between samples from the group SFR, FR and CH, in comparison to values between samples from VIE. Multiple approaches generated similar patterns of this differentiation, e.g. GD and sHua between populations (Table 3).
PCoA provided a visualization of the genetic distance patterns between individuals of the four sample groups. Samples from the groups SFR, FR and CH formed one discrete cluster. Samples from Vienna were separated from the remaining samples by relatively high distances (Fig. 2). No genetic structure was obvious in PCoA between the three sample groups SFR, FR and CH (n = 65), excluding the samples assigned to Vienna. Structure and Structure Harvester found that Delta K is highest for a K value of two, meaning that the likelihood of two different big genetic clusters is the highest (Fig. 2). The second highest likelihood is for K = 5. Generally, structure analyses were consistent with the outputs of PCoA, indicating two distinctive groups, one for the VIE samples and one for the rest, at K = 2 (Fig. 3). A higher K output indicates different clusters, most diverse in the group FR (cluster 2) followed by CH, and SFR. When cluster assignment was used as a group criterion in the PCoA, these samples grouped to some extend together. This was most clearly found for a group of six individuals from the group FR.

Genetic structure bar plots for Megachile sculpturalis based on cluster assignment probability calculations in Structure for the best K values K = 2 and K = 5 (1 = Switzerland, 2 = France, 3 = South-France and 4 = Vienna)

Principal coordinates analyses from the four sample groups. For the above panel individuals are colored according to the sample groups: Switzerland (CH), France (FR), South France (SFR) and Vienna (VIE). The panel below based on cluster assignment probability calculations in Structure for K = 4. A sample was assigned to a certain cluster if the Q score was above 50%, otherwise was considered to be admixed
Overall, pairwise FST values between populations were high between Vienna and the other groups (0.33–0.36), suggesting different populations, and low between the groups from France and Switzerland (0.04–0.07; Table 3).
In an AMOVA, 21% of variation was explained by differences between geographical groups and 59% within groups and 20% within individuals. When grouping was done according to the structure analysis, 37% of the variation was described between clusters, 45% within clusters and 18% within individuals. The latter values were derived from K = 4 because of the lower number of admixed individuals. Despite the genetic structure apparent in Structure analysis and PCoA, the sign tests for bottlenecks were significant for all regions with He > Heq as well as the one-tailed Wilcoxon tests (Table 4).
Discussion
Besides the general threats of invasive species (i.e. translocated pathogens, overcompetition), introduced bees have the potential to impact the indigenous biodiversity by altering pollination networks. Invasive pollinators can have effects on the native flora by modifying the pollen transfer patterns, which affects the seed set of indigenous plants [58]. In addition, introduced plant species lacking a suitable pollinator, can develop from so-called ‘sleeper weeds’ to expanding populations once a suitable pollinator is introduced [59]. There are already 80 bee species around the world that occur in non-native areas. The genus Megachile is most frequently represented in this group [7]. Therefore, understanding introduction events, colonization pathways and promoting factors is crucial in the effort to conserve biodiversity.
Species introduction events begin at their origins, where efficient vectors for single or multiple colonization events are required. Humans most often act as vectors and transport species via commerce and trading networks [60, 61]. With ongoing globalisation, the frequency of species introductions has increased from year to year on a global scale [12]. Human-mediated dispersal is influenced by the likelihood and frequency for a certain species to infiltrate human transport and arrive alive and able to reproduce, which depends on several factors: population size, nesting behavior, biotic interactions and phenology [62,63,64,65,66]. In the case of M. sculpturalis, only long-distance transportation can explain its invasion [26, 67]. As an example, one specimen was found previously on an airplane in Hawaii arriving from Japan, although it never established there [14]. More likely, M. sculpturalis found its way to North America and Europe on ships following maritime trading routes [26, 30]. As the species is a cavity-nesting bee, it is assumed that wood packaging material provided shelter for overwintering larvae during transportation, like for most insects translocated between continents [65, 68]. After arrival, the species was unintentionally released in its new environment, with establishment then dependent on the balance of available resources vs. the species’ requirements. Besides long-distance introdcutions to their new environment, some invaders experience range expansions over large longitudinal distances. This stepwise dispersal pattern are in most cases man-made promoting invasion success rates [10, 69]. Humans act as ecological filters by translocating species around the world, accidentally or intentionally, whereby the likelihood of human-vectored dispersal increases with human movement and contact zones, e.g. transport networks for roads and ships, trading points and urban areas [66, 70, 71].
This theoretical framework is reflected in the location where M. sculpturalis was first observed in Europe. Marseille constitutes an important maritime trading point in southern France. Ships for maritime trading were also most likely the vector delivering the species unintentionally to European islands, e.g. the most recent finding on Mallorca (Balearic island, Spain; ) [72]. Similarly, M. disjunctiformis was found near an international trading hub (Interporto) near Bologna, Italy [29]. This second invasive wild bee recognized in Europe presents very similar ecological requirements to M. sculpturalis in sharing the same natural biogeography, East-Asia, and it is even a member of the same subgenus (Callomegachile). This supports the assumption that invaders are not random samples of the global species pool [7, 66, 73, 74]. Marker development of the founder specimen of M. disjunctiformis based on the method described is currently in progress.
Introduced species often require a demographic lag phase immediately following the initial colonization event, prior to full establishment and subsequent range expansion [4]. Although M. sculpturalis was already recognized in 2008 [26], for many years the species was reported by single or very few observations restricted to certain areas. During this lag phase, it can be challenging to gather a sufficient sample size to develop and test microsatellites to investigate their genetic structure.
By implementing SSR-GBAS, fine-scale ecological questions can be answered [45], e.g. the small-scale genetic differences found within samples from France. Further, low genetic variation for the sample group of SRF, its putative point of origin, was observed according to the unbiased expected heterozygosity. Invaders typically show low genetic variation, as only fractions of the original gene pool were translocated within the founder specimen(s). Reduced heterozygosity and effective size of the gene pool result in genetic drift fostered by inter-mating of founding individuals [75,76,77]. Nevertheless, organisms with haplo-diploid systems generally have the ability to overcome extreme bottlenecks. Within several hymenopterans, haploid unfertilised eggs develop to males and diploid fertilised eggs mostly into females. In the honey bee sex is determined by the csd locus that is heterozygous in females and homozygous in males in what is described as single-locus complementary sex determination [78]. In small groups, likewise founder populations, as a result of the decreased overall heterozygosity, diploid males are more common since diploid eggs are more likely to be homozygote for this locus and develop as males. Diploid male production was found to produce less viable and sterile males driving the population towards the edge of extinction [79, 80]. However, studies have shown that these systems have the ability to overcome the genetic load by a balanced selection on the csd locus, which acts as a coping strategy circumventing the extinction vortex [80, 81]. Besides a smaller effective population size and heterozygosity levels, males are under strong purging selection of exposed deleterious alleles. Therefore, even a small number of founder individuals in haplo-diploid organisms like M. sculpturalis can successfully invade new areas [74, 77, 82,83,84]. Regardless, three out of four males sampled in Vienna in 2018 were found to be diploid (Additional file 1: Fig. S1) indicating a high percentage of diploid males in the species at least in expanding or founding populations. During future investigations more specimens morphologically assigned as males will be included to investigate this context.
A polyandrous mating system is another possible explanation for the observed invasion success despite severe founder effects. Even moderate polyandry, with more than two mates, can maintain genetic diversity and enable populations to overcome founder effects [85, 86]. But in contrast to many social hymenopterans, polyandry in solitary bees is reportedly scarce, with few exceptions (e.g. Megachilidae: Anthidium manicatum) [87], and a female was seen to mate singly, but only once (personal observation, Lanner 2020).
The striking differences detected between Vienna and all other regions represents our most unexpected finding. The genetic structure of both females and all samples, as well as the principal coordinate analysis, differentiated these two groups clearly: a western sample group (South-France, France and Switzerland) and a second, eastern group from Vienna. Vienna contains the lowest number of effective alleles and polymorphic loci. The observed heterozygosity of the sample group VIE exceeded expected heterozygosity. However, specimens from Vienna also had the highest number of private alleles. Given these findings, it seems highly unlikely that the population in Vienna spread from the 2008 origin in France. Instead, we hypothesize that they were transported independently, meaning that Europe was invaded multiple times by this species. This would explain both the discordance between eastern and western genotypes and the remarkably fast and wide-spread distribution of M. sculpturalis in Europe [23]. In addition, specimens from the remaining sample groups show genetic structure indicated by the assignment likelihood using Structure, the higher between group variation in the AMOVA (when groups are defined not geographically but according to the inferred clusters) and by the respective comparisons of pairwise distances as shown in the PCoA. The origin of these distinct genetic clusters from one colonization event is highly unlikely [88,89,90]. Although we could not assign all individuals to a location, this shows that, besides the split between a western lineage and an eastern lineage (represented by Vienna), additional lineages might have been independently introduced. Considering the recent report of the closely related M. disjunctiformis in Italy, the potential of frequent recurrent introduction of this species should be high. This might be one factor for its high invasive potential [23].
Multiple introductions can facilitate successful establishment via an increased number of individuals, a broader spatial range and increased genetic diversity [40, 41, 91, 92]. Thereby, with an increasing number of introductions and individuals, the chance of establishment and range expansion also increases. With multiple sources, the established population should also be more diverse, buffering the negative impacts of genetic drift [76, 93]. Moreover, genetically diverse invasive species benefit from higher adaptive capacity, and can react more flexibly when facing challenges in their new environment [41, 94]. In some cases, with sufficient independent introductions, genetic diversity in the invaded range may be even higher than in its indigenous range [40].
Conclusion
Microsatellite loci enabled the first insights on the demographic dynamics of this introduced wild bee. By better quantifying population genetic structuring, indirect measurements of gene flow revealed striking differences between populations within Europe. As the SSR-GBAS was applied in a relatively early invasion stage before the expected admixture between European populations took place, we were able to find tremendous genetic patterns. This east–west differentiation as well as fine-scale genetic patterns are best explained by multiple colonization events in Middle Europe. Such multiple colonization events can partly explain its invasion success in Europe. Further, it is a possible explanation for its adaptation potential to its new environment. To confidently determine the number of colonization events, future studies with larger sampling from all invaded regions of Europe will be necessary. By implementing genetic landscape modelling, we are planning to estimate the probability of migration between geographic separated populations and to test the degree of connectivity more explicitly. Furthermore, specimens from its native origin and North America will be included to produce a synthetic view of its global invasion patterns and potential. By including North American samples for genetic structure analyses, we will be able to test if European individuals are bridgehead populations, those from a previously-invaded region rather than the native range, as are often found in other hymenopterans [61]. If the primary exotic habitat contains substantial genetic diversity, it may act as a particularly potent source for another, second translocation [40, 49, 73, 95], making it crucial that we quickly better our understanding of this species ‘ invasive dynamics.
Methods
Sample collection
Specimens for the present study were gathered from commercially-circulated trap nests (in France and Switzerland) and an international initiated citizen science project aiming to investigate its distribution in Switzerland, Liechtenstein and Austria [23]. Samples provided by citizen scientists were collected on private properties. In Fig. 4, all published localities are indicated until 2018 [16, 22, 23, 26, 30,31,32,33,34,35,36,37, 96,97,98,99,100,101,102,103] as well as localities mentioned in public nature platforms and communication tools related to nature topics (i.e. iNaturalist, observations.org, naturgucker.de). For tests of genetic variation, samples were organized into four sample groups reflecting geographic origins, which were used as populations in subsequent analyses (South-France = SFR, France = FR, Switzerland = CH, Vienna = VIE; Fig. 4). Specimens from trap nests are representatives from localities close to their point of first recognition (SFR, n = 7), other parts in France (n = 40), Switzerland (n = 22) and Vienna (n = 11). Coordinates from vouchers were available from 28 specimens. Samples were included as adults (n = 20) and larvae (n = 61) collected from trap nests in the study, whereas 42 adults and larvae were assigned as females and 39 individuals were assigned as males (Additional file 2: Table S1). Genetic tests were performed for all samples and due to the haplo-diploid system of hymenopterans, we segregated female larvae by their size and position within the nests from all others (as a protandrous solitary bee, females emerge later than males and were positioned at the back of the nests).

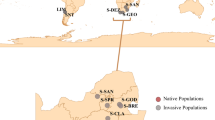
Reported European occurrences of Megachile sculpturalis until 2018. Records were derived from literature and verified citizen science programs (iNaturalist ©, info fauna CSCF, observation.org, GBIF©, naturgucker.de, insecte.org), including indicated sample groups from South-France (yellow circle), France (green circle), Switzerland (blue circle) and Vienna (brown circle). The map was created with QGIS for the present study
Marker design and Illumina amplicon sequencing
Marker development was conducted according to Tibihika [51] and Curto et al. [50]. We prepared a whole DNA library (approx. insert length 400–500 bp according to Nextera XT Library Prep., Illumina, USA) and used it for a low coverage run on the Miseq platform, targeting to produce about 300,000 to 1,000,000 paired reads. Whole DNA Library construction and sequencing was performed by Service (LMU Sequencing center) based on a specimen from the founder population in South-France.
In total, 83 samples were available for DNA isolation. To extract DNA from larval samples, approximately 1 mm of tissue was used. Tissue samples from adult bees were taken by cutting off the third leg. Bee legs were ground with 5 Zirkonium beads (Zirkonoxide beads, Type ZY-P, 2.7–3.3 mm, Sigmund Lindner, Germany) per tube in a mill at maximum force (700 rpm) for 20 min. Isolation was done using the DNA tissue Kit by Macherey–Nagel GmbH & Co KG, Germany with according to the manufacturers protocol, with few modifications as follows: In each tube, 180 μL Pre-lysis buffer T1 and 10 μL Proteinase K (10 mg/mL) was added to the ground tissue, vortexed, and incubated at 56 °C overnight on a Mixing Block MB-102 (Bioer, China). Next, 10 μL RNAse (10 mg/mL) was added and the mixture incubated at 37 °C for 10 min, and subsequently at 70 °C for 10 min with an additional 180 μL Lysis buffer B3. After lysis, the samples were centrifuged with for one minute each at 1000, 2000, 4000, 8000 rpm in a Eppendorf 5430 R centrifuge (Eppendorf, Germany). The last step was carried out at 11,000 rpm for 7 min. After centrifugation, 360 μL supernatant was mixed with 180 μL Ethanol abs. and loaded onto an EconoSpinTM columns with silica membranes (Epoch Life Science, USA) and centrifuged at 8000 rpm for 1 min. The membrane was washed with 600 μL of 80% Ethanol at 12,000 rpm for 1 min and dried afterwards via centrifugation for 2 min and at 12,000 rpm after discarding the flow through. DNA was eluted at 12,000 rpm for 1 min with 50 μL with 10 Tris (pH of 8) preheated to 65 °C and incubated at least 3 min. The resulting DNA solution was used as the template for amplification.
DNA amplification was carried out by a two-step PCR approach with four sets of primer mixes (Table 1), which were multiplexed for PCR following the procedure described in Curto et al. 50. During the first PCR, the target region is amplified using specific primer pairs. The primer pairs contain additional bases at the 5′-ends. In the second PCR reaction the adapters bind to their complementary oligonucleotides in the primer pair of the first PCR reaction [104,105,106]. Five μL Master mix (Qiagen Multiplex PCR Kit), 1 μL primer mix with a concentration of 1 μM (forward and reverse combined), 3 μL H2O and 1 μL DNA were mixed. PCR run with the following program: 95 °C for fifteen minutes, 95 °C denaturation temperature for 30 s, annealing at 55 °C for one minute and elongation at 72 °C for one minute, repeating for 30 cycles. For the second PCR, 1.5 μL from each PCR product from the four multiplex reactions were pooled per sample and cleaned by mixing it with 4.3 μL AMPure XP magnetic beads (Beckman Coulter Life Sciences, USA). Washing steps as well as the second PCR elongating the PCR strands with unique indexes for sequencing, pooling the amplicons and purification followed Curto et al. 50. The libraries were sequenced in one Illumina MiSeq run targeting around 3,000,000 paired reads of 300 bp, corresponding to an average depth of 800 per sample per marker.
Data analyses
Data analysis was done using scripts described previously and available at github.com/ mcurto/SSR‐GBS‐pipeline [51]. Sequences were processed by merging the paired reads, quality control used PEAR [107], identification of the primer and sorting the sequences by loci. Allele calls according to length and SNPs, as well as determination of heterozygote genotypes from the consensus sequences and stutter control followed the approach of Curto et al. 50. Manual control of the raw data, which is possible with these scripts, was deemed unnecessary. The pipeline results were transformed into a co-dominant matrix in GenAlex [108] format, from which it can be transformed to other population genetic software. Expected heterozygosity (He), observed heterozygosity (Ho), and number of alleles (Na), Mean Shannon Values over loci (sHua) using Log Base = 2, number of migrants, AMOVA and G-statistics were calculated with GenAlex. We also calculated Joest’s estimate of differentiation with GenAlex, as for highly polymorphic loci the Fst value depends on the allele number per locus. Pairwise Fst [109] was tested with 9999 permutations. Allelic richness was tested with the program Hp-rare [110]. Demographic changes such as population expansion or bottlenecks were evaluated using the program Bottleneck [111]. Genetic structure was tested using Structure [112] and principal coordinate analysis based on pairwise genetic distances (PCoA) between individuals as implemented in GenAlex. Structure ran for 50.000 generations of burnin followed by 100,000 generations for K values ranging from 1 to 9 with 10 replicates each. The optimal K value was accessed using the DeltaK method as implemented in Structure Harvester [113]. For a second PCoA, clusters were assigned according to the Structure results for the most frequent lineages and with a likelihood > 50%.
Finally, we tested for genetic bottlenecks with the hypothesis of bottlenecked populations showing a higher than expected heterozygosity based on Hardy–Weinberg Equilibrium (HWI) than under the mutation-drift equilibrium (He > Heq) [114, 115]. Number of loci with He > Heq were counted and tested if regions showed signs of an increase in heterozygosity above zero by calculating sign tests and carried out one-tailed Wilcoxon tests, as in Neophytou et al. (2019). Tests were carried out in the program BOTTLENECK under the infinite allele model (IAM) [116].
Availability of data and materials
The datasets generated and/or analysed during the current study are available in the NCBI repository with the following BioProject number: PRJNA680990 and submission number SUB8642671.
Change history
20 February 2021
A Correction to this paper has been published: https://doi.org/10.1186/s12862-021-01765-1
Abbreviations
- SSR-GBAS:
-
Short sequence repeat–genotyping by amplicon sequencing
- PCR:
-
Polymerase chain reaction
- SRF:
-
Sample group South-France
- FR:
-
Sample group France
- CH:
-
Sample group Switzerland
- VIE:
-
Sample group Vienna
- uHeL:
-
Unbiased expected heterozygosity
- Na:
-
Number of alleles
- He:
-
Expected heterozygosity
- Ho:
-
Observed heterozygosity
- PCoA:
-
Principal coordinates analysis
- N:
-
Number of samples
- Ne:
-
Effective number of alleles
- uHe :
-
Unbiased expected heterozygosity
- Heq :
-
Expected heterozygosity under mutation-drift equilibrium
- Fst:
-
Pairwise F statistics
- Nm:
-
Number of effective migrants
- GD:
-
Genetic distance
- sHua:
-
Mean Shannon Values over loci
References
Groom SVC, Tuiwawa MV, Stevens MI, Schwarz MP. Recent introduction of an allodapine bee into Fiji: a new model system for understanding biological invasions by pollinators. Insect Sci. 2015;22:532–40.
Colautti RI, Richardson DM. Subjectivity and flexibility in invasion terminology: too much of a good thing? Biol Invasions. 2009;11:1225–9.
Holt RD. Up against the edge: invasive species as testbeds for basic questions about evolution in heterogeneous environments. Mol Ecol. 2009;18:4347–8.
Sakai AK, Allendorf FW, Holt JS, Lodge DM, Molofsky J, With KA, et al. The population biology of invasive species. Annu Rev Ecol Syst. 2001;32:305–32.
Bellard C, Cassey P, Blackburn TM. Alien species as a driver of recent extinctions. Biol Lett. 2016;12:1–4.
Potts SG, Biesmeijer JC, Kremen C, Neumann P, Schweiger O, Kunin WE. Global pollinator declines: trends, impacts and drivers. Trends Ecol Evol. 2010;25:345–53.
Russo L. Positive and negative impacts of non-native bee species around the world. Insects. 2016;7:69.
Marzoli F, Forzan M, Bortolotti L, Pacini MI, Rodríguez‐Flores MS, Felicioli A, et al. Next generation sequencing study on RNA viruses of Vespa velutina and Apis mellifera sharing the same foraging area. Transboundary and Emerging Diseases. 2020;:tbed.13878.
Poulsen NR, Rasmussen C. Island bees: do wood nesting bees have better island dispersal abilities? Apidologie. 2020;:1–12.
Baumann JM, Walker K, Threlfall C, Williams NSG. African Carder Bee, Afranthidium (Immanthidium) repetitum (Hy- menoptera: Megachilidae) - a New Exotic Species for Victoria. The Victorian Naturalist. 2016;133:21–4.
Meimberg H, Hammond JI, Jorgensen CM, Park TW, Gerlach JD, Rice KJ, et al. Molecular evidence for an extreme genetic bottleneck during introduction of an invading grass to California. Biol Invasions. 2006;8:1355–66.
Seebens H, Blackburn TM, Dyer EE, Genovesi P, Hulme PE, Jeschke JM, et al. No saturation in the accumulation of alien species worldwide. Nature Commun. 2017;8:14435.
Michener C. The Bees of the World. 2nd ed. Baltimore: The Johns Hopkins University Press; 2007.
Mangum WA, Brooks RW. First Records of Megachile (Callomegachile) sculpturalis Smith (Hymenoptera: Megachilidae) in the Continental United States. J Kansas Entomol Soc. 1997;70:140–2.
Lee H-S, Ryu D-P. Insect Fauna of Korea. Arthropoda: Insecta: Hymenoptera: Megachilidae Leafcutter Bees. 4th edition. Korea: National Institute of Biological Resources, Ministry of Environment; 2013.
Quaranta M, Sommaruga A, Balzarini P, Felicioli A. A new species for the bee fauna of Italy: Megachile sculpturalis continues its colonization of Europe. Bull Insectol. 2014;67:287–93.
Westrich P. Faszination Wildbienen. Forschungsprojekte: Megachile sculpuralis. wildbienen.info; 2018.
Magnum WA, Sumner S. A survey of the North American range of Megachile (Callomegachile) sculpturalis, an adventive species in North America. J Kansas Entomol Soc. 2003;76:658–62.
Parys K, Tripodi A, Sampson B. The Giant Resin Bee, Megachile sculpturalis Smith: New distributional records for the Mid- and Gulf-south USA. Biodiversity Data J. 2015;3:e6733.
Hinojosa-Diaz I, Yanez-Ordonez O, Chen G, Perterson T, Engel M. The North American invasion of the Giant Resin Bee (Hymenoptera: Megachilidae). J Hymenoptera Res. 2005;14:69–77.
Hinojosa-Díaz I. The giant resin bee making its way west: First record in Kansas (Hymenoptera: Megachilidae). ZooKeys. 2008;1:67–71.
Aguado O, Hernández-Castellano C, Bassols E, Miralles M, Navarro D, Stefanescu C, et al. Megachile (Callomegachile) sculpturalis Smith, 1853 (Apoidea: Megachilidae): a new exotic species in the Iberian Peninsula, and some notes about its biology. Butlletí de laInstitució Catalana d’Història Natural. 2018;82:157–62.
Lanner J, Huchler K, Pachinger B, Sedivy C, Meimberg H. Dispersal patterns of an introduced wild bee, Megachile sculpturalis Smith, 1853 (Hymenoptera: Megachilidae) in European alpine countries. PLoS ONE. 2020;15:e0236042.
Laport RG, Minckley RL. Occupation of active Xylocopa virginica nests by the recently invasive Megachile sculpturalis in upstate New York. J Kansas Entomol Soc. 2012;85:384–6.
Geslin B, Gachet S, Deschamps-Cottin M, Flacher F, Ignace B, Knoploch C, et al. Bee hotels host a high abundance of exotic bees in an urban context. Acta Oecologica. 2020;105:103556.
Vereecken PNJ. Premières données sur la présence de l’abeille asiatique Megachile (Callomegachile) sculpturalis Smith (Hymenoptera, Megachilidae) en Europe. Osmia. 2009;3:4–6.
Villemant C, Haxaire J, Streito J-C. Premier bilan de l’invasion de Vespa velutina Lepeletier en France (Hymenoptera, Vespidae). Bulletin de la Société entomologique de France. 2006;111:535–8.
Villemant C, Barbet-Massin M, Perrard A, Muller F, Gargominy O, Jiguet F, et al. Predicting the invasion risk by the alien bee-hawking Yellow-legged hornet Vespa velutina nigrithorax across Europe and other continents with niche models. Biol Cons. 2011;144:2142–50.
Bortolotti L, Luthi F, Flaminio S, Bogo G, Sgolastra F. First record of the Asiatic bee Megachile disjunctiformis in Europe. Bull Insectol. 2018;71:143–9.
Le Féon V, Aubert M, Genoud D, Andrieu-Ponel V, Westrich P, Geslin B. Range expansion of the Asian native giant resin bee Megachile sculpturalis(Hymenoptera, Apoidea, Megachilidae) in France. Ecol Evol. 2018;00:1–9.
Bila-Dubaić J, Lanner J, Plećaš M, Raičević J, Ćetković A. Asian bee Megachile sculpturalis (Hymenoptera: Megachilidae) jumps further eastwards through Europe: confirmed establishment in Serbia after a short lag phase. in prep.
Plećaš M, Cetkovic A. Dalje širenje alohtone azijske pčele u Evropi: prvi nalaz Megachile sculpturalis na Balkanu. Serbia; 2017. p. 17–21.
Kovács T. Megachile sculpturalis Smith, 1853 in Hungary (Hymenoptera, Megachilidae). Folio Historico-Naturalia Musei Matraensis. 2015;39:73–6.
Gogala A, Zadravec B. First record of Megachile sculpturalis Smith in Slovenia (Hymenoptera: Megachilidae). Acta entomologica Slovenica. 2018;26:79–82.
Ivanov SP, Fateryga AV. First record of the invasive giant resin bee Megachile (Callomegachile) sculpturalis Smith, 1853 (Hymenoptera: Megachilidae) in the Crimea. Far Eastern Entomologist. 2019;395:7–13.
Westrich P, Knapp A, Berney I. Megachile sculpturalis Smith 1853 (Hymenoptera, Apidae), a new species for the bee fauna of Germany, now north of the Alps. Eucera. 2015;9:3–10.
Westrich P. Neues zur Ausbreitung der Mörtelbiene Megachile sculpturalis Smith 1853 (Hymenoptera: Anthophila) in Deutschland – Stand Oktober 2019. Eucera. 2020;14:12–5.
Westrich P. Forschungsprojekte - Megachile sculpturalis. wildbienen.info. 2017. https://www.wildbienen.info/forschung/projekte_17.php. Accessed 13 Oct 2020.
Garnas J, Auger-Rozenberg M-A, Roques A, Bertelsmeier C, Wingfield MJ, Saccaggi DL, et al. Complex patterns of global spread in invasive insects: eco-evolutionary and management consequences. Biol Invasions. 2016;18:935–52.
Kolbe JJ, Glor RE, Rodríguez Schettino L, Lara AC, Larson A, Losos JB. Genetic variation increases during biological invasion by a Cuban lizard. Nature. 2004;431:177–81.
Meimberg H, Milan NF, Karatassiou M, Espeland EK, McKAY JK, Rice KJ. Patterns of introduction and adaptation during the invasion of Aegilops triuncialis (Poaceae) into Californian serpentine soils: invasion of Aegilops into serpentine soils. Mol Ecol. 2010;19:5308–19.
Nagoshi RN, Fleischer S, Meagher RL. Texas is the overwintering source of Fall Armyworm in Central Pennsylvania: implications formigration into the northeastern United States. Environ Entomol. 2009;38:1546–54.
Wang IJ, Shaffer HB. Population genetic and field-ecological analyses return similar estimates of dispersal over space and time in an endangered amphibian. Evol Appl. 2017;10:630–9.
Cayuela H, Rougemont Q, Prunier JG, Moore J-S, Clobert J, Besnard A, et al. Demographic and genetic approaches to study dispersal in wild animal populations: a methodological review. Mol Ecol. 2018;27:3976–4010.
Selkoe KA, Toonen RJ. Microsatellites for ecologists: a practical guide to using and evaluating microsatellite markers. Ecol Lett. 2006;9:615–29.
Kim KS, Sappington TW. Microsatellite Data Analysis for Population Genetics. Microsatellites. 2013;271–95.
Putman AI, Carbone I. Challenges in analysis and interpretation of microsatellite data for population genetic studies. Ecol Evol. 2014;n/a-n/a.
Guichoux E, Lagache L, Wagner S, Chaumeil P, Léger P, Lepais O, et al. Current trends in microsatellite genotyping. Molecular Ecology Resources. 2011;11:591–611.
Lesieur V, Lombaert E, Guillemaud T, Courtial B, Strong W, Roques A, et al. The rapid spread of Leptoglossus occidentalis in Europe: a bridgehead invasion. J Pest Sci. 2019;92:189–200.
Curto M, Winter S, Seiter A, Schmid L, Scheicher K, Barthel LMF, et al. Application of a SSR-GBS marker system on investigation of European Hedgehog species and their hybrid zone dynamics. Ecol Evol. 2019;9:2814–32.
Tibihika PD, Curto M, Dornstauder-Schrammel E, Winter S, Alemayehu E, Waidbacher H, et al. Application of microsatellite genotyping by sequencing (SSR-GBS) to measure genetic diversity of the East African Oreochromis niloticus. Conserv Genet. 2019;20:357–72.
Šarhanová P, Pfanzelt S, Brandt R, Himmelbach A, Blattner FR. SSR-seq: Genotyping of microsatellites using next-generation sequencing reveals higher level of polymorphism as compared to traditional fragment size scoring. Ecol Evol. 2018;8:10817–33.
Vartia S, Villanueva-Cañas JL, Finarelli J, Farrell ED, Collins PC, Hughes GM, et al. A novel method of microsatellite genotyping-by-sequencing using individual combinatorial barcoding. Royal Society Open Science. 2016;3:150565.
Darby BJ, Erickson SF, Hervey SD, Ellis-Felege SN. Digital fragment analysis of short tandem repeats by high-throughput amplicon sequencing. Ecol Evol. 2016;6:4502–12.
De Barba M, Miquel C, Lobréaux S, Quenette PY, Swenson JE, Taberlet P. High-throughput microsatellite genotyping in ecology: improved accuracy, efficiency, standardization and success with low-quantity and degraded DNA. Mol Ecol Resourc. 2017;17:492–507.
Farrell ED, Carlsson JEL, Carlsson J. Next Gen Pop Gen: implementing a high-throughput approach to population genetics in boarfish ( Capros aper). Royal Soc Open Sci. 2016;3:160651.
Tibihika PD, Curto M, Alemayehu E, Waidbacher H, Masembe C, Akoll P, et al. Molecular genetic diversity and differentiation of Nile tilapia (Oreochromis niloticus, L. 1758) in East African natural and stocked populations. BMC Evol Biol. 2020;20:16.
Goulson D. Effects of introduced bees on native ecosystems. Annu Rev Ecol Evol Syst. 2003;34:1–26.
Groves RH. Are some weeds sleeping? Some concepts and reasons. Euphytica. 2006;148:111–20.
Seebens H, Essl F, Dawson W, Fuentes N, Moser D, Pergl J, et al. Global trade will accelerate plant invasions in emerging economies under climate change. Glob Change Biol. 2015;21:4128–40.
Bertelsmeier C, Keller L. Bridgehead effects and role of adaptive evolution in invasive populations. Trends Ecol Evol. 2018;33:527–34.
Carrasco LR, Mumford JD, MacLeod A, Harwood T, Grabenweger G, Leach AW, et al. Unveiling human-assisted dispersal mechanisms in invasive alien insects: Integration of spatial stochastic simulation and phenology models. Ecol Model. 2010;221:2068–75.
Wills BD, Moreau CS, Wray BD, Hoffmann BD, Suarez AV. Body size variation and caste ratios in geographically distinct populations of the invasive big-headed ant, Pheidole megacephala (Hymenoptera: Formicidae): Size Variation and Caste Ratios in Ants. Biol J Linn Soc Lond. 2014;113:423–38.
Valdovinos FS, Berlow EL, de Espanés P, Ramos-Jiliberto R, Vázquez DP, Martinez ND. Species traits and network structure predict the success and impacts of pollinator invasions. Nature Commun. 2018. https://doi.org/10.1038/s41467-018-04593-y.
Meurisse N, Rassati D, Hurley BP, Brockerhoff EG, Haack RA. Common pathways by which non-native forest insects move internationally and domestically. J Pest Sci. 2019;92:13–27.
Gippet JM, Liebhold AM, Fenn-Moltu G, Bertelsmeier C. Human-mediated dispersal in insects. Curr Opin Insect Sci. 2019;35:96–102.
Shigesada N, Kawasaki K. Chapter 17 Invasion and the range expansion of species: effects of long-distance dispersal. In: Dispersal Ecology. Blackwell Science; 2002. p. 350–73.
Liebhold AM, Brockerhoff EG, Garrett LJ, Parke JL, Britton KO. Live plant imports: the major pathway for forest insect and pathogen invasions of the US. Front Ecol Environ. 2012;10:135–43.
Prendergast KS. First records of the introduced African carder bee, Pseudoanthidium (Immanthidium) repetitum (Hymenoptera:Megachilidae), in Western Australia. Pacific Conser Biol. 2020. https://doi.org/10.1071/PC20041.
Renault D, Laparie M, McCauley SJ, Bonte D. Environmental adaptations, ecological filtering, and dispersal central to insect invasions. Annu Rev Entomol. 2018;63:345–68.
Bullock JM, Bonte D, Pufal G, da Silva CC, Chapman DS, García C, et al. Human-mediated dispersal and the rewiring of spatial networks. Trends Ecol Evol. 2018;33:958–70.
Ribas-Marquès E, Díaz-Calafat J, The invasive Asian giant resin bee Megachile sculpturalis Smith, . (Hymenoptera: Apoidea: Megachilidae), a new exotic species for the bee fauna of Mallorca. Spain: Balearic Islands; 1853. (in rev).
Hurley BP, Garnas J, Wingfield MJ, Branco M, Richardson DM, Slippers B. Increasing numbers and intercontinental spread of invasive insects on eucalypts. Biol Invasions. 2016;18:921–33.
Liebhold AM, Yamanaka T, Roques A, Augustin S, Chown SL, Brockerhoff EG, et al. Global compositional variation among native and non-native regional insect assemblages emphasizes the importance of pathways. Biol Invasions. 2016;18:893–905.
Baker AJ, Moeed A. Rapid genetic differentiation and founder effect in colonizing populations of common Mynas (acridotheres Tristis). Evolution. 1987;41:525–38.
Lockwood JL, Cassey P, Blackburn T. The role of propagule pressure in explaining species invasions. Trends Ecol Evol. 2005;20:223–8.
Auger-Rozenberg M-A, Boivin T, Magnoux E, Courtin C, Roques A, Kerdelhué C. Inferences on population history of a seed chalcid wasp: invasion success despite a severe founder effect from an unexpected source population. Mol Ecol. 2012;21:6086–103.
Cho S, Huang ZY, Green DR, Smith DR, Zhang J. Evolution of the complementary sex-determination gene of honey bees: Balancing selection and trans-species polymorphisms. Genome Res. 2006;16:1366–75.
Zayed A, Packer L. Complementary sex determination substantially increases extinction proneness of haplodiploid populations. Proc Natl Acad Sci. 2005;102:10742–6.
van Wilgenburg E, Driessen G, Beukeboom L. Single locus complementary sex determination in Hymenoptera: an “unintelligent” design? Front Zool. 2006. https://doi.org/10.1186/1742-9994-3-1.
Gloag R, Ding G, Christie JR, Buchmann G, Beekman M, Oldroyd BP. An invasive social insect overcomes genetic load at the sex locus. Nat Ecol Evol. 2017;1:0011.
Gerloff CU, Schmid-hempel P. Inbreeding depression and family variation in a social insect, Bombus terrestris (Hymenoptera. Apidae), Oikos. 2005;:67–80.
Schmid-Hempel P, Schmid-Hempel R, Brunner PC, Seeman OD, Allen GR. Invasion success of the bumblebee, Bombus terrestris, despite a drastic genetic bottleneck. Heredity. 2007;99:414–22.
Lye GC, Lepais O, Goulson D. Reconstructing demographic events from population genetic data: the introduction of bumblebees to New Zealand. Mol Ecol. 2011;20:2888–900.
Arca M, Mougel F, Guillemaud T, Dupas S, Rome Q, Perrard A, et al. Reconstructing the invasion and the demographic history of the yellow-legged hornet, Vespa velutina, in Europe. Biol Invasions. 2015;17:2357–71.
Ding G, Xu H, Oldroyd BP, Gloag RS. Extreme polyandry aids the establishment of invasive populations of a social insect. Heredity (Edinb). 2017;119:381–7.
Danforth BN, Minckley RL, Neff J. The Solitary Bees: Biology, Evolution, Conservation. 1st ed. Princeton, Woodstock: Princeton University Press; 2019.
Darling JA, Bagley MJ, Roman J, Tepolt CK, Geller JB. Genetic patterns across multiple introductions of the globally invasive crab genus Carcinus. Mol Ecol. 2008;17:4992–5007.
Ficetola GF, Bonin A, Miaud C. Population genetics reveals origin and number of founders in a biological invasion: estimating founders in biological invasions. Mol Ecol. 2008;17:773–82.
Gariepy TD, Haye T, Fraser H, Zhang J. Occurrence, genetic diversity, and potential pathways of entry of Halyomorpha halys in newly invaded areas of Canada and Switzerland. J Pest Sci. 2015;88:451–60.
Ahlroth P, Alatalo RV, Holopainen A, Kumpulainen T, Suhonen J. Founder population size and number of source populations enhance colonization success in waterstriders. Oecologia. 2003;137:617–20.
Roman J, Darling J. Paradox lost: genetic diversity and the success of aquatic invasions. Trends Ecol Evol. 2007;22:454–64.
Blackburn TM, Duncan RP. Determinants of establishment success in introduced birds. Nature. 2001;414:195–7.
Novak S, Mack R. Allozyme diversity in the apomictic vineBryonia alba (cucurbitaceae): potential consequences of multiple introductions. Am J Bot. 1995;82:1153–62.
Lombaert E, Guillemaud T, Cornuet J-M, Malausa T, Facon B, Estoup A. Bridgehead Effect in the Worldwide Invasion of the Biocontrol Harlequin Ladybird. PLoS ONE. 2010;5:e9743.
Amiet F. Die Blattschneiderbiene Megachile sculpturalis Smith, 1853 (Hymenoptera, Apidae) nun auch in der Schweiz. Entomo Helvetica. 2012;5:157–9.
Eingeschleppte D-X, Megachile AM, sculpturalis Smith, . (Hymenoptera, Apidae) erstmals nördlich der Alpen gesichtet. Entomo Helvetica. 1853;2016:153–6.
Guariento E, Lanner J, Staggl MA, Kranebitter P, Megachile sculpturalis (Smith, . (Hymenoptera: Megachilidae), the giant resin bee new for South Tyrol with a newly described plant species interaction. Gredleriana. 1853;2019:19.
Wiesbauer H. Wilde Bienen. Biologie- Lebensraumdynamik am Beispiel Österreich-Artenporträts. 1st edition. Stuttgart: Ulmer Verlag; 2017.
Zandigiacomo P, Grion M. First finding of Megachile sculpturalis Smith (Hymenoptera, Megachilidae) in Friuli Venezia Giulia (North-Eastern Italy). Gortania Botanica, Zoologica. 2017;39:37–40.
Poggi R, Tavano ML, Bonifacino M. Reperti liguri di Megachile (Callomegachile) sculpturalis Smith, 1853 (Hymenoptera, Megachilidae). Annali del museo civico di storia naturale “G Doria” Genova. 2020;9:1–6.
Ruzzier E, Menchetti M, Bortolotti L, Selis M, Monterastelli E, Forbicioni L. Updated distribution of the invasive Megachile sculpturalis (Hymenoptera: Megachilidae) in Italy and its first record on a Mediterranean island. Biodiversity Data J. 2020;8:e57783.
Ortiz-Sánchez FJ, Navarro JF, Taeger U. Megachile (Callomegachile) sculpturalis Smith, 1853, nueva especie para la fauna ibercia (Hymenoptera, Megachilidae). Boletin de la Sociedad Entomologica Aragonesa. 2018;63:259–61.
Kozarewa I, Ning Z, Quail MA, Sanders MJ, Berriman M, Turner DJ. Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of (G+C)-biased genomes. Nat Methods. 2009;6:291–5.
Meimberg H, Schachtler C, Curto M, Husemann M, Habel JC. A new amplicon based approach of whole mitogenome sequencing for phylogenetic and phylogeographic analysis: An example of East African white-eyes (Aves, Zosteropidae). Mol Phylogenet Evol. 2016;102:74–85.
van Dijk EL, Jaszczyszyn Y, Thermes C. Library preparation methods for next-generation sequencing: Tone down the bias. Exp Cell Res. 2014;322:12–20.
Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics. 2014;30:614–20.
Peakall R, Smouse PE. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research – an update. Bioinformatics. 2012;28:2537–9.
Hedrick PW. A Standardized Genetic Differentiation Measure Evolution. 2005;59:1633–8.
Kalinowski ST. hp-rare 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes. 2005;5:187–9.
Luikart G, Cornuet J-M. Empirical evaluation of a test for identifying recently bottlenecked populations from allele frequency data. Conserv Biol. 1998;12:10.
Hubisz MJ, Falush D, Stephens M, Pritchard JK. Inferring weak population structure with the assistance of sample group information. Mol Ecol Resourc. 2009;9:1322–32.
Earl DA, vonHoldt BM. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resourc. 2012;4:359–61.
Cornuet J-M, Luikart G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics. 1996;144:2001–14.
Neophytou C, Pötzelsberger E, Curto M, Meimberg H, Hasenauer H. Population bottlenecks have shaped the genetic variation of Ailanthus altissima (Mill.) Swingle in an area of early introduction. Forestry. 2019;1:1–10.
Piry S, Luikart G, Cornuet J-M. Computer note BOTTLENECK: a computer program for detecting recent reductions in the effective size using allele frequency data. J Heredity. 1999;90:502–3.
Acknowledgements
We thank all citizen scientists for providing samples: Merz M., Jung P., Steinmann P., Huber M., Cigler H., Treitner S. and Gerber S. from the Naturmuseum Wallis and the company “Wildbiene+Partner”. Donstauder-Schrammel E. provided technical assistance during lab work and Jovana Bila Dubaić with QGIS. Further, we want to thank Meyer P. for two pictures of this species.
Funding
The study was partly funded as JL is recipient of a DOC Fellowship of the Austrian Academy of Sciences at the Institute of Integrative Nature Conservation Research at the University of Natural Resources and Life Sciences. The funder provided support in the form of salaries for authors [JL], but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The specific roles of these authors are articulated in the ‘author contributions’ section.
Author information
Authors and Affiliations
Contributions
The experiment was planned and designed by HM, MC and JL. Parts of the study formed the base of a master thesis, FG. Lab work was carried out by FG and JL. JL, CS, BP, LB, BG, MCO provided and organized the samples. KH organized species distribution data. Genotypes were analyzed by HM, MC and JL. JL, MCO and HM led the writing of the manuscript with important contributions from all authors. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
No requirements or permissions are needed to sample the invasive wild bee in France, Switzerland and Austria as the invasive bee is no subject to any particular status of protection in the mentioned countries and were sampled outside nature reserves.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict ofinterests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Following publication of the original article, we were notified that the legends for Figure 2 and Figure 3 were swapped
Supplementary information
Additional file 1: Fig. S1.
Box plot of the proportion of heterozygous loci found in females from the sample groups CH (Switzerland), SRF (South-France), VIE (Vienna) and males collected in Vienna (VIE male1–4), whereas VIE males 2–4 were found to be diploid in several loci.
Additional file 1: Table S1.
Detailed information of the samples included in the present study. Adult specimens were assigned as males = m, or females = f. As protandric species, females emerge later than males and are positioned at the cavity end: L1 = first larvae positioned at the cavity back, L2 = second larvae positioned nest to L1; Ln = larvae were counted and labeled according to their nest position.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Lanner, J., Gstöttenmayer, F., Curto, M. et al. Evidence for multiple introductions of an invasive wild bee species currently under rapid range expansion in Europe. BMC Ecol Evo 21, 17 (2021). https://doi.org/10.1186/s12862-020-01729-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-020-01729-x