
Abstract
Background
The Qinghai-Tibetan Plateau (QTP) is the world’s highest and largest plateau, but the role of its uplift in the evolution of species or biotas still remains poorly known. Toad-headed lizards of the reproductively bimodal genus Phrynocephalus are a clade of agamids, with all viviparous species restricted to the QTP and adjacent regions. The eastern part of the range of the viviparous taxa is occupied by three closely related but taxonomically controversial species, P. guinanensis, P. putjatia and P. vlangalii. Here, we combined genetic (mitochondrial ND4 gene and nine microsatellite loci), morphological (11 mensural and 11 meristic variables), and ecological (nine climatic variables) data to explore possible scenarios that may explain the discordance between genetic and morphological patterns, and to test whether morphological divergence is associated with local adaptation.
Results
We found weak genetic differentiation but pronounced morphological divergence, especially between P. guinanensis and P. vlangalii. Genetically, the species boundary was not so clear between any species pair. Morphologically, the species boundary was clear between P. guinanensis and P. vlangalii but not between other two species pairs. Body size and scale characters accounted best for morphological divergence between species. Morphological divergence was related to habitat types that differ climatically.
Conclusions
Our study provides evidence for genetic and morphological divergence among the three closely related viviparous species of Phrynocephalus lizards, and supports the idea that natural selection in spatially heterogeneous environments can lead to population divergence even in the presence of gene flow. Our study supports the hypothesis that the evolutionary divergence between viviparous Phrynocephalus species was a consequence of environmental change after the uplift of the QTP.
Similar content being viewed by others


Background
Genetic divergence and speciation can occur in different parts of an ancestral species’ range and even within habitats [1]. Genetic divergence within and among species is not always accompanied by clear phenotypic (morphological, anatomical, physiological, and/or behavioral) differences due to silent mutations or phenotypic convergence [2]. However, it can give rise to significant phenotypic changes due to novel adaptations via selection that drives local adaptation [2]. Depending on its relationship to the environment, phenotypic variation may be either adaptive or non-adaptive. Adaptive phenotypic variation often occurs between populations that live in different environments and is associated with local adaptation [1, 3]. Phenotype-environment correlations have been documented in a wide variety of taxa from plant [4] to invertebrates [5, 6] and vertebrates [7,8,9], particularly with respect to the morphology-environment correlation. Functionally important morphological traits that are highly associated with reproductive success, heat exchange, water transfer and locomotion are particularly suitable to studies of speciation and population evolution [10].
Integrative analyses that combine molecular phylogeny, phylogenetic biogeography and phenotypic evolution represent a powerful approach to identify divergent clades with or without phenotypic differentiation, to detect population genetic structure, and to assess early stages of the speciation process [5, 11, 12]. Studies on lizards have showed that use of different habitats may lead to divergent selection on traits that define body size, body shape, coloration pattern and/or scale characteristics (size, number and scutellation), resulting in morphological diversification among populations or species [13,14,15]. However, to date, few studies have used an integrative approach to address morphological and species diversification of lizards in the Old World.
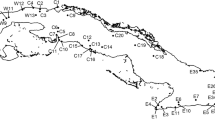
Toad-headed lizards of the reproductively bimodal genus Phrynocephalus (Agamidae) inhabit desert, arid and semiarid regions in Central and West Asia and North-Northwest China, with all viviparous species restricted to the Qinghai-Tibetan Plateau (QTP) and adjacent regions (Fig. 1) [16]. The eastern part of the range of the viviparous taxa is occupied by a group of three closely related but taxonomically controversial species, P. guinanensis, P. putjatia and P. vlangalii [17,18,19,20]. Phrynocephalus vlangalii is the most widespread species and inhabits arid and semiarid habitats in the western part of the group’s range across an altitudinal range from 2200 to 4500 m, P. putjatia is the oldest species restricted to steppe desert habitats at relatively low altitudes (2200–3300 m) around Qinghai Lake, and P. guinanensis is the most narrowly distributed species restricted to sand dunes (2700–3500 m) in the south of Qinghai Lake (Fig. 1) [17, 19,20,21,22]. The ranges of P. putjatia and P. vlangalii overlap around Qinghai Lake, but neither occurs in the range of P. guinanensis [17, 20]. Morphological data support a valid species of P. guinanensis, but genetic data do not support that distinction [17, 19]. So the currently accepted status of P. guinanensis is an ecotype of P. putjatia [19].

Map of mainland China [upper-left, showing the distribution (shaded area) of six viviparous Phrynocephalus species], map of northwestern China [lower-left, showing the distribution (shaded area) of the species studied herein], sampling localities (middle), and typical habitats used by P. guinanensis (PG, upper-right), P. putjatia (PP, middle-right), and P. vlangalii (PV, lower-right). See Table 1 for detailed information on sampling localities
However, as the ecotype hypothesis has yet to be empirically tested, a knowledge gap remains. In order to fill the gap we collected specimens from 28 localities (Fig. 1), we downloaded climatic data form WorldClim and trimmed to each sampling locality, took morphological measurements and used molecular markers (mitochondrial ND4 gene and nine microsatellite loci) to assess the structure and clustering of specimens. We then calculated distances based on all of them and compared the dissimilarity matrices. We aim to explore possible scenarios that may explain the discordance between genetic and morphological patterns, and to test whether morphological divergence among these species is associated with local adaptation. We predict that, if the ecotype hypothesis were true, the morphology should be well correlated with climate, or at least more than with genetics.
Results
Genetic polymorphism
We obtained a sequence of 684 base pairs (bp) of the mitochondrial ND4 gene, which contained nine singleton variable sites and 133 parsimony informative sites. Eight haplotypes were shared by two species (four by PG and PP, two by PP and PV, and two by PG and PV), and only one haplotype was shared by all three species (Additional file 5: Figure S1). Within individual localities, haplotype diversity varied from 0 to 0.82, and nucleotide diversity from 0 to 0.052 (Table 1). Haplotype diversity (h ± SD) was 0.92 ± 0.01 in PG, 0.94 ± 0.01 in PP, and 0.86 ± 0.02 in PV; nucleotide diversity [(π ± SD) × 103] was 7.48 ± 4.04 in PG, 23.07 ± 11.44 in PP and 64.23 ± 31.19 in PV (Table 1).
A total of 462 lizards were genotyped and scored at nine microsatellite loci. The number of alleles per locus varied from 14 to 60, with a mean of 37. The mean observed heterozygosity was 0.582, and the mean expected heterozygosity was 0.916 (Additional file 2: Table S2).
Relationships among mtDNA haplotypes
A clade of PV included individuals from PV16, PV20 and PV29; individuals from PV17 formed a clade; individuals from PV18 were admixed in branch. Because of low support values at several nodes, there was a polytomy consisting of PP, PV17, and all other groups of the admixed section, within which a clade of PP included individuals from seven localities (PP22–27) northeast of Qinghai Lake, PV17 and the remaining haplotypes of the three species. Approximately 89% (25/28) of PG haplotypes formed a PG subclade (Fig. 2). Median-joining network based on ND4 haplotypes showed similar grouping patterns to the gene tree and all clades and subclades were recovered (Additional file 5: Figure S1). The mean pairwise distance was 2.0% between PG and PP, 7.1% between PG and PV, and 7.3% between PP and PV. The mean pairwise distance within species was 0.8% for PG, 2.4% for PP, and 7.1% for PV.

A Bayesian 50% concensus phylogenetic tree based on ND4 haplotypes. Numbers under the tree branches are posterior probabilities
Population structure
Assignment tests based on nine microsatellite loci identified two distinct genetic clusters (Fig. 3a). One (red) groups individuals from all three species together, and the other (green) groups individuals from PP and PV. Two major genetic clusters were revealed in PP with one including individuals from localities northeast of Qinghai Lake, and the other including individuals mostly from localities south of the lake. Individuals of PG showed a pure genetic cluster, while individuals of PV had admixed assignment (Fig. 3b). At larger values (3–4) of K, additional clusters appeared. When STRUCTURE was run under the assumption that the data represented three separate populations (K = 3), individuals from localities south of Qinghai Lake were still assigned to their respective clusters (green), but individuals from localities south of the lake and PG individuals were assigned to two groups with moderate probability (red and blue), PV individuals were assigned to a distinct, third cluster (blue) (Fig. 3b).

Results of Bayesian model-based clustering in STRUCTURE based on nine microsatellite markers. a The plot of the mean posterior probability LnP(D) and values of ΔK against K values (number of clusters) resulting from 10 runs. b Bar plots showing Bayesian assignment probabilities from the software STRUCTURE 2.3.2 for two, three, and four clusters. Each bar represents an individual and its probabilities of being assigned to clusters. See Fig. 1 for definitions for PG, PP and PV
Morphological divergence
All examined morphological variables except tail length differed among the three species (Table 2). All 11 mensural variables differed between the sexes; only two (superciliaris and dorsal scales) of 11 meristic variables differed between the sexes (Table 2). PCAs on the body dimensions and scalation characters performed separately for each sex showed that mean scores on the first two axes differed among the three species in both sexes (Additional file 3: Table S3, Additional file 6: Figure S2). Mean scores on the first axis differed among the three species, and in both sexes mean scores were greatest in PG and smallest in PV (Table 3, Fig. 4). Differences were also found between mean scores on the second axis in females, with mean scores being greatest in PV and smallest in PG (Table 3, Fig. 4).

Positions of the species studied herein in a two-dimension space defined by the first two axes of a principal component analysis based on 22 adult morphological variables. Size effects were removed when necessary using residuals from the regressions of the corresponding variables on SVL. Mean values (±SE) for factor scores on the first two axes of the populations measured are given in the figure. Green dots and lines: P. guinanensis; red dots and lines: P. putjatia; blue dots and lines: P. vlangalii
Climatic differences
A PCA of nine climatic variables for 28 localities revealed that the first two components accounted for 80% of the variance (Additional file 4: Table S4). Mean PC scores on the first axis (F2, 25 = 10.20, P < 0.001; PGa, PPb, PVb) differed significantly among the three species, while mean PC scores on the second axis did not (F2, 25 = 1.29, P = 0.29). Overall, climatic differences were more evident between PP and PG than between any other pairs of species (Fig. 5).

Positions of the species studied herein in a two-dimension space defined by the first two axes of a principal component analysis based on nine climatic variables
Distance correlation analysis
The first morphology PC axis (M1) was positively related to the first climate PC axis (C1) in males (F1, 19 = 18.22, P < 0.001), and so was in females (F1, 19 = 18.24, P < 0.001) (Fig. 6). The single Mantel tests for the combined data matrix showed that: (1) geographic distance was significantly related to C1 and M1 in both sexes, and to genetic distance inferred from the ND4 gene; and (2) the first climate PC axis was significantly related to the first morphology PC axis in both sexes (Table 4). In both sexes M1 was significantly related to genetic distance inferred from the ND4 gene. Morphological divergence and genetic distance were spatially patterned and both were climatically dependent (Table 4). Holding C1 constant with the partial Mantel test, we found that the coefficient of a correlation between morphological divergence and genetic distance was 0.187 for males and 0.176 for females, and in both sexes the correlation was not statistically significant (Table 4). Holding geographic distance constant, we found once again that C1 significantly correlated with M1 in both sexes (Table 4).

Relationship between mean scores of the first morphology PC axis (M1) and the first climate PC axis (C1)
Discussion
Lineage separation and divergence form a temporal process in which populations may accumulate genetic, ecological, and/or morphological changes which make organisms better adapt to their environments, until eventually they are reproductively isolated and form separate species [8, 23]. Viviparous Phrynocephalus species form a monophyletic lineage that diverged from the oviparous taxa 9.78 Ma, with the most recent common ancestor of viviparous species dated to 5.04 Ma [24]. The species studied herein do not occur in syntopy (Fig. 1). Of these species, only PP has a range overlapping with oviparous congenerics, largely because it evolved earlier than did other viviparous Phrynocephalus species currently found on the QTP and at relatively low altitudes allowing oviparous reproduction [22, 24, 25]. The divergence of these species from other viviparous Phrynocephalus species on the QTP is dated to 3.79 ± 0.67 Ma, while the earliest speciation event within the complex is dated to 3.09 ± 0.61 Ma [22, 24], following the recent uplift of the QTP (3.6–0.01 Ma). It is therefore likely that environmental changes accompanied by the uplift of the QTP, imposed strong selective forces on local Phrynocephalus populations, and promoted morphological and species diversification. In this study, we found weak genetic differentiation but pronounced morphological divergence between species, and that the morphological diagnoses of species boundaries were not supported by genetic evidence. From this study we can draw the following conclusions. First, PG, PP and PV are not reciprocally monophyletic (Fig. 2). Second, morphological divergence is climatically (ecologically) rather than genetically dependent (Fig. 6). Third, PG is genetically and morphologically more similar to PP than to PV (Figs. 2, 3 and 4).
Weak genetic divergence
Genetic divergence inferred from the ND4 gene was correlated not only with the first climate PC axis (C1) but also with geographic distance (Table 4). This finding allows us to conclude that geographic distance and environmental humidity (or aridity) have major roles in driving genetic divergence between species. In the mtDNA tree, although each species is wildly polyphyletic, we can see similar genetic distance corresponding to the groups identified by morphological characters (Fig. 2). Results of the single Mantel test show a significant correlation between morphological divergence and genetic divergence inferred from the ND4, and significant climatic correlates of morphological and genetic divergence (Table 4). It is worth noting, however, that the morphological-genetic correlation disappeared when holding C1 constant (Table 4). This finding, together with the result that M1 was significantly correlated with C1 in both sexes, indicates that climatic (ecological) dissimilarity rather than genetic divergence has a key role in inducing morphological variation in this group of Phrynocephalus species.
Microsatellite-based population genetic analyses showed considerable population level admixture. Twenty-nine out of 462 individuals could be assigned to one of the two identified groups with lower than 70% probability, which supports the occurrence of historical introgressive hybridization at the nuclear genetic level. The unclear assignment between PP individuals from south of Qinghai Lake and PG might result from a lack of geographical barriers, fast and recent population expansion, relatively homogeneous habitat, or a combined effect of these factors.
We found two main monophyletic mtDNA clades that separate populations of PV16, PV20 and PV29 to the rest populations. Noble and his colleagues [20] also found two deeply diverged clades in both mtDNA and nuclear markers that were largely congruent with PV and PP; however, there are many individuals with a nuclear genome composition from one species while with mtDNA haplotype from another in ten sampling sites. The admixed mtDNA clade and the individuals sharing the same haplotypes between species confirm the occurrence of historical introgressive hybridization events between species [20]. Two major genetic clusters in PP were found, respectively corresponding to the Qinghai Lake Basin and the southeast of this basin [19]. PG is genetically very close to PP, as revealed by the fact that the mean pairwise distance between PG and PP was only 2.0% (Figs. 2 and 3). In the mtDNA tree, the lack of resolution of star-shaped clade suggests that these groups diverged quite rapidly. Low genetic diversity and clear pure genetic clustering suggest that PG divergence was a very recent event, presumably as a consequence of adapting to desert environments resulting from the uplift of the plateau. High haplotype diversity and low nucleotide diversity indicate rapid recent population expansion in PG. Additionally, the PCA of climatic variables revealed significant climatic niche separation between species (Fig. 5). This result supports the idea that spatially heterogeneous natural selection can lead to population divergence and ecological speciation even in the presence of gene flow [23].
The admixed mtDNA clade and the individuals sharing the same haplotypes between species imply the occurrence of historical introgressive hybridization events between species. Many hybrids (29/462) with an admixed nuclear genome were detected, and we can expect the presence of individuals with admixed or hybrid genomes as a consequence of hybridization events. In addition, high levels of gene flow between three species suggest that these species may suffer from hybridization. Taken together, the three lines of genetic analyses (mtDNA, STRUCTURE and microsatellite based estimations of migration) all suggest ongoing gene flow between species.
Adaptive morphological evolution
Species inhabiting different habitats may experience phenotypic divergence in a suite of traits as a result of adaptation to divergent environments [26]. Using different habitats may lead to divergent selection on a number of fitness-related morphological traits, and the morphology-environment correlation has been identified in a number of lizard species [13, 14]. For instance, on the gypsum sand dunes of White Sands, data across three different lizard species show that morphological traits are under strong and multifarious selection, and present evidence of the essential factors for divergence [27].
In this study, morphological differences are evident and show adaptive divergence in response to local environments (habitat type in particular). Similar to the pattern of variation in scale number or size reported for lizards of the genera Anolis [28] and Sceloporus [14], our data show that species in more arid environments have fewer larger (inferred from the inverse relationship between scale size and number) scales to reduce skin exposure and thus the amount of evaporative water loss (Table 2). Scale number is a heritable trait that is likely to respond to ecologically-based natural selection pressures along environmental gradients, with the complexity of scale hinges, the surface area of skin, and thus the capacity of heat and water exchange increasing with scale number [14, 27, 29]. In agreement with earlier studies of the species studied herein [17, 21], our data show that these species differ morphologically from each other, with body size and scale characters accounting best for morphological divergence between species. Of the three morphological groups, PG and PV are most completely separated, with PP in between (Table 3, Fig. 4). Morphologically, all specimens could be clearly assigned to the species recently described [17, 21]. However, morphological divergence is incongruent with genetic divergence inferred from both mitochondrial and microsatellite DNA data sets, as revealed by the fact that the three species do not form any clear lineages or genetic clusters that can be assigned to individual species already described [17, 21].
Climatic PC scores differed among the three species. Holding geographic distance constant using the partial Mantel test, we found a significant correlation between ecological divergence (climate PC1) and morphological divergence (morphology PC1) (Table 4). These findings suggest that morphological differences between species result from local adaptation. Different habitats can generate strong divergent selection and allow adaptive divergence in space even if gene flow is initially substantial [9, 11,12,13]. It is therefore likely that these species exhibit morphological divergence due to their differences in habitat preference. Morphological divergence could restrict gene flow, such as by sexual selection linked to morphological traits or coloration [23, 30]. Initial restriction on gene flow could enhance further divergence, and then generate reproductive barriers [31]. Ecological divergence acts as a legitimate isolating mechanism reducing the rate of recombination between divergent habitat types [2], and can therefore drive the evolution of additional intrinsic isolating mechanisms through reinforcement [30].
Species differentiation process
Environmental changes on the QTP, especially desertification and landcover change, have likely driven population divergence and promoted the speciation of the previously so called P. vlangalii (including PP) at 2.29 Ma. After the uplift of the QTP about 1.7 Ma, many areas on the plateau rapidly became arid and some lakes began to disappear [32]. Qinghai Lake became very large during the Middle Pleistocene, caused by the violent lift of the plateau, and then remained stable for a long period due to a drying climate in the Late Pleistocene and declination of its water level since the Holocene [33]. The spreading deserts might have forced P. guinanensis to adapt to sand dunes colonized the adjacent area, resulting in the secondary contact of these three species and potential hybridization. Subsequently, recent gene flow may result in convergence at neutral loci, whereas divergent ecology and selection maintain adaptive differences in morphology.
Conclusions
Our data show that body size and scale characters account best for morphological divergence between species in this group of Phrynocephalus lizards. Morphological divergence is related to habitat types that differ climatically. Morphologically, the species boundary is clear between P. guinanensis and P. vlangalii but not between other two pairs of species. Weak genetic differentiation and pronounced morphological divergence could have resulted from high levels of gene flow and historical introgressive hybridization between species that live in different environments. Our study supports the idea that natural selection in spatially heterogeneous environments can lead to population divergence even in the presence of gene flow. Our study provides a better understanding of genetic, morphological and ecological divergence among closely related species using different habitats, and reveals the initial adaptation to different environments.
Methods
Animal collection and treatment
All procedures described in this study were approved by the Animal Care and Use Committee of Nanjing Normal University (2011–04-008). We collected lizards between May and July 2011 from 28 localities around Qinghai Lake (Fig. 1, Table 1). Nine of these localities are occupied only by P. guinanensis (PG), 14 by P. putjatia (PP), and five by P. vlangalii (PV). We identified species based on diagnostic characters reported for these three species [17, 21, 34]. A total of 175 PG (89 females and 86 males), 195 PP (95 females and 100 males) and 106 PV (54 females and 52 males) adults were used for the collection of morphological (11 mensural and 11 meristic characters) data (Table 2). Morphological information for each species × sex × sampling locality combination with a sample size ≥5 was provided in Additional file 1: Table S1. Following the collection of morphological data, the most distal 2–3 mm of the tail tip was excised from each lizard. Lizards were then released at their site of capture. Tissue samples preserved in absolute ethanol were deposited at Nanjing Normal University.
Mitochondrial DNA amplification and sequencing
Total genomic DNA was extracted from each of 426 individuals using standard phenol-chloroform methods [35]. A partial sequence of the mitochondrial ND4 gene was amplified using forward (ND4) and reverse (Leu) primers [36]. Thermal cycling was performed with initial denaturation for 5 min at 95 °C followed by 35 cycles for 50 s at 95 °C, 45 s at 58 °C, 1 min at 72 °C and a final extension for 10 min at 72 °C. PCR products were purified and sequenced with each of the PCR primers on an ABI 377 sequencer.
Microsatellite genotyping
We amplified nine microsatellite DNA loci previously developed for P. vlangalii (PVMS32, PVMS35, PVMS38 and PVMS39) [37], or for the congeneric P. przewalskii (Phr51, Phr75, Phr78, Phr79 and Phr81) [38]. Reactions took place in a thermocycler with an initial denaturation for 5 min at 95 °C followed by 35 cycles for 45 s at 95 °C, 30 s at 57 °C, 40 s at 72 °C, and a final extension for 5 min at 72 °C [39, 40]. Fragment lengths were analyzed with the internal size marker GeneScan-500 ROX (Applied Biosystems), and scored with GeneMarker 2.2.0 (SoftGenetics, LLC, CA, USA).
Genetic polymorphism
For mitochondrial DNA data we calculated the number of segregating sites, haplotype diversity and nucleotide diversity for each population (locality) and all populations combined using DnaSP 5.10.1 [41]. Fu’s (1997) Fs [39] and Tajima’s (1989) D [40] were used to detect departures from the mutation-drift equilibrium that could indicate past demographic changes or selection.
For microsatellite DNA data, parameters such as the number of alleles per locus, average allelic richness, observed heterozygosity, expected heterozygosity, the Hardy-Weinberg equilibrium, and exact tests of linkage disequilibrium between pairs of loci for each population were calculated using ARLEQUIN 3.5 [42] and FSTAT 2.9.3.2 [43].
Phylogeography and population structure
Sequences were aligned using Clustal_X 1.81 [44] with default parameters, and then optimized by eye in MEGA 5 [45]. Mean sequence divergences among major clades were calculated using MEGA 5 and the pairwise Kimura two-parameter (K2P). Bayesian phylogenetic analyses were performed using MrBayes 3.1.2 [46]. We used two oviparous Phrynocephalus species as outgroups: P. albolineatus (GenBank Accession No. AY054002) and P. axillaris (HM235646). Three partitions (the three codon positions of the ND4 sequence; 1st: HKY + G, 2nd: HKY + G and 3rd: GTR + I) were applied to the data and models of molecular evolution were selected for each partition using MrModeltest 2.3 [47]. Four Markov Chains Monte Carlo (MCMC) chains were run for 2.0 × 107 generations. Two independent runs were performed to allow additional confirmation of the convergence of MCMC runs. Two runs from random starting trees resulted in the same topology with negligible differences in clade credibility values. We used NETWORK 4.6.1.0 [48] to generate a median-joining network for all individuals of the three species. To facilitate data presentation and interpretation, we used an initial star-contraction procedure with a star connection limit of two to reduce the data set [49].
We examined each population’s demographic changes by calculating the raggedness index of the observed mismatch distribution according to the population expansion model implemented in ARLEQUIN 3.5 [42]. We used parametric bootstrapping (1000 replicates) in ARLEQUIN 3.5 to test the goodness-of-fit of the observed mismatch distribution. Whether regional or pooled samples matched the spatial expansion model was estimated by the sum of squared deviations statistic.
We used STRUCTURE 2.3.2 [50] to identify genetically distinct groups among microsatellite genotypes with a burn-in of 5 × 107 and 5 × 108 iterations without prior population information, following the admixture model. We conducted 10 replicate runs for each specified value of K (the most likely number of populations) from 1 to 20. Individual assignment probability, LnP(D) and convergence between runs were used to assess the most likely value of K, and the most likely number of clusters was estimated according to Evanno and his colleagues [51].
Morphological analyses
We used two-way ANOVA [for snout-vent length (SVL) and all meristic variables] or ANCOVA (for other mensural variables with SVL as the covariate) to examine morphological differences between sexes and among species. Prior to parametric analyses, data were tested for the homogeneity of variances using the Bartlett’s test, and for the normality of data using the Kolmogorov-Smirnov test. Tukey’s post hoc test was performed on the traits that differed among species. We performed a principal components analysis (PCA) on 22 morphological variables to show positions of three groups (PG, PP and PV; see Fig. 1 for abbreviation definitions) on a two-dimension plane. For the variables that were related to body size, we removed the size effect by using residuals from the regressions against SVL. All statistical analyses were performed with Statistica 10.0 (Tulsa, OK, USA). Throughout this paper, descriptive statistics are presented as mean ± SE and range, and the significance level is set at P < 0.05.
Ecological divergence
In order to evaluate ecological distinctiveness among groups, we used ArcGIS 10.1 to extract values of the 19 climatic variables available in the WorldClim database (http://worldclim.org/version2) at 30 arc-seconds resolution [52]. In order to remove the effect of colinearity, we performed pairwise correlation comparisons between 19 bioclimatic variables and used nine variables that were not highly correlated (r < 0.85) in subsequent analyses. We performed a PCA on the nine climatic variables to reduce the number of predictor variables in our data set, and plotted PC scores on a two-dimension plane. We used linear regression analysis to test if morphology PC scores were correlated with climate PC scores.
Distance correlation tests
We performed a series of single and partial Mantel tests using the ‘vegan’ package 2.3–5 in R [53] with significance determined using 1000 permutations, testing the correlation between various dissimilarity matrices: (1) geographic distance, (2) climatic PC scores, (3) morphological PC scores, and (4) genetic distance based on mtDNA (genetic distance 1) and microsatellites (genetic distance 2). The climatic and morphological PC scores were converted into distance matrices in R. Only samples that had data for all variables were included in single and partial Mantel tests. Euclidian distance matrices (matrix 2 and 3) were compiled in Statistica 10.0 using the clade means calculated from PC scores for each lizard on a two-dimension plane. Pairwise Fst values [54] were estimated as the matrix of genetic distance using both mitochondrial and microsatellite data, with the procedure implemented in ARLEQUIN 3.5 [42]. Specifically, we tested the following two hypotheses after accounting for geographic distance between localities: (1) climate predicts morphological differences; and (2) climate predicts genetic divergence.
Abbreviations
- Ar:
-
Allelic richness
- BSP:
-
Bayesian skyline plot
- F is :
-
Inbreeding coefficients
- H e :
-
Expected heterozygosity
- H o :
-
Observed heterozygosity
- H s :
-
Genetic diversity
- MCMC:
-
Markov Chains Monte Carlo
- N a :
-
Total number of alleles
- PCA:
-
Principal components analysis
- PG:
-
Phrynocephalus guinanensis
- PP:
-
Phrynocephalus putjatia
- PV:
-
Phrynocephalus vlangalii
- QTP:
-
Qinghai-Tibetan Plateau
- SVL:
-
Snout-vent length
References
Schluter D. Ecology and the origin of species. Trends Ecol Evol. 2001;16:372–80.
Nosil P. Ecological speciation. Oxford: Oxford University Press; 2012.
Rundell RJ, Price TD. Adaptive radiation, nonadaptive radiation, ecological speciation and nonecological speciation. Trends Ecol Evol. 2009;24:394–9.
Ribeiro PC, Souza ML, Muller LAC, Ellis VA, Heuertz M, Lemos-Filho JP, Lovato MB. Climatic drivers of leaf traits and genetic divergence in the tree Annona crassiflora: a broad spatial survey in the Brazilian savannas. Glob Chang Biol. 2016;22:3789–803.
Ahrens D, Fabrizi S, Šipek P, Lago PK. Integrative analysis of DNA phylogeography and morphology of the European rose chafer (Cetonia aurata) to infer species taxonomy and patterns of postglacial colonisation in Europe. Mol Phylogenet Evol. 2013;69:83–94.
Puillandre N, Meyer CP, Bouchet P, Olivera BM. Genetic divergence and geographic variation in the deep-water Conus orbignyi complex (Mollusca: Conoidea). Zool Scr. 2011;40:350–63.
Faulks L, Svanbäck R, Eklöv P, Östeman Ö. Genetic and morphological divergence along the littoral-pelagic axis in two common and sympatric fishes: perch, Perca fluviatilis (Percidae) and roach, Rutilus rutilus (Cyprinidae). Biol J Linn Soc. 2015;114:929–40.
Leaché AD, Koo MS, Spencer CL, Papenfuss TJ, Fisher RN, McGuire JA. Quantifying ecological, morphological, and genetic variation to delimit species in the coast horned lizard species complex (Phrynosoma). Proc Natl Acad Sci U S A. 2009;106:12418–23.
Wilson RE, Peters JL, McCracken KG. Genetic and phenotypic divergence between lowand high-altitude populations of two recently diverged cinnamon teal subspecies. Evolution. 2013;67:170–84.
Losos JB. Lizards in an evolutionary tree: ecology and adaptive radiation of anoles. Berkeley: University of California Press; 2009.
Mathews LM, Adams L, Anderson E, Basile M, Gottardi E, Buckholt MA. Genetic and morphological evidence for substantial hidden biodiversity in a freshwater crayfish species complex. Mol Phylogenet Evol. 2008;48:126–35.
Satler JD, Carstens BC, Hedin M. Multilocus species delimitation in a complex of morphologically conserved trapdoor spiders (Mygalomorphae, Antrodiaetidae, Aliatypus). Syst Biol. 2013;62:805–23.
Muñoz MM, Crawford NG, McGreevy TJ Jr, Messana NJ, Tarvin RD, Revell LJ, Zandvliet RM, Hopwood JM, Mock E, Schneider AL, Schneider CJ. Divergence in coloration and ecological speciation in the Anolis marmoratus species complex. Mol Ecol. 2013;22:2668–82.
Oufiero CE, Gartner GE, Adolph SC, Garland T. Latitudinal and climatic variation in body size and dorsal scale counts in Sceloporus lizards: a phylogenetic perspective. Evolution. 2011;65:3590–607.
Wollenberg KC, Wang IJ, Glor RE, Losos JB. Determinism in the diversification of hispaniolan trunk-ground anoles (Anolis cytbotes species complex). Evolution. 2013;67:3175–90.
Barabanov AV, Ananjeva NB. Catalogue of the available scientific species group names for lizards of the genus Phrynocephalus Kaup, 1825 (Reptilia, Sauria, Agamidae). Zootaxa. 2007;1399:1–57.
Ji X, Wang YZ, Wang Z. New species of Phrynocephalus (Squamata, Agamidae) from Qinghai, Northwest China. Zootaxa. 2009;1988:61–8.
Jin YT, Brown RP, Liu NF. Cladogenesis and phylogeography of the lizard Phrynocephalus vlangalii (Agamidae) on the Tibetan plateau. Mol Ecol. 2008;7:1971–82.
Jin YT, Yang ZS, Brown RP, Liao PH, Liu NF. Intraspecific lineages of the lizard Phrynocephalus putjatia from the Qinghai-Tibetan plateau: impact of physical events on divergence and discordance between morphology and molecular markers. Mol Phylogenet Evol. 2014;71:288–97.
Noble D, Qi Y, Fu JZ. Species delineation using Bayesian model-based assignment tests: a case study using Chinese toad-headed agamas (genus Phrynocephalus). BMC Evol Biol. 2010;10:197.
Wang YZ, Zeng XM, Fang ZL, Wu GF, Liu ZJ, Papenfuss TJ, Macey JR. A valid species of the genus Phrynocephalus: P. putjatia and a discussion on taxonomy of Phrynocephalus hongyuanensis (Sauria: Agamidae). Acta Zootaxon Sin. 2002;27:372–83.
Guo XG, Wang YZ. Partitioned Bayesian analyses, dispersal-vicariance analysis, and the biogeography of Chinese toad-headed lizards (Agamidae: Phrynocephalus): a re-evaluation. Mol Phylogenet Evol. 2007;45:643–62.
Coyne JA, Orr HA. Speciation. Sunderland: Sinauer Associates; 2004.
Jin YT, Brown RP. Species history and divergence times of viviparous and oviparous Chinese toad-headed sand lizards (Phrynocephalus) on the Qinghai-Tibetan plateau. Mol Phylogenet Evol. 2013;68:259–68.
Wang Z, Lu HL, Ma L, Ji X. Viviparity in high-altitude Phrynocephalus lizards is adaptive because embryos cannot fully develop without maternal thermoregulation. Oecologia. 2014;174:639–49.
Langerhans RB, Layman CA, Shokrollahi A, DeWitt TJ. Predator-driven phenotypic diversification in Gambusia affinis. Evolution. 2004;58:2305–18.
Rosenblum EB, Harmon LJ. “Same same but different”: replicated ecological speciation at White Sands. Evolution. 2011;65:946–60.
Wegener JE, Gartner GEA, Losos JB. Lizard scales in an adaptive radiation: variation in scale number follows climatic and structural habitat diversity in Anolis lizards. Biol J Linn Soc. 2014;113:570–9.
Sanders KL, Malhotra A, Thorpe RS. Ecological diversification in a group of Indomalayan pitvipers (Trimeresurus): convergence in taxonomically important traits has implications for species identification. J Evol Biol. 2004;17:721–31.
Rundle HD, Nosil P. Ecological speciation. Ecol Lett. 2005;8:336–52.
Rice WR, Hostert EE. Laboratory experiments on speciation: what have we learned in 40 years? Evolution. 1993;47:1637–53.
Rieser AB, Neubauer F, Liu YJ, Ge XH. Sandstone provenance of north-western sectors of the intracontinental Cenozoic Qaidam Basin, western China: tectonic vs. climatic control. Sediment Geol. 2005;177:1–18.
Chang H, Jin ZD, An ZS. Sedimentary evidence of the uplift of the Qinghai Nanshan (the mountains south to Qinghai lake) and its implication for structural evolution of the Lake Qinghai-Gonghe basin. Geol Rev. 2009;55:49–57.
Zhao KT. Phrynocephalus Kaup, 1825. In: Zhao EM, Zhao KT, Zhou KY, editors. Fauna Sinica, Reptilia, vol. 2. Beijing: Science Press; 1999. p. 151–93.
Sambrook J, Russell DW. Molecular cloning: a laboratory manual, vol. 3. New York: Cold Spring Harbor Laboratory Press; 1989.
Arèvalo E, Davis SK, Sites JW. Mitochondrial DNA sequence divergence and phylogenetic relationships among eight chromosome races of the Sceloporus grammicus complex (Phrynosomatidae) in Central Mexico. Syst Biol. 1994;43:387–418.
Zhan AB, Fu JZ. Microsatellite DNA markers for three toad-headed lizard species (Phrynocephalus vlangalii, P. przewalskii and P. guttatus). Mol Ecol Resour. 2009;9:535–9.
Urquhart J, Bi K, Gozdzik A, Fu JZ. Isolation and characterization of microsatellite DNA loci in the toad-headed lizards, Phrynocephalus przewalskii. Mol Ecol Notes. 2005;5:928–30.
Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–25.
Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–95.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and windows. Mol Ecol Resour. 2010;10:564–7.
Goudet J. FSTAT (version 1.2): a computer program to calculate F-statistics. J Hered. 1995;86:485–6.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–82.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9.
Ronquist F, Teslenko M, van der Mark P, Ayres DJ, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42.
Nylander JAA. MrModeltest v2. Program distributed by the author. Sweden: Evolutionary Biology Centre, Uppsala University; 2004.
Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48.
Forster P, Torroni A, Renfrew C, Röhl A. Phylogenetic star contraction applied to Asian and Papuan mtDNA evolution. Mol Biol Evol. 2001;18:1864–81.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59.
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14:2611–20.
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A. Very high resolution interpolated climate surfaces for global land areas. Int J Climatol. 2005;25:1965–78.
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR. Package vegan. Community ecology package version 2; 2013. p. 3–5.
Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–70.
Acknowledgments
We thank Ce Chen, Tian-Bao Fu (deceased), Ya-Qing Liu, Fei Mao, Hong-Liang Lu and Zhu-Yuan Zhang for their invaluable assistance in the field. We thank Hong Li, Yan-Fu Qu and Zheng Wang for helpful discussion. We thank Duarte V. Gonçalves, Philipp Wagner and other two anonymous reviewers for their helpful comments.
Funding
This work was supported by grants from the National Natural Science Foundation of China (31870390, 31672277, 31470471 and 31071910) and the Priority Academic Program Development of Jiangsu Higher Education Institutions to XJ. The funders had no role in the study design, data collection, analysis and interpretation, decision to publish, and preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
XJ and CCH conceived and designed the study. CCH, YQW, LM, YJC, and XJ conducted fieldwork. CCH, YQW, LM, and YJC conducted laboratory and data analysis. CCH and XJ wrote the manuscript. All authors reviewed and contributed to editing of the manuscript and approved of its final publication.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Lizards were collected under the permit (2011–04-011) issued by Nanjing Normal University (NNU), and our experimental procedures were approved by the Animal Research Ethics Committee of NNU.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Mensural and meristic data, expressed as mean ± SE and range, collected from adults of three viviparous Phrynocephalus species. (XLS 60 kb)
Additional file 2:
Table S2. Summary of genetic variation at nine microsatellite loci. Ho: observed heterozygosity; He: expected heterozygosity; Na: total number of alleles; Hs: genetic diversity; Ar: allelic richness; Fis: inbreeding coefficients. (DOC 111 kb)
Additional file 3:
Table S3. Loading of the first two axes of a principal components (PC) analysis on 22 morphological variables for both sexes. Size effects are removed in all cases by using residuals from the regressions on SVL. Variables with the main contribution to each factor are in bold. Species with different superscripts differ significantly (Tukey’s test, α = 0.05; a > b > c). (XLSX 14 kb)
Additional file 4:
Table S4. Loading of the first two axes of a principal component (PC) analysis on nine climatic variables. (DOC 38 kb)
Additional file 5:
Figure S1. Median-joining network based on ND4 haplotypes. Red dots in network represent the corresponding mutation steps. Green: P. guinanensis; red: P. putjatia; blue: P. vlangalii. (TIF 5524 kb)
Additional file 6:
Figure S2. Position of three viviparous species of Phrynocephalus lizards in two-dimension space defined by the four principal component analysis according to the mensural and meristic data for both sexes. Green dots and lines: P. guinanensis; red dots and lines: P. putjatia; blue dots and lines: P. vlangalii. (TIF 3711 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hu, CC., Wu, YQ., Ma, L. et al. Genetic and morphological divergence among three closely related Phrynocephalus species (Agamidae). BMC Evol Biol 19, 114 (2019). https://doi.org/10.1186/s12862-019-1443-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-019-1443-y