
Abstract
Background
Long noncoding RNAs (lncRNAs) have dense linkages with a plethora of important cellular activities. lncRNAs exert functions by linking with corresponding RNA-binding proteins. Since experimental techniques to detect lncRNA-protein interactions (LPIs) are laborious and time-consuming, a few computational methods have been reported for LPI prediction. However, computation-based LPI identification methods have the following limitations: (1) Most methods were evaluated on a single dataset, and researchers may thus fail to measure their generalization ability. (2) The majority of methods were validated under cross validation on lncRNA-protein pairs, did not investigate the performance under other cross validations, especially for cross validation on independent lncRNAs and independent proteins. (3) lncRNAs and proteins have abundant biological information, how to select informative features need to further investigate.
Results
Under a hybrid framework (LPI-HyADBS) integrating feature selection based on AdaBoost, and classification models including deep neural network (DNN), extreme gradient Boost (XGBoost), and SVM with a penalty Coefficient of misclassification (C-SVM), this work focuses on finding new LPIs. First, five datasets are arranged. Each dataset contains lncRNA sequences, protein sequences, and an LPI network. Second, biological features of lncRNAs and proteins are acquired based on Pyfeat. Third, the obtained features of lncRNAs and proteins are selected based on AdaBoost and concatenated to depict each LPI sample. Fourth, DNN, XGBoost, and C-SVM are used to classify lncRNA-protein pairs based on the concatenated features. Finally, a hybrid framework is developed to integrate the classification results from the above three classifiers. LPI-HyADBS is compared to six classical LPI prediction approaches (LPI-SKF, LPI-NRLMF, Capsule-LPI, LPI-CNNCP, LPLNP, and LPBNI) on five datasets under 5-fold cross validations on lncRNAs, proteins, lncRNA-protein pairs, and independent lncRNAs and independent proteins. The results show LPI-HyADBS has the best LPI prediction performance under four different cross validations. In particular, LPI-HyADBS obtains better classification ability than other six approaches under the constructed independent dataset. Case analyses suggest that there is relevance between ZNF667-AS1 and Q15717.
Conclusions
Integrating feature selection approach based on AdaBoost, three classification techniques including DNN, XGBoost, and C-SVM, this work develops a hybrid framework to identify new linkages between lncRNAs and proteins.
Similar content being viewed by others


Introduction
Motivation
RNA-protein interactions regulate many cellular processes including splicing, polyadenylation, stability, transportation and translation [1, 2]. Recently, an increasing knowledge about RNA-binding proteins is shifting towards long non-coding RNAs (lncRNAs) [3, 4]. lncRNAs are a class of transcribed RNA molecules with the length of more than 200 nucleotides [5, 6]. The class of molecules are densely associated with a plethora of cellular activities and play vital roles in regulating gene expression [7]. The dysregulations of lncRNAs may result in various diseases, particularly cancers [8, 9]. For example, lncRNA-protein complex may influence severity degree of human pancreatic cancer phenotype. lncRNAs have been validated to closely link with poorer prognosis in lymphoma, colon cancer, and breast cancer [10].
Despite of abundant information about lncRNA-disease associations, their mechanisms still remain enigmatic. Researches found that lncRNAs exert their regulation roles through associations with the homologous RNA-binding proteins, that is, lncRNA-protein interactions (LPIs) [10,11,12]. Therefore, identification of LPIs will be beneficial to complex disease research and can thus advance diagnosis and treatment procedures [11]. Considering the time-consuming and laborious nature of laboratory methods, researchers pay more attention to computational intelligence [13]. Computation methods for LPI prediction can be roughly grouped into two categories: network-based approaches and machine learning-based approaches.
Network-based approaches took advantage of known LPIs to find unknown LPIs [14,15,16]. Li et al. [17] explored a random walk with restart algorithm (LPIHN) to propagate labels of LPIs on a heterogeneous lncRNA-protein network. Ge et al. [18] used a two-step algorithm (LPBNI) on a bipartite network. Hu et al. [19] delineated a semi-supervised lncRNA-protein linkage inference framework called LPI-ETSLP. Deng et al. [20] integrated diffusion and HeteSim features on the heterogeneous lncRNA-protein network (PLIPCOM). Zheng et al. [21] fused sequences, domains, GO terms of proteins and the STRING database and built a more informative model. Zhang et al. [22] proposed a linear neighborhood propagation method (LPLNP) for LPI mining. Zhou et al. [23] developed a similarity kernel fusion-based algorithm, LPI-SKF. Zhang et al. [24] adopted a network distance analysis technique. Network-based approaches uncovered many linkages between lncRNAs and proteins, however, they are out of the LPI prediction problem for a new lncRNA or protein.
Machine learning-based approaches including ensemble learning-based approaches [25,26,27] and deep learning-based approaches have increasingly achieved more attentions. Muppirala et al. [28] combined support vector machine (SVM) and random forest and proposed an LPI identification algorithm (RPISeq). Wang et al. [29] used an extended naive Bayes model to find hidden LPIs. Suresh et al. [30] built an SVM-based LPI inference model with sequence and structure information. Zhao et al. [31] and Liu et al. [32] proposed two neighborhood regularized matrix factorization-based methods, IRWNRLPI and LPI-NRLMF. Hu et al. [19] adopted an eigenvalue transformation-based semi-supervised LPI prediction approach.
Ensemble learning-based models demonstrated powerful performance in various association prediction area [26]. Zhang et al. [33] designed a sequence feature projection-based ensemble learning framework for predicting LPIs. Hu et al. [19] adopted an ensemble strategy for LPI discovery. Wekesa et al. [34] combined an innovative feature selection technique and an ordered boosting algorithm [35] (LPI-XGBoost) to mine new LPIs. Yi et al. [36] presented a learning distributed representation algorithm based on RNA and protein sequences.
Deep learning has been widely applied to capture unobserved LPIs and obtained remarkable performance [37]. Pan et al. [38] made use of stacked ensembling model (IPMiner) to mine underlying ncRNA-protein interaction sequential patterns. Zhang et al. [39] designed a hybrid deep learning architecture combining convolutional neural network (CNN) and recurrent neural network for LPI detection. Pan et al. [40] proposed a deep learning-based method (iDeepS) to identify RNA-binding proteins based on CNNs and a bidirectional long short term memory network (Bi-LSTM). Deng et al. [41] presented a deep neural network-based inference framework (PLIPCOM) through distributed representations of RNA sequences and structures. Fan et al. [42] trained a broad learning-based stacked ensemble classifier. Zhang et al. [43] used a CNN combing the copy-padding trick (LPI-CNNCP). Song et al. [44] and Li et al. [45] exploited capsule network-based prediction techniques (AC-caps and Capsule-LPI).
Previous studies significantly searched the interplays between lncRNAs and proteins, however, several problems still remain to solve: (1) The majority of models were measured on one unique dataset, and it is difficult to investigate their generalization performance. (2) Most algorithms were validated the prediction performance based on Cross Validation (CV) on lncRNA-protein pairs, fail to report the measurements under other CVs, for example, CVs on lncRNAs, proteins, and independent lncRNAs and independent proteins. (3) There are abundant biological information about lncRNAs and proteins. How to effectively integrate these biological characteristics to improve the prediction performance must be considered.
Study contributions
In this manuscript, a hybrid framework (LPI-HyADBS) is presented to identify LPI candidates. This framework takes advantages of diverse biological information acquisition, feature selection, and ensemble learning. The study has three main contributions:
-
1
A feature selection algorithm based on AdaBoost is proposed to select the most representative biological features from the originally acquired lncRNA and protein features.
-
2
A hybrid framework combining deep neural network (DNN), extreme gradient boost (XGBoost), and SVM with a penalty coefficient of misclassification (C-SVM) is developed to capture unobserved LPIs.
-
3
Four different CVs, especially for CV on independent lncRNAs and independent proteins, and five different LPI datasets are applied to further evaluate the generalization ability of the proposed LPI-Hybrid framework.
Materials and methods
Data preparation
In this study, we arrange five different LPI datasets. Each dataset contains lncRNA sequences, protein sequences, and an LPI network. Datasets 1, 2, and 3 were from human and were provided by Li et al. [17], Zheng et al. [21], and Zhang et al. [22], respectively. We preprocess the three datasets by removing lncRNAs and proteins involved in one associated protein (or lncRNA) or without sequence or expression information in UniProt [46], NPInter [47], NONCODE [48], and SUPERFAMILY [49]. Datasets 4 and 5 were from plant Arabidopsis thaliana and Zea mays, respectively. The two datasets were provided by Bai et al. [50]. Sequences of lncRNAs and proteins can be achieved from PlncRNADB [50]) and known LPIs can be downloaded from http://bis.zju.edu.cn/PlncRNADB/. The details are shown in Table 1.
Each LPI network is defined as a matrix Y where
Overview of LPI-HyADBS
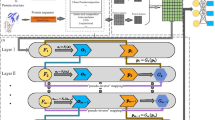
In this manuscript, we propose a hybrid framework for LPI identification (LPI-HyADBS). Figure 1 illustrates the pipeline of LPI-HyADBS after data arrangement. As shown in Fig. 1, the LPI-HyADBS method contains the following five procedures: (1) Data arrangement. Five LPI datasets are obtained and preprocessed. Each dataset contains lncRNA sequences, proteins sequences, and an LPI matrix. (2) Initial feature acquisition. lncRNA and protein features are characterized using Pyfeat [51] and concatenated to characterize each lncRNA-protein pair. (3) Feature selection. The concatenated features are reduced based on AdaBoost. (4) LPI classification. DNN, XGBoost, and C-SVM are designed to classify unknown lncRNA-protein pairs, respectively. (5) Ensemble. A hybrid framework is developed to integrate the classification results from the three classifiers.

The Pipeline of the LPI-HyADBS framework. (1) Initial feature acquisition. lncRNA and protein features are acquired with Pyfeat [51] and concatenated to depict each lncRNA-protein pair. (2) Feature selection. The concatenated features are reduced based on AdaBoost. (3) LPI classification. DNN, XGBoost, and C-SVM are designed to capture unobserved LPIs, respectively. (4) Ensemble. An ensemble framework is proposed to combine prediction results from the three classifiers
Initial feature acquisition
Pyfeat [51] is used to acquire initial numerical features of lncRNAs and proteins based on their sequences. We set k as 5 in all kgap-related features. The obtained lncRNA features include ATGC Ratio (1 feature), CumulativeSkew (2 features), diDiKGap (\(256 \times 5=1280\) features), diMonoKGap (\(64 \times 5= 320\) features), diTriKGap (\(1024 \times 5=5120\) features), gcContent (1 feature), monoDiKGap (\(64 \times 5=320\) features), monoMonoKGap (\(16 \times 5=80\) features), monoTriKGap (\(256 \times 5=1280\) features), Chou’s pseudoKNC (84 features), triMonoKGap (\(256 \times 5=1280\) features), tri-DiKGap (\(1024 \times 5=5120\) features), and zCurve (3 features). Each lncRNA is represented as a 14,891-dimensional vector based on the above features.
The obtained protein features include pseudoKNC (8420 features) and monoMonoKGap (\(400 \times 5=2000\) features). Each protein is denoted as a 10,420-dimensional vector based on the pseudoKNC and monoMonoKGap features.
Feature selection
Feature selection has been broadly applied to eliminate redundant features and plays an important role in classification. To delete irrelevant features, Gao et al. [52] presented two novel feature selection approaches, that is, linear feature selection method based on class-specific mutual information variation and multilabel feature selection method with constrained latent structure shared term [53]. The two methods obtained the best performance in corresponding application area and are the most representative feature selection techniques.
During the feature acquisition process in the above section, the obtained lncRNA and protein features are highly redundant, which severely increases computational time and affects prediction performance. AdaBoost has good generalization ability, better performance and low computational complexity, and has thus become one of the most effective classifiers [54]. In this manuscript, inspired by the two feature selection methods proposed by Gao et al. [52, 53], we utilize AdaBoost and develop a feature selection algorithm to select the most informative features for lncRNAs and proteins.
Based on initial feature acquisition, the obtained two feature vectors are first concatenated and each lncRNA-protein pair is represented as a 25, 313-dimensional vector \(\varvec{x}\). The concatenated vector is then used as the input of the feature selection algorithm to select the representative LPI features. The process can be divided into three parts.
Part I Initialization.
For given n LPI samples \(\varvec{X}=\{(x_{1}^{1}, x_{1}^{2}, \ldots , x_{1}^{m}),\ldots ,(x_{n}^{1}, x_{n}^{2}, \ldots , x_{n}^{m})\}\) where \(x_i^{j}\) denotes the jth feature of the ith sample and the labels \(\varvec{Y}=\{y_{1}, y_{2}, \ldots , y_{n}\}\), the weight coefficient for each LPI sample is initialized: \(D(x_{i}^{j})=1 / n\).
Part II Iteration and updating.
At each iteration, conducting the following six steps.
Step 1 For each feature j, a weak classifier \(h_{j}\) is trained to evaluate its importance.
Step 2 Set the corresponding hypothetical relationship between features and labels: \(h_{t}=\{x_{i}^{j} \rightarrow Y\}\).
Step 3 The error corresponding to \(D(x_{j}^{i})\) is expressed as Eq. (2):
Step 4 For one feature f with a minimum error \(\varepsilon _{t}\), delete f from initial feature set \(\varvec{x}\) and add it to the optimal target feature subset \(f_o\) by Eq. (3):
Step 5 Update the weight for each weak classifier based on the error from the best classifier \(h_{t}\) by Eq. (4):
Step 6 Update \(D(x_{i}^{j})\) by Eq. (5):
where \(N_{t}\) is a regularized constant term satisfying:
Part III Normalization of features.
We select the optimal k LPI features by iteratively updating LPI descriptions based on the performance from multiple weak classifiers. For the obtained k optimal features \(F=\{(x_{1}^{1}, x_{1}^{2}, \ldots , x_{1}^{k}),(x_{2}^{1}, x_{2}^{2}, \ldots , x_{2}^{k}), \ldots , (x_{n}^{1}, x_{n}^{2}, \ldots , x_{n}^{k})\}\), we normalize each feature:
where \(\max (x_1^j,x_2^j,\ldots ,x_n^j)\) and \(\min (x_1^j,x_2^j,\ldots ,x_n^j)\) denote the maximum and minimum values in one column, respectively.
To boost the tiny difference between a few classifiers, we used decision trees as weak classifiers based on threshold values. Through ensemble of multiple weak classifiers, the feature selection algorithm based on AdaBoost can add the most appropriate features to the optimal target feature subset.
For a given LPI dataset with n LPI examples and the selected k LPI features \({\mathcal {D}} = \{({\mathbf {x}}_{i}, y_{i})\}(|{\mathcal {D}}| = n, {\mathbf {x}}_{i} \in {\mathbb {R}}^{k}, y_{i} \in \{+1,-1\})\), we aim to classify unknown lncRNA-protein pairs based on DNN, C-SVM, and XGBoost, respectively.
Deep neural network
To build a standard neural network, researchers utilize neurons to generate real-valued activations and adjust the weights. However, training a neural network needs to take long causal chains in the phase of computation. Therefore, a new training method called layer-wise greedy learning was proposed and marked the birth of deep learning [55]. In contrast to traditional artificial intelligence methods, deep learning techniques have been progressing massively broad application in various areas. Given enough labeled data and appropriate models, the deep learning technologies can more accurately map functions [56].

Flowchart of LPI prediction method based on deep neural network
DNNs, employing deep architectures in neural networks, can effectively depict functions with higher complexity when the numbers of layers and neurons in a single layer are increased [57]. DNNs are available to more training data, can improve learning procedures, and demonstrate more computing power and better software engineering [58]. More importantly, it is relatively easy to control overfitting problems during the training of DNNs [59]. Therefore, DNNs have obtained wide applications in various complex machine learning tasks. In this manuscript, the architecture of DNN is illustrated in Fig. 2. It is divided into three main layers, that is, input layer, hidden layers, and output layer. The input layer feeds each LPI sample \(\varvec{x}\) into the network. Thus the number of neurons in the input layer is the same as one of the selected LPI features based on AdaBoost. Given an LPI sample \(\varvec{x}\), the input layer with k inputs is denoted as Eq. (8):
where \(x_i\) denotes the ith feature in an LPI sample \(\varvec{x}\).
The following layers are the hidden layers. A deep learning framework consists of more than one hidden layer. The hidden layers map each LPI sample \(\varvec{x}\) from the input layer. The input in the hidden layers are denoted as Eq. (9):
where \(w_i\) denotes the weight of \(x_i\) which are continuing updated to minimize the training errors, j indicates the number of hidden layers in the DNN, and \(b_j\) denotes the bias in the jth hidden layer.
In each hidden layer, there is an activation function. The ReLU function can solve the vanishing and exploding gradient problem, accelerate the training process, and thus demonstrates better performance. Therefore, we use ReLU as an activation function for classifying unlabeled lncRNA-protein pairs.
The output in the jth hidden layer are denoted as Eq. (10):
where \(f(h_j)=ReLU(h_j)\).
Finally, the output layer takes the outputs from the hidden layer as input and produces the output h by an activation function. In the output layer, we use sigmoid as an activation function for LPI classification. The output of DNN is represented as Eq. (11):
An LPI is classified to positive class when the output in the output layer is larger than 0.5; otherwise, the LPI is classified to negative class.
Extreme gradient boost
XGBoost has high efficiency in both balanced and imbalanced datasets. It is extremely fast due to it parallel computation ability [60]. In known five LPI datasets, there are several positive LPI samples and a large number of unknown lncRNA-protein pairs. That is, known LPI datasets are imbalanced. Considering the imbalanced characteristics of data, we utilize XGBoost to detect underlying LPIs.
Regularized learning
Gradient tree boosting techniques obtain widespread applications on the area of bioinformatics [35]. In this study, we use XGBoost to classify unlabeled lncRNA-protein pairs. For a given data set with n LPI examples and k LPI features \({\mathcal {D}} = \{({\mathbf {x}}_{i}, y_{i})\}(|{\mathcal {D}}| = n, {\mathbf {x}}_{i} \in {\mathbb {R}}^{k}, y_{i} \in \{+1,-1\})\), a tree ensemble model with M additive functions can be applied to score each unknown lncRNA-protein pair by Eq. (12).
where \(f_{j}\) denotes the jth tree with structure q and leaf weights w, \({\mathcal {F}}=\{f({\mathbf {x}})=w_{q({\mathbf {x}})}\}(q: {\mathbb {R}}^{k} \rightarrow T, w \in {\mathbb {R}}^{T})\) indicates the space composed of k regression trees, q denotes the structure of each tree mapping an LPI sample to corresponding leaf index, and T represents the number of leaves in the tree.
For an unknown lncRNA-protein pair, we utilize the decision rules obtained from q to compute its final classification result by summing up the interaction scores in the corresponding leaves obtained by w. To train the model in Eq. (12), we minimize the following objective function with regularization term by Eq. (13):
where l denotes a loss function applied to quantify the difference between the predicted label \({\hat{y}}_{i}\) and the real label \({y}_{i}\), and \(\Omega\) is used to penalize the complexity of the model. In Eq. (13), the regularization term contributes to reduce overfitting by smoothing the final learned weights. Inspired by the regularized greedy forest model proposed by [61], we set the regularization parameter to zero, and thus the objective function in Eq. (13) is transformed to a gradient tree boosting model.
Gradient tree boosting
The model in Eq. (13) is difficult be optimized by the traditional optimization algorithms in Euclidean space. Instead, an additive term is introduced to solve the model Eq. (13). Let \({\hat{y}}_{i}^{(t)}\) denote the predicted label of the ith LPI sample at the tth iteration, we add \(f_{t}\) to the model (13) to minimize the objective function defined by Eq. (14):
By Eq. (14), we gradually add \(f_{t}\) to improve the classification capability. The second-order approximation algorithm [62] can be then applied to optimize the model (14) by Eq. (15):
where \(g_{i}=\partial _{{\hat{y}}^{(t-1)}} l(y_{i}, {\hat{y}}^{(t-1)})\) and \(h_{i}=\partial _{{\hat{y}}^{(t-1)}}^{2} l(y_{i}, {\hat{y}}^{(t-1)})\) denote first-order and second-order gradient statistics on the cost function, respectively. A simplified objective function denoted by Eq. (16) can be obtained after removing the constant terms at step t:
Let \(I_{j}=\{i \mid q({\mathbf {x}}_{i})=j\}\) indicate LPI sample set in leaf j, Eq. (16) can be rewritten as Eq. (17) by expanding \(\omega\):
For a fixed structure \(q({\mathbf {x}})\), the optimal weight \(w_{j}^{*}\) in leaf j can be defined by Eq. (18):
and corresponding optimal value can be computed to evaluate the quality of a structure q by Eq. (19):
However, it is difficult to enumerate all potential tree structures. We thus use a greedy algorithm to iteratively add branches to a tree starting from a single leaf. Let \(I=I_{L} \cup I_{R}\) where \(I_L\) and \(I_R\) denote LPI sample sets on left and right nodes of a tree after splitting, respectively, we build the loss reduction by Eq. (20):
C-support vector machine
SVM is independent of feature dimensionality of data and thus avoids from “curse of dimensionality”. It has better robustness against variation of all vectors except for its support vectors [63]. Considering that the powerful classification ability of SVM, in this section, we utilize C-SVM to capture unobserved LPIs.
Given a LPI training dataset \(X=\{\varvec{x}_1, \varvec{x}_2,\ldots , \varvec{x}_n\}\) where each LPI sample \({\varvec{x}}_{i} \in R^{k}\), and a label dataset \({\varvec{y}} \in R\) where \(\varvec{y}_{i} \in \{1,-1\}\), we use an C-SVM provided by Cortes et al. [64] to classify unlabeled lncRNA-protein pairs. When C is bigger, that is, the degree of penalty on the misclassified samples is bigger, the computed accuracy is higher on the training set, however, its generalization ability may decrease, that is, the computed accuracy decreases on the test set. On the contrast, smaller C can tolerate some misclassified LPI samples on the training set and the generalization ability of the model thus is stronger. Let the misclassified LPIs are denoted as noises, C-SVM can be defined by Eq. (21):
where \(C>0\) is a penalty coefficient of misclassified LPI samples. \(\xi _i\) is a slack variable used to measure the degree of misclassification of data, \(\phi \left( {\varvec{x}}_{i}\right)\) is used to map \({\varvec{x}}_{i}\) into a higher-dimensional space and b denotes a bias. Considering the high dimensional characteristics of vector variable \({\varvec{w}}\), Cortes at al. [64] solve the model (21) based on Eq. (22):
where \({\varvec{e}}=[1, \ldots , 1]^{T}\) denotes a vector with all elements of 1, Q is an \(l \times l\) positive semidefinite matrix where \(Q_{i j} = y_{i} y_{j} K\left( {\varvec{x}}_{i}, {\varvec{x}}_{j}\right)\), and \(K\left( {\varvec{x}}_{i}, {\varvec{x}}_{j}\right) = \phi \left( {\varvec{x}}_{i}\right) ^{T} \phi \left( {\varvec{x}}_{j}\right)\) denotes a kernel function.
The optimal \({\varvec{w}}\) can be obtained based on the primal-dual relationship by the model Eq. (23):
Thus LPI classification function can be denoted by Eq. (24).
A hybrid framework
In the above sections, DNN, XGBoost, and C-SVM efficiently capture potential LPIs. However, DNNs need to train more parameters [65], XGBoost may lead to an overfitting state when hyperparameters are not appropriately tuned [59], C-SVM needs abundant labeled training data [63]. Ensemble learning demonstrates better classification ability compared to one single classifier [26]. To reduce overfitting and obtain optimal prediction performance, we integrate the three classifiers and develop a hybrid framework for LPI identification by Eq. (25):
where \(C_{DNN}\), \(C_{XGBoost}\), and \(C_{C-SVM}\) represent the classification results of an unlabeled lncRNA-protein pair from DNN, XGBoost, and C-SVM, respectively. \(\alpha\), \(\beta\), and \(\theta\) indicate the corresponding weights.
Results
Evaluation metrics
We use six evaluation metrics to measure the classification ability of our proposed LPI-HyADBS framework. That is, precision, recall, accuracy, F1-score, AUC and AUPR. For the six measurements, higher values indicate better prediction performance. The experiments are repeatedly performed 20 times and the average performance for the 20 experiments is taken as the final results.
Experimental settings
Pyfeat is applied to extract lncRNA and protein features. The parameters in Pyfeat for lncRNA initial feature acquisition are set as: kGap = 5, kTuple = 3, opti-mumDataset = 1, pseudoKNC = 1, zCurve = 1, gcContent = 1, cumulativeSkew = 1, atgcRatio = 1, monoMono = 1, monoDi = 1, monoTri = 1, diMono = 1, diDi = 1, diTri = 1, triMono = 1, triDi = 1.
The parameters in Pyfeat for protein initial feature acquisition are set as: kGap = 5, kTuple = 3, opti-mumDataset = 1, pseudoKNC = 1, zCurve = 0, gcContent = 0, cumulativeSkew = 0, atgcRatio = 0, monoMono = 1, monoDi = 0, monoTri = 0, diMono = 0, diDi = 0, diTri = 0, triMono = , triDi = 0.
To tune parameters and avoid overfitting, we perform the following experimental settings in DNN: (1) Original settings: an original neural network with one hidden layer is built, where learning rate, epoch, and batch size are originally set to 0.1, 200, and 64, respectively. The number of intermediate layers is selected based on the classification results on dataset 1. (2) Loss function: mean absolute deviation, mean square error, and binary cross-entropy loss [66] are used as loss functions to evaluate the performance of DNN, respectively. Finally, binary cross-entropy loss is selected as loss function because DNN computes better performance using binary cross-entropy loss function. (3) Optimizer: stochastic gradient descent, average stochastic gradient descent, adaptive gradient, and adaptive moment estimation [67] are used as optimizer, respectively. Finally, adaptive moment estimation is selected as optimizer due to the optimal classification ability of DNN. (4) Learning rate, epoch, and batch size: the three parameters are set to corresponding optimal values by grid research. (5) Activation function: LPI classification capability of DNN based on tanh and ReLU is compared and ReLU is selected as activation function in the hidden function where DNN calculates better performance. (6) Dropout: LPI identification accuracy of DNN does not significantly change when dropout is set as 0.2, 0.25, 0.3, and 0.5, therefore, dropout is selected as 0.25 where DNN obtains slightly better performance on dataset 1. (7) Iteration termination: during training, the iteration will be terminated when accuracy is greater than or equal to 0.99 to avoid overfitting.
In SVM, each LPI features are standardized because the selected features based on AdaBoost have multiple dimensions and scales. In addition, SVM is not sensitive to selection of kernel functions on five LPI datasets. Radial basis function (RBF), polynomial function, and sigmoid function are taken as kernel functions to measure LPI classification ability of SVM, respectively. After comparison, SVM with RBF gains slightly better prediction accuracy, therefore, RBF is selected as kernel function.
In XGBoost, parameters are originally set as defaults. Because there are many parameters in XGBoost, the parameters are combined in pairs. And the optimal parameter combination can be obtained by grid search for each group. In the training process, validation set is used to achieve the early stop mechanism of XGBoost and effectively avoid overfitting.
LPI-NRLMF, Capsule-LPI, LPI-CNNCP, LPLNP, and LPI-HyADBS obtain the best performance when they select the optimal parameter combinations by grid search. The optimal parameter combinations for the five methods are shown in Table 2. The parameters in LPI-SKF and LPBNI are set to corresponding values provided by Zhou et al. [23] and Ge et al. [18], respectively.
Four different 5-fold CVs are implemented to investigate the performance of LPI-HyADBS.
-
1.
5-fold CV on lncRNAs (\(CV_l\)): random rows in Y are hidden for testing, that is, 80% of lncRNAs are randomly screened as the train set and the remaining are applied to the test set.
-
2.
5-fold CV on proteins (\(CV_p\)): random columns in Y are hidden for testing, that is, 80% of proteins are randomly screened as the train set and the remaining are applied to the test set.
-
3.
5-fold CV on lncRNA-protein pairs (\(CV_{lp}\)): random lncRNA-protein pairs in Y are hidden for testing, that is, 80% of lncRNA-protein pairs are randomly screened as the train set and the remaining are applied to the test set.
-
4.
5-fold CV on independent lncRNAs and independent proteins (\(CV_{ind}\)) [68]: First, 20% of lncRNAs and 20% of proteins are randomly screened to construct the “node test set”. Second, the remaining nodes, which contain lncRNAs and proteins, are used as the “node train set”. Third, all edges linking a node from the node train set with a node from the node test set are removed. Finally, one classification model is trained only on edges linking two nodes within the node train set to infer edges linking two nodes within the node test set.
The above four CVs correspond to potential LPI identification for (1) new (unknown) lncRNAs without linkages with any protein, (2) new proteins without linkages with any lncRNA, (3) new lncRNA-protein pairs, and (4) the constructed independent lncRNA-independent protein pairs.
More importantly, negative samples (non-LPIs) are randomly screened from unknown lncRNA-protein pairs. The number of negative samples is set to the same as that of positive samples (LPIs).
Comparison with six state-of-the-art LPI prediction methods
We compare the proposed LPI-HyADBS framework with six classical LPI inference models, that is, LPI-SKF, LPI-NRLMF, Capsule-LPI, LPI-CNNCP, LPLNP, and LPBNI to investigate the classification ability of LPI-HyADBS. LPI-SKF, LPLNP, and LPBNI are three network-based methods, LPI-NRLMF is a logistic matrix factorization-based approach with neighbor regularization, Capsule-LPI and LPI-CNNCP are two deep learning-based models.

The ROC curves and the PR curves of seven methods under \(CV_{l}\)
Table I in Additional File 1 show the precision, recall, accuracy, F1-score, AUC and AUPR values obtained from LPI-SKF, LPI-NRLMF, Capsule-LPI, LPI-CNNCP, LPLNP, LPBNI, and LPI-HyADBS on five datasets under \(CV_l\). Figure 3 illustrates the ROC and PR curves of the seven LPI prediction methods under \(CV_l\). From Table I, we can observe that LPI-HyADBS computes the best average precision, AUC, and AUPR on five datasets under \(CV_l\). In particular, LPI-HyADBS computes the best average AUC of 0.8514, better 2.29%, 1.96%, 3.56%, 7.81%, 24.86%, and 7.12% than LPI-SKF, LPI-NRLMF, Capsule-LPI, LPI-CNNCP, LPLNP, and LPBNI, respectively. LPI-HyADBS obtains the highest average AUPR of 0.8412, outperforming 3.79%, 3.21%, 3.24%, 8.25%, 93.94%, and 45.82% compared the above six models, respectively. Although the average F1-score calculated by LPI-HyADBS is lower than one from Capsule-LPI, the difference is very small. For example, Capsule-LPI computes the average F1-score of 0.7570, while LPI-HyADBS obtains the average F1-score of 0.7535, which is only lower 0.46% than Capsule-LPI. Although LPLNP and LPBNI computes better average recall and accuracy than LPI-HyADBS, respectively, LPI-HyADBS markedly outperforms the two methods in terms of average AUC and AUPR. More importantly, AUC and AUPR can more precisely depict the prediction performance of LPI identification techniques compared to the other four evaluation metrics. LPI-HyADBS obtains better AUCs and AUPRs, and can thus accurately find proteins interacting with a new lncRNA.

The ROC curves and the PR curves of seven methods under \(CV_{p}\)
Table II in Additional File 2 illustrates the precision, recall, accuracy, F1-score, AUC and AUPR values calculated by LPI-SKF, LPI-NRLMF, Capsule-LPI, LPI-CNNCP, LPLNP, LPBNI, and LPI-HyADBS on five datasets under \(CV_p\). Figure 4 describes the ROC and PR curves of the seven LPI prediction methods under \(CV_p\). From Table II, we can find that LPI-HyADBS computes the best average precision, F1-score, AUC, and AUPR. In particular, there are only 59, 84, 27, 35, and 42 proteins on five datasets, respectively. Under \(CV_p\), only 80% samples (proteins) are used to train the model on five datasets, respectively. That is, the number of samples is relatively smaller. However, LPI-HyADBS outperforms the other six methods and significantly boosts the performance of LPI prediction. For example, the average AUC computed by LPI-HyADBS exceeds 4.73% and 11.23% than the best and the second-best methods (LPI-NRLMF and Capsule-LPI), respectively. AUPR from LPI-HyADBS is better 6.16% and 15.57% than the best two methods (LPI-NRLMF and LPI-SKF). Although LPBNI computes better accuracy, its calculated AUC and AUPR are obviously smaller than ones from LPI-HyADBS. The results suggest that LPI-HyADBS is a more robust classifier even under relatively smaller samples.

The ROC curves and the PR curves of seven methods under \(CV_{lp}\)

The ROC curves and the PR curves of seven methods under \(CV_{ind}\)
Table III in Additional File 3 depicts the precision, recall, accuracy, F1-score, AUC and AUPR values achieved from LPI-SKF, LPI-NRLMF, Capsule-LPI, LPI-CNNCP, LPLNP, LPBNI, and LPI-HyADBS on five datasets under \(CV_{lp}\). Figure 5 characterizes the ROC and PR curves of the seven LPI prediction methods under \(CV_{lp}\). Under \(CV_{lp}\), LPI-HyADBS computes the best average performance in terms of precision, recall, F1-score, and AUPR. In particular, LPI-HyADBS calculates the best F1-score on all five datasets. It still obtains the highest average F1-score of 0.8715, outperforming 19.46%, 11.50%, 2.56%, 32.46%, 97.92%, and 54.54% than LPI-SKF, LPI-NRLMF, Capsule-LPI, LPI-CNNCP, LPLNP, and LPBNI, respectively. In addition, LPI-HyADBS calculates the best AUPRs on datasets 1, 2, 4, and 5. The average AUPR is 0.9166, better 5.99%, 3.41%, 4.15%, 9.76%, 38.44%, and 41.33% than the above six approaches, respectively. The results bring out the optimal LPI classification ability of the proposed LPI-HyADBS under \(CV_{lp}\).
Table IV in Additional File 4 reveals the precision, recall, accuracy, F1-score, AUC and AUPR values acquired from LPI-SKF, LPI-NRLMF, Capsule-LPI, LPI-CNNCP, LPLNP, LPBNI, and LPI-HyADBS on five datasets under \(CV_{ind}\). Figure 6 displays the ROC and PR curves of the seven LPI prediction methods under \(CV_{ind}\). Under \(CV_{ind}\), the performance of all seven classifiers drastically declines on five datasets. However, LPI-HyADBS achieves better average precision and AUPR than the other six models even under \(CV_{ind}\). The average AUPR calculated by LPI-HyADBS is higher 7.64%, 27.97%, 9.22%, 10.25%, 83.56%, and 15.16% than the above six approaches, respectively. Although the AUC, accuracy, and recall values from LPLNP are better than LPI-HyADBS, its precision, F1-score and AUPR values are abnormally behind our method. The performance of LPI-HyADBS is much more stable compared to LPLNP. The results from \(CV_{ind}\) again demonstrate the superior LPI identification capability of LPI-HyADBS.
Performance comparison of single classifiers and hybrid framework
In this section, each single classifier is compared with the proposed LPI-HyADBS framework to measure the performance of a single classifier with a hybrid method. LPI-HyADBS is a hybrid framework composed of DNN, XGBoost, and C-SVM. Figure 7 illustrates the precisions, recalls, accuracies, F1-scores, AUCs, and AUPRs from the three classifiers and LPI-HyADBS. From Fig. 7, we can observe that LPI-HyADBS obtains better precision, F1-score, AUC, and AUPR compared to the other three approaches under all four CVs. In particular, Under \(CV_p\) and \(CV_{ind}\), LPI-HyADBS is significantly superior to the other three methods. The results suggest that LPI-HyADBS, ensemble of DNN, XGBoost, and C-SVM, can improve LPI prediction performance.

The performance of three single classifiers and a hybrid framework under four cross validations
Performance comparison of single classifiers based on deep learning
In the proposed LPI-HyADBS framework, DNN, as one single classifier based on deep learning, gains better LPI prediction performance. To investigate the performance of the other deep learning-based models on LPI discovery, we compare DNN with two classical deep learning-based methods, that is, Text-attentional CNN (Text-CNN) [69] and Bi-LSTM [40]. Text-CNN [69] focused on extracting text-related features from image components and effectively detected highly challenging text patterns. Bi-LSTM [40] revealed underlying long range dependencies between RNA binding sequences and structure motifs from RNA sequences. The two methods computed better performance on corresponding applications. Figure 8 describes the comparison results of DNN with Text-CNN and Bi-LSTM. From Fig. 8, we can observe that DNN significantly outperforms Text-CNN and Bi-LSTM in terms of recalls, accuracies, F1-scores, AUCs and AUPRs on five datasets in the vast majority of cases. The results demonstrate that DNN may be more appropriate for underlying LPI detection.

Performance comparison of single classifiers based on deep learning
Case study
In this section, we investigate the application of the proposed LPI-HyADBS method.
Finding possible proteins for a new lncRNA
RNase MRP RNA is an abundant and essential noncoding RNA. The functions of RNase MRP RNA are still incompletely understood in humans. Mutations on RNase MRP RNA genes may cause a recessively inherited developmental disorder, that is, cartilage-hair hypoplasia [70]. Cartilage-hair hypoplasia is highly human pleiotropic. It has dense associations with defective cellular immunity and short stature. More importantly, it may cause multiple cancers [71].
In human datasets 1–3, RNase MRP RNA (its name is NONHSAT130962, n5543, NONHSAT130962, respectively) interacts with 3, 13, and 10 proteins, respectively. To infer possible proteins linking with RNase MRP RNA, all its associated proteins are hidden and it is regarded as a new lncRNA. LPI-HyADBS together with the other six comparison methods are applied to infer the relevances between RNase MRP RNA and proteins. The predicted top 5 proteins linking with RNase MRP RNA are shown in Table 3. In dataset 1, P35367, O00425, Q9Y6M1, and Q9NZI8 are predicted to have high association probabilities with RNase MRP RNA. P35637 is known to interact with RNase MRP RNA in dataset 2, O00425 and Q9NZI8 have been confirmed to associate with RNase MRP RNA in dataset 3, and Q9Y6M1 is reported association information with RNase MRP RNA in datasets 2 and 3. Although interactions between Q9NZI8 and and RNase MRP RNA, and between P35367 and RNase MRP RNA are unknown in datasets 2 and 3, respectively, they have been validated in datasets 3 and 2, respectively. In summary, the predicted top 5 proteins interacting with RNase MRP RNA in one human dataset can be confirmed in the other two datasets.
Finding possible lncRNAs for a new protein
P35637 involves in multiple cellular processes. The processes include transcription regulation, DNA repair and damage response, RNA splicing and transport [72]. In neuronal cells, P35367 plays crucial roles in RNA transport, mRNA stability, dendritic spine formation and stability, and synaptic homeostasis [46].
P35637 may interact with 935, 885, and 990 lncRNAs on datasets 1–3, respectively. We hide all linkage data for P35367 and utilize the proposed LPI-HyADBS framework to infer lncRNAs related to P35367. The predicted top 5 relevant lncRNAs on three human datasets are shown in Table 4. In dataset 2, interaction between hTR and P35367 is known in dataset 3; interaction between 7SL and P35367 has been confirmed in datasets 1 and 3. However, interactions between P35367 and two lncRNAs (RPI001_1039837 and RN7SK) can not been validated. RN7SK is a small nuclear RNA involved in cellular senescence [73] and neuronal differentiation [74], it regulates macrophage polarization and innate immune responses [75]. The interaction between RN7SK and P35367 is ranked as 4 and 2 by LPI-HyADBS and LPI-NRLMF, respectively. We infer RN7SK may interact with P35367 and need further validation.
Finding possible LPIs based on observed LPIs
We score each lncRNA-protein pair on datasets 1–5. Figures 9, 10, 11, 12 and 13 illustrate the discovered top 50 lncRNA-protein pairs with the highest interaction probabilities. In the figures, black solid lines and red dotted lines represent known and unknown LPIs obtained from LPI-HyADBS, respectively. Deep sky blue diamonds represent lncRNAs. Yellow ellipses denote proteins.

The predicted top 50 LPIs on Dataset 1

The predicted top 50 LPIs on Dataset 2

The predicted top 50 LPIs on Dataset 3

The predicted top 50 LPIs on Dataset 4

The predicted top 50 LPIs on Dataset 5
On five datasets, there are separately 55,165, 74,340, 26,730, 3815, and 71,568 lncRNA-protein pairs, respectively. Unknown lncRNA-protein pairs between NONH-SAT048052(RP11-561C5.4) and Q15717, n383560(ZNF667-AS1) and Q15717, NONHSAT006085(RPI0-01_1004095) and Q9NUL5, AthlncRNA296(TCONS_0004-9605) and F4JLJ3, and ZmalncRNA1655 and B8A0M3 show the highest interaction probabilities, respectively. The five pairs are rank as 3, 10, 22, 15, and 1619 among all lncRNA-protein pairs, respectively.
ZNF667-AS1 play important roles in aberrant methylation and downregulation [76]. The lncRNA can inhibit inflammatory response [77], proliferation of cervical cancer [78], and progression of colorectal cancer [79], reduce tumor invasion and metastasis in cervical cancer [80], and promote recovery of spinal cord injury [77]. Q15717 has close relevance with embryonic stem cells differentiation. The protein interacts with ZNF385A to control nuclear export induced by CDKN2A and mediate in part the CDKN2A anti-proliferative activity. Both ZNF667-AS1 and Q15717 densely link with the inhibition of proliferation, and interaction between ZNF667-AS1 and Q15717 need experimental validation.
Discussion and conclusion
lncRNAs have dense connections with multiple physiological and pathological processes by interacting with proteins. In this manuscript, we develop an LPI inference framework combining an LPI feature selection algorithm based on AdaBoost and an ensemble learning model composed of DNN, XGBoost, and C-SVM. To observe the performance of the proposed LPI-HyADBS framework, we compare it with six representative LPI prediction approaches on five datasets under four different CVs. The six methods are LPI-SKF, LPI-NRLMF, Capsule-LPI, LPI-CNNCP, LPLNP, and LPBNI. LPI-SKF, LPLNP, and LPBNI are three representative network-based LPI prediction models. LPI-NRLMF is a classical matrix factorization-based LPI identification approach. Capsule-LPI and LPI-CNNCP are two state-of-the-art deep learning-based LPI classification models.
Under all four different CVs, LPI-HyADBS achieves better prediction performance, significantly outperforming the other six approaches. The results demonstrate the strong classification ability of LPI-HyADBS. In particular, under \(CV_{p}\), only smaller samples are applied to train the model in each round. However, LPI-HyADBS still computes the best performance, showing its robustness under small samples. More importantly, \(CV_{ind}\) is conducted on independent lncRNAs and independent proteins. Under \(CV_{ind}\), all edges connecting a node from the node train set with another node from the node test set are removed. And seven LPI identification approaches are trained only on edges connecting two nodes within the node train set to infer interactions between two nodes within the node test set. \(CV_{ind}\) reduces the overfitting problem of the classification models. LPI-HyADBS obtains better performance than the other six approaches even under \(CV_{ind}\). The results again show the robustness of LPI-HyADBS.
Capsule-LPI and LPI-CNNCP are two deep learning-based LPI prediction algorithms. From Tables I–IV in the Supplementary Materials and Figs. 3, 4, 5 and 6, we can find that LPI-HyADBS outperforms the two deep learning-based LPI inference models. More importantly, LPI-HyADBS integrates DNN, XGBoost, and C-SVM. Figure 7 illustrates that LPI-HyADBS improves LPI prediction ability compared to the three basic classifiers. The results indicate that deep ensemble-based models may more accurately find possible interplays between lncRNAs and proteins. In addition, LPI-HyADBS calculates the best performance on datasets 1, 2, 4, and 5. On dataset 3, LPI-HyADBS achieves relatively lower performance. It may be resulted in by different structures of data.
LPI-HyADBS can precisely predict the relevances between lncRNAs and proteins. It may be attributed to the following advantages. First, LPI-HyADBS fuses various biological characteristics for LPI prediction. Second, the feature selection algorithm based on AdaBoost selects the informative LPI features. Finally, an ensemble learning framework, composed of DNN, XGBoost, and C-SVM, integrates the merits of the three basic classifiers and can more effectively classify unlabeled lncRNA-protein pairs.
Although LPI-HyADBS computes the best performance on three human datasets and two plant datasets, considering other species more relative to human may more accurately evaluate LPI prediction models. Therefore, in the future, we will integrate existing data sources and construct LPI datasets for other species closer to human.
Availability of data and materials
Source codes and datasets are freely available for download at https://github.com/plhhnu/LPI-HyADBS.
Abbreviations
- LPI-HyADBS:
-
A hybrid framework integrating feature selection based on AdaBoost, and classification models including DNN, XGBoost, and SVM used to predict LPIs
- lncRNA:
-
Long noncoding RNA
- LPI:
-
lncRNA-protein interaction
- CVs:
-
Cross validations
- AdaBoost:
-
Adaptive boosting
- XGBoost:
-
eXtreme gradient boosting
- SVM:
-
Support vector machine
- C-SVM:
-
SVM with a penalty coefficient of misclassification
- DNN:
-
Deep neural network
- CNN:
-
Convolutional neural network
- Text-CNN:
-
Text-attentional CNN
- Bi-LSTM:
-
Bidirectional long short term memory network
References
Ramanathan M, Porter DF, Khavari PA. Methods to study RNA-protein interactions. Nat Methods. 2019;16(3):225–34.
Chen X, Yin J, Qu J, Huang L. Mdhgi: matrix decomposition and heterogeneous graph inference for miRNA-disease association prediction. PLoS Comput Biol. 2018;14(8):1006418.
Ferre F, Colantoni A, Helmer-Citterich M. Revealing protein-lncRNA interaction. Brief Bioinform. 2016;17(1):106–16.
Chen X, Xie D, Zhao Q, You Z-H. Micrornas and complex diseases: from experimental results to computational models. Brief Bioinform. 2019;20(2):515–39.
Suravajhala R, Gupta S, Kumar N, Suravajhala P. Deciphering lncRNA–protein interactions using docking complexes. J Biomol Struct Dyn. 2020;1–8.
Wang Y, Li H, Kuang L, Tan Y, Li X, Zhang Z, Wang L. Iclrbbn: a tool for accurate prediction of potential lncRNA disease associations. Mol Ther-Nucleic Acids. 2021;23:501–11.
Li J, Zhao H, Xuan Z, Yu J, Feng X, Liao B, Wang L. A novel approach for potential human lncRNA-disease association prediction based on local random walk. IEEE/ACM Trans Comput Biol Bioinform. 2019.
Zhang Y, Jia C, Kwoh CK. Predicting the interaction biomolecule types for lncRNA: an ensemble deep learning approach. Brief Bioinform. 2020.
Gutiérrez-Cárdenas J, Wang Z. Classification of breast cancer and breast neoplasm scenarios based on machine learning and sequence features from lncRNAs–miRNAs-diseases associations. Interdiscip Sci Comput Life Sci. 2021;1–10.
Chen X, Sun Y-Z, Guan N-N, Qu J, Huang Z-A, Zhu Z-X, Li J-Q. Computational models for lncRNA function prediction and functional similarity calculation. Brief Funct Genomics. 2019;18(1):58–82.
Philip M, Chen T, Tyagi S. A survey of current resources to study lncRNA-protein interactions. Non-Coding RNA. 2021;7(2):33.
Xu X, Hao Y, Xiong S, He Z. Comprehensive analysis of long non-coding RNA-associated competing endogenous RNA network in Duchenne muscular dystrophy. Interdiscip Sci Comput Life Sci. 2020;12(4):447–60.
Shen C, Ding Y, Tang J, Guo F. Multivariate information fusion with fast kernel learning to kernel ridge regression in predicting lncRNA-protein interactions. Front Genet. 2019;9:716.
Zhou Y-K, Shen Z-A, Yu H, Luo T, Gao Y, Du P-F. Predicting lncRNA-protein interactions with miRNAs as mediators in a heterogeneous network model. Front Genet. 2020;10:1341.
Peng L, Liu F, Yang J, Liu X, Meng Y, Deng X, Peng C, Tian G, Zhou L. Probing lncRNA-protein interactions: data repositories, models, and algorithms. Front Genet. 2020;10:1346.
Liu H, Ren G, Chen H, Liu Q, Yang Y, Zhao Q. Predicting lncRNA-miRNA interactions based on logistic matrix factorization with neighborhood regularized. Knowl-Based Syst. 2020;191:105261.
Li A, Ge M, Zhang Y, Peng C, Wang M. Predicting long noncoding RNA and protein interactions using heterogeneous network model. BioMed Res. Int. 2015;2015.
Ge M, Li A, Wang M. A bipartite network-based method for prediction of long non-coding RNA-protein interactions. Genomics Proteomics Bioinform. 2016;14(1):62–71.
Hu H, Zhu C, Ai H, Zhang L, Zhao J, Zhao Q, Liu H. Lpi-etslp: lncrna-protein interaction prediction using eigenvalue transformation-based semi-supervised link prediction. Mol BioSyst. 2017;13(9):1781–7.
Deng L, Wang J, Xiao Y, Wang Z, Liu H. Accurate prediction of protein-lncrNA interactions by diffusion and hetesim features across heterogeneous network. BMC Bioinform. 2018;19(1):1–11.
Zheng X, Wang Y, Tian K, Zhou J, Guan J, Luo L, Zhou S. Fusing multiple protein–protein similarity networks to effectively predict lncRNA-protein interactions. BMC Bioinform. 2017;18(12):11–8.
Zhang W, Qu Q, Zhang Y, Wang W. The linear neighborhood propagation method for predicting long non-coding RNA-protein interactions. Neurocomputing. 2018;273:526–34.
Zhou Y-K, Hu J, Shen Z-A, Zhang W-Y, Du P-F. LPI-SKF: predicting lncRNA-protein interactions using similarity kernel fusions. Front Genet. 2020;11:1554.
Zhang L, Yang P, Feng H, Zhao Q, Liu H. Using network distance analysis to predict lncRNA-miRNA interactions. Interdiscip Sci Comput Life Sci. 2021;13(3):535–45.
Zhang P, Meng J, Luan Y, Liu C. Plant miRNA-lncRNA interaction prediction with the ensemble of CNN and indRNN. Interdiscip Sci Comput Life Sci. 2020;12(1):82–9.
Chen X, Zhu C-C, Yin J. Ensemble of decision tree reveals potential miRNA-disease associations. PLoS Comput Biol. 2019;15(7):1007209.
Kang Q, Meng J, Shi W, Luan Y. Ensemble deep learning based on multi-level information enhancement and greedy fuzzy decision for plant miRNA–lncRNA interaction prediction. Interdiscip Sci Comput Life Sci. 2021;1–12.
Muppirala UK, Honavar VG, Dobbs D. Predicting RNA-protein interactions using only sequence information. BMC Bioinform. 2011;12(1):1–11.
Wang Y, Chen X, Liu Z-P, Huang Q, Wang Y, Xu D, Zhang X-S, Chen R, Chen L. De novo prediction of RNA-protein interactions from sequence information. Mol BioSyst. 2013;9(1):133–42.
Suresh V, Liu L, Adjeroh D, Zhou X. RPI-pred: predicting ncRNA-protein interaction using sequence and structural information. Nucleic Acids Res. 2015;43(3):1370–9.
Zhao Q, Zhang Y, Hu H, Ren G, Zhang W, Liu H. IRWNRLPI: integrating random walk and neighborhood regularized logistic matrix factorization for lncRNA-protein interaction prediction. Front Genet. 2018;9:239.
Liu H, Ren G, Hu H, Zhang L, Ai H, Zhang W, Zhao Q. LPI-NRLMF: lncRNA-protein interaction prediction by neighborhood regularized logistic matrix factorization. Oncotarget. 2017;8(61):103975.
Zhang W, Yue X, Tang G, Wu W, Huang F, Zhang X. SFPEL-LPI: sequence-based feature projection ensemble learning for predicting lncRNA-protein interactions. PLoS Comput Biol. 2018;14(12):1006616.
Wekesa JS, Meng J, Luan Y. Multi-feature fusion for deep learning to predict plant lncRNA-protein interaction. Genomics. 2020;112(5):2928–36.
Chen X, Huang L, Xie D, Zhao Q. EGBMMDA: extreme gradient boosting machine for miRNA-disease association prediction. Cell Death Dis. 2018;9(1):1–16.
Yi H-C, You Z-H, Cheng L, Zhou X, Jiang T-H, Li X, Wang Y-B. Learning distributed representations of RNA and protein sequences and its application for predicting lncRNA-protein interactions. Comput Struct Biotechnol J. 2020;18:20–6.
Pan X, Yang Y, Xia C-Q, Mirza AH, Shen H-B. Recent methodology progress of deep learning for RNA-protein interaction prediction. Wiley Interdiscip Rev RNA. 2019;10(6):1544.
Pan X, Fan Y-X, Yan J, Shen H-B. IPMINER: hidden ncRNA-protein interaction sequential pattern mining with stacked autoencoder for accurate computational prediction. BMC Genomics. 2016;17(1):1–14.
Zhang K, Xiao Y, Pan X, Yang Y. Prediction of RNA-protein interactions with distributed feature representations and a hybrid deep model. In: Proceedings of the 10th international conference on internet multimedia computing and service. 2018. pp. 1–5.
Pan X, Rijnbeek P, Yan J, Shen H-B. Prediction of RNA-protein sequence and structure binding preferences using deep convolutional and recurrent neural networks. BMC Genomics. 2018;19(1):1–11.
Deng L, Liu Y, Shi Y, Liu H. A deep neural network approach using distributed representations of RNA sequence and structure for identifying binding site of RNA-binding proteins. In: 2019 IEEE international conference on bioinformatics and biomedicine (BIBM). IEEE. 2019. pp. 12–17.
Fan X-N, Zhang S-W. LPI-BLS: Predicting lncRNA-protein interactions with a broad learning system-based stacked ensemble classifier. Neurocomputing. 2019;370:88–93.
Zhang S-W, Zhang X-X, Fan X-N, Li W-N. LPI-CNNCP: prediction of lncRNA-protein interactions by using convolutional neural network with the copy-padding trick. Anal Biochem. 2020;601:113767.
Song J, Tian S, Yu L, Xing Y, Yang Q, Duan X, Dai Q. AC-caps: attention based capsule network for predicting RBP binding sites of lncRNA. Interdiscip Sci Comput Life Sci. 2020;12(4):414–23.
Li Y, Sun H, Feng S, Zhang Q, Han S, Du W. Capsule-LPI: a lncRNA-protein interaction predicting tool based on a capsule network. BMC Bioinform. 2021;22(1):1–19.
Consortium U. Uniprot: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47(D1):506–15.
Yuan J, Wu W, Xie C, Zhao G, Zhao Y, Chen R. Npinter v2. 0: an updated database of ncRNA interactions. Nucleic Acids Res. 2014;42(D1):104–8.
Xie C, Yuan J, Li H, Li M, Zhao G, Bu D, Zhu W, Wu W, Chen R, Zhao Y. Noncodev4: exploring the world of long non-coding RNA genes. Nucleic Acids Res. 2014;42(D1):98–103.
Pandurangan AP, Stahlhacke J, Oates ME, Smithers B, Gough J. The superfamily 2.0 database: a significant proteome update and a new webserver. Nucleic Acids Res. 2019;47(D1):490–4.
Bai Y, Dai X, Ye T, Zhang P, Yan X, Gong X, Liang S, Chen M. PlncRNADB: a repository of plant lncRNAs and lncRNA-RBP protein interactions. Curr Bioinform. 2019;14(7):621–7.
Muhammod R, Ahmed S, Md Farid D, Shatabda S, Sharma A, Dehzangi A. PyFeat: a python-based effective feature generation tool for DNA, RNA and protein sequences. Bioinformatics. 2019;35(19):3831–3.
Gao W, Hu L, Zhang P. Class-specific mutual information variation for feature selection. Pattern Recogn. 2018;79:328–39.
Gao W, Li Y, Hu L. Multilabel feature selection with constrained latent structure shared term. IEEE Trans Neural Netw Learn Syst. 2021.
Vezhnevets A, Vezhnevets V. Modest adaboost-teaching adaboost to generalize better. In: Graphicon, vol. 12; 2005. pp. 987–997.
Hinton GE. Training products of experts by minimizing contrastive divergence. Neural Comput. 2002;14(8):1771–800.
Liu W, Wang Z, Liu X, Zeng N, Liu Y, Alsaadi FE. A survey of deep neural network architectures and their applications. Neurocomputing. 2017;234:11–26.
Devan P, Khare N. An efficient XGBoost–DNN-based classification model for network intrusion detection system. Neural Comput Appl. 2020;1–16.
Deng L, Hinton G, Kingsbury B. New types of deep neural network learning for speech recognition and related applications: an overview. In: 2013 IEEE international conference on acoustics, speech and signal processing. IEEE. 2013. pp. 8599–8603.
Guidi G, Pettenati MC, Miniati R, Iadanza E. Heart failure analysis dashboard for patient’s remote monitoring combining multiple artificial intelligence technologies. In: 2012 Annual international conference of the IEEE engineering in medicine and biology society. IEEE. 2012. pp. 2210–2213.
Abdu-Aljabar RD, Awad OA. A comparative analysis study of lung cancer detection and relapse prediction using XGBoost classifier. In: IOP conference series: materials science and engineering, vol. 1076. IOP Publishing. 2021. p. 012048.
Johnson R, Zhang T. Learning nonlinear functions using regularized greedy forest. IEEE Trans Pattern Anal Mach Intell. 2013;36(5):942–54.
Friedman J, Hastie T, Tibshirani R. Special invited paper. additive logistic regression: a statistical view of boosting. Ann Stat. 2000;337–374.
Li R, Bhanu B, Krawiec K. Hybrid coevolutionary algorithms vs. SVM algorithms. In: Proceedings of the 9th annual conference on genetic and evolutionary computation. 2007. pp. 456–463.
Cortes C, Vapnik V. Support-vector networks. Mach Learn. 1995;20(3):273–97.
Cheng Y, Wang D, Zhou P, Zhang T. A survey of model compression and acceleration for deep neural networks. arXiv:1710.09282. 2017.
Ates GC, Gorguluarslan RM. Two-stage convolutional encoder-decoder network to improve the performance and reliability of deep learning models for topology optimization. Struct Multidiscip Optim. 2021;63(4):1927–50.
Dogo E, Afolabi O, Nwulu N, Twala B, Aigbavboa C. A comparative analysis of gradient descent-based optimization algorithms on convolutional neural networks. In: 2018 International conference on computational techniques, electronics and mechanical systems (CTEMS). IEEE. 2018. pp. 92–99.
Park Y, Marcotte EM. Flaws in evaluation schemes for pair-input computational predictions. Nat Methods. 2012;9(12):1134.
He T, Huang W, Qiao Y, Yao J. Text-attentional convolutional neural network for scene text detection. IEEE Trans Image Process. 2016;25(6):2529–41.
Martin AN, Li Y. RNase MRP RNA and human genetic diseases. Cell Res. 2007;17(3):219–26.
Ridanpää M, van Eenennaam H, Pelin K, Chadwick R, Johnson C, Yuan B, Pruijn G, Salmela R, Rockas S, Mäkitie O, et al. Mutations in the RNA component of RNase MRP cause a pleiotropic human disease, cartilage-hair hypoplasia. Cell. 2001;104(2):195–203.
Yamaguchi A, Takanashi K. Fus interacts with nuclear matrix-associated protein safb1 as well as matrin3 to regulate splicing and ligand-mediated transcription. Sci Rep. 2016;6(1):1–14.
Musavi M, Kohram F, Abasi M, Bolandi Z, Ajoudanian M, Mohammadi-Yeganeh S, Hashemi SM, Sharifi K, Fathi HR, Ghanbarian H. Rn7sk small nuclear RNA is involved in cellular senescence. J Cell Physiol. 2019;234(8):14234–45.
Bazi Z, Bertacchi M, Abasi M, Mohammadi-Yeganeh S, Soleimani M, Wagner N, Ghanbarian H. Rn7sk small nuclear RNA is involved in neuronal differentiation. J Cell Biochem. 2018;119(4):3174–82.
Ahmad I, Valverde A, Naqvi RA, Naqvi AR. Long non-coding RNAs rn7sk and gas5 regulate macrophage polarization and innate immune responses. Front Immunol. 2020;11.
Meng W, Cui W, Zhao L, Chi W, Cao H, Wang B. Aberrant methylation and downregulation of znf667-as1 and znf667 promote the malignant progression of laryngeal squamous cell carcinoma. J Biomed Sci. 2019;26(1):1–14.
Li J, Kuang Y, Chen L, Wang J. Lncrna znf667-as1 inhibits inflammatory response and promotes recovery of spinal cord injury via suppressing jak-stat pathway. Eur Rev Med Pharmacol Sci. 2018;22(22):7614–20.
Zhao L, Li R, Han D, Zhang X, Nian G, Wu M, Feng Y, Zhang L, Sun Z. Independent prognostic factor of low-expressed lncRNA ZNF667-AS1 for cervical cancer and inhibitory function on the proliferation of cervical cancer. Eur Rev Med Pharmacol Sci. 2017;21(23):5353–60.
Zhuang L, Ding W, Ding W, Zhang Q, Xu X, Xi D. lncRNA ZNF667-AS1 (NR_036521. 1) inhibits the progression of colorectal cancer via regulating ANK2/JAK2 expression. J Cell Physiol. 2021;236(3):2178–93.
Li Y-J, Yang Z, Wang Y-Y, Wang Y. Long noncoding RNA ZNF667-AS1 reduces tumor invasion and metastasis in cervical cancer by counteracting microrna-93-3p-dependent peg3 downregulation. Mol Oncol. 2019;13(11):2375–92.
Acknowledgements
We would like to thank all authors of the cited references.
Funding
This research was funded by the National Natural Science Foundation of China (Grant 62072172, 61803151, 62172158), the Natural Science Foundation of Hunan province (Grant 2021JJ30219), scientific research project of Hunan Provincial Department of Education (20C0636), scientific research and innovation Foundation of Hunan University of Technology (Grant CX2031).
Author information
Authors and Affiliations
Contributions
Conceptualization: L-HP, QD and L-QZ; Funding acquisition: L-HP, L-QZ, J-XT; Investigation: L-HP and QD; Methodology: L-HP and QD; Project administration: L-HP, L-QZ, J-XT; Software: QD, HX; Validation: QD, X-FT; Writing original draft: L-HP; Writing-review and editing: L-HP and QD. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1
: Table SI. The performance of seven LPI prediction methods on CVl, the precision, recall, accuracy, F1-score, AUC and AUPR values obtained from LPI-SKF, LPI-NRLMF, Capsule-LPI, LPI-CNNCP, {LPLNP, LPBNI,} and LPI-HyADBS on five datasets under CVl.
Additional file 2
: Table SII The performance of seven LPI prediction methods on CVp, the precision, recall, accuracy, F1-score, AUC and AUPR values obtained from LPI-SKF, LPI-NRLMF, Capsule-LPI, LPI-CNNCP, LPLNP, LPBNI, and LPI-HyADBS on five datasets under CVp.
Additional file 3
: Table SIII. The performance of seven LPI prediction methods on CVlp, the precision, recall, accuracy, F1-score, AUC and AUPR values obtained from LPI-SKF, LPI-NRLMF, Capsule-LPI, LPI-CNNCP, LPLNP, LPBNI, and LPI-HyADBS on five datasets under CVlp.
Additional file 4
: Table SIV. The performance of seven LPI prediction methods on CVind, the precision, recall, accuracy, F1-score, AUC and AUPR values obtained from LPI-SKF, LPI-NRLMF, Capsule-LPI, LPI-CNNCP, LPLNP, LPBNI, and LPI-HyADBS on five datasets under CVind.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhou, L., Duan, Q., Tian, X. et al. LPI-HyADBS: a hybrid framework for lncRNA-protein interaction prediction integrating feature selection and classification. BMC Bioinformatics 22, 568 (2021). https://doi.org/10.1186/s12859-021-04485-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-021-04485-x