
Abstract
Background
Gene regulatory networks (GRNs) can be inferred from both gene expression data and genetic perturbations. Under different conditions, the gene data of the same gene set may be different from each other, which results in different GRNs. Detecting structural difference between GRNs under different conditions is of great significance for understanding gene functions and biological mechanisms.
Results
In this paper, we propose a Bayesian Fused algorithm to jointly infer differential structures of GRNs under two different conditions. The algorithm is developed for GRNs modeled with structural equation models (SEMs), which makes it possible to incorporate genetic perturbations into models to improve the inference accuracy, so we name it BFDSEM. Different from the naive approaches that separately infer pair-wise GRNs and identify the difference from the inferred GRNs, we first re-parameterize the two SEMs to form an integrated model that takes full advantage of the two groups of gene data, and then solve the re-parameterized model by developing a novel Bayesian fused prior following the criterion that separate GRNs and differential GRN are both sparse.
Conclusions
Computer simulations are run on synthetic data to compare BFDSEM to two state-of-the-art joint inference algorithms: FSSEM and ReDNet. The results demonstrate that the performance of BFDSEM is comparable to FSSEM, and is generally better than ReDNet. The BFDSEM algorithm is also applied to a real data set of lung cancer and adjacent normal tissues, the yielded normal GRN and differential GRN are consistent with the reported results in previous literatures. An open-source program implementing BFDSEM is freely available in Additional file 1.
Similar content being viewed by others


Background
GRNs visually reflect the gene-gene interactions, which are significant for understanding gene functions and biological activities. In the past few years, a series of inference algorithms have been proposed to reconstruct topology structures of GRNs. Some computational methods were only developed to infer GRNs from gene expression data, such as Boolean networks [1], mutual information models [2, 3], Gaussian Graphical models [4, 5], Bayesian networks [6, 7] and linear regression models [8, 9]; several other methods were also built to improve the accuracy of inference by integrating genetic perturbations with gene expression data, among which the algorithms based on SEMs [10–13] are one of the most popular approaches.
Most of the existing computational methods mainly focus on inferring GRNs under one single condition, but can not jointly identify changes in GRN structures when the condition (e.g. environments, tissues, diseases) changes. However, the differential analysis of GRNs under different conditions is also of significant importance to identify gene functions, discover biological mechanisms of different tissues and find genes related to diseases [14–16].
Intuitively, a naive approach for identifying the structure difference between GRNs under different conditions is to separately infer GRNs with existing methods and identify the difference by comparing the resulted GRNs. However, in this way, the similarity between GRNs are not taken into consideration, so the accuracy is probably unsatisfactory. Recently, several algorithms were developed to jointly infer GRNs from gene expression data under different conditions. For example, Mohan et al. [17] and Danaher et al. [18] proposed penalized algorithms based on multiple Gaussian graphical models to jointly infer GRNs under different conditions exploiting the similarities and differences between them. Wang et al. [19] developed an efficient proximal gradient algorithm to jointly infer GRNs modeled with linear regression models and identify the changes in the structure. However, the Gaussian graphical models can not identify directed networks, and the above algorithms were all developed for inferring GRNs from a single data source. Zhou and Cai [20] modeled GRNs with SEMs to integrate genetic perturbations with gene expression data, and developed a fused sparse SEM (FSSEM) algorithm to make joint inference. Ren and Zhang [21] proposed a re-parametrization based differential analysis algorithm for SEMs (ReDNet), they re-parameterized the pair-wise SEMs as one integrated SEM incorporating the averaged GRN and differential GRN, and then identified the difference GRN directly from the integrated model. Both FSSEM and ReDNet made joint differential analysis for directed GRNs modeled with SEMs, their simulation studies demonstrated that FSSEM and ReDNet significantly outperformed naive approaches based on SML [13] and 2SPLS [22], respectively.
In this paper, we propose a Bayesian Fused Differential analysis algorithm for GRNs modeled with SEMs (BFDSEM) to jointly infer pair-wise GRNs under different conditions. Following the fact that GRNs under different conditions differ slightly from each other, the sparsity of separate GRNs and differential GRN are both taken into consideration. In addition, there is no limitation on the structure of GRNs, that is, both directed acyclic GRNs (DAGs) and directed cyclic GRNs (DCGs) are supported. Computer simulations are run to compare the performance of our proposed BFDSEM to FSSEM and ReDNet, the results demonstrate that BFDSEM has somewhat consistent results with FSSEM and has better performance than ReDNet.
Preliminaries
The Bayesian Fused Lasso for linear regression models
Linear regression models can be represented as follows:
where X=[x1,x2,⋯,xp] is the design matrix including p predictor variables, y=[y1,y2,⋯,yn]T denotes the response vector and β=[β1,β2,⋯,βp]T is the coefficient vector to be estimated.
Tibshirani [28] proposed Lasso with l1 penalty on parameters to realize variable selection and parameter estimation simultaneously, the Lasso estimator of Eq. (1) is given by
In a Bayesian framework, the Lasso can be interpreted as the Bayesian posterior mode under independent Laplace priors [28, 29]. As suggested by Park and Casella in [29], the conditional Laplace prior of β can be represented as a scale mixture of normals with an exponential mixing density
where σ2 could be assign a noninformative prior or any conjugate Inverse-Gamma prior, and ψ is equivalent to the tuning parameter λ as in Eq. (2) that controls the degree of sparsity. After integrating out \(\tau _{1}^{2},\tau _{2}^{2},\cdots,\tau _{p}^{2}\), the conditional prior on β has the desired Laplace form [34]. From this relationship, the Bayesian formulation of Lasso as given in [29] is given by the following hierarchical prior.
where Np(μ,Σ) denotes p-variate normal distribution with mean vector μ and covariance matrix Σ, and Exp(ψ) denotes exponential distribution with rate parameter ψ.
A series of extensions of Lasso such as SCAD [30], Elastic net [31], fused Lasso [32], adaptive Lasso [33] were developed for various applications. The fused Lasso penalizes both the coefficients and the differences between adjacent coefficients with l1-norm, the estimator of fused Lasso for Eq. (1) is given by
Kyung et al. proposed the Bayesian interpretation of fused Lasso in [34]. The conditional prior can be expressed as
where λ1 and λ2 are tuning parameters. They provide the theoretical asymptotic limiting distribution and a degrees of freedom estimator. Following the way of Bayesian Lasso, this prior can be represented as the following hierarchical form:
where \(\tau _{1}^{2},\tau _{2}^{2},\cdots,\tau _{p}^{2}, \omega _{1}^{2},\omega _{2}^{2},\cdots,\omega _{p-1}^{2}\) are mutually independent, and Σβ is a tridiagonal matrix with main diagonal=\(\left \{\frac {1}{\tau _{j}^{2}}+\frac {1}{\omega _{j-1}^{2}}+\frac {1}{\omega _{j}^{2}},j=1,2,\cdots,p\right \}\) and off diagonal \(\left \{-\frac {1}{\omega _{k}^{2}},k=1,2,\cdots,p-1\right \}\), \(\frac {1}{\omega _{0}^{2}}\) and \(\frac {1}{\omega _{p}^{2}}\) are defined as 0 for convenience.
As suggested by Park and Casella [29], there are two common approaches to estimate the tuning parameters: one is to estimate them through marginal likelihood implemented with an EM/Gibbs algorithm [36]; another way is to assign a Gamma hyperprior on each tuning parameter, and put them into the hierarchical models to estimate it with a Gibbs sampler.
GRNs modeled with SEMs
As in [10–13], genetic perturbations can be incorporated into SEMs to infer GRNs and result in better performance. The perturbations could be various, such as the expression Quantitative Trait Loci (eQTLs) and the Copy Number Variants (CNVs). In this paper we consider the variations observed on the cis-eQTLs. Suppose we have expression levels of p genes and genotypes of q cis-eQTLs observed from n individuals. Let Y=[y1,y2,⋯,yp] be an n×p gene expression matrix, X=[x1,x2,⋯,xq] be an n×q cis-eQTL matrix. Then the GRN can be modeled with the following SEM:
where the p×p matrix B is the adjacency matrix defining the structure of a GRN, Bij represents the regulatory effect of the ith gene on the jth gene; and the q×p matrix F is composed of the regulatory effects of cis-eQTLs, in which Fkm denotes the effect of the kth cis-eQTL on the mth gene. It is often assumed that every gene has no effect on itself, which implies Bii=0 for i = 1, ⋯,p. To ensure the identifiable of GRNs, we assume there is at least one unique cis-eQTL for each gene.
Let yi=[y1i,y2i,⋯,yni]T,i=1,⋯,p be the ith column of Y, denoting expression levels of the ith gene observed from n individuals. And let Bi,i=1,⋯,p be the ith column of B. As mentioned before, the ith gene is considered to have no effect on itself, meaning that the ith entry of Bi is known to be zero, so this entry can be removed before inference to reduce the computation complexity. Correspondingly, the ith column of Y needs also to be removed. Then we can split Eq. (8) into p SEMs, in which the ith SEM as follows describes how much other genes and corresponding cis-eQTLs affect the ith gene.
where n×1 vector yi is the ith column of Y and n×(p−1) matrix Y−i refers to Y excluding the ith column; (p−1)×1 vector bi is the ith column of B excluding the ith row; q×1 vector fi denotes the ith column of F; n×1 vector ei represents the residual error vector, in which all entries are modeled as independent and identical normal distributions with zero mean and variance σ2.
GRNs under different conditions
In this paper, we mainly focus on the joint inference of GRNs under different conditions. We denote the expression levels of p genes under two different conditions as \(\mathbf {Y}^{(k)} = \left [\mathbf {y}^{(k)}_{1}, \mathbf {y}^{(k)}_{2},\cdots, \mathbf {y}^{(k)}_{p}\right ], k=1, 2\). Similarly, the genotypes of cis-eQTLs under two conditions are represented as \(\mathbf {X}^{(k)} = \left [\mathbf {x}^{(k)}_{1}, \mathbf {x}^{(k)}_{2},\cdots, \mathbf {x}^{(k)}_{q}\right ], k=1,2\). Based on the SEM introduced in the previous subsection, we can represent two pair-wise GRNs as
and further represent the sub-models as
where B(k) depict the structures of two GRNs under different conditions, which contain coefficients for the direct causal effects of the genes on each other.
As discussed above, \(\mathbf {f}^{(k)}_{i}\) is sparse and the locations of nonzero entries have been obtained via pretreatment. We assume the row index set of nonzero entries of \(\mathbf {f}^{(k)}_{i}\) as \(S^{(k)}_{i}\), so in the ith model of Eq. (11), X(k) can be reduced to a matrix \(\mathbf {X}^{(k)}_{S^{(k)}_{i}}\) that only contains the columns whose indices are in \(S^{(k)}_{i}\). Accordingly, \(\mathbf {f}^{(k)}_{i,S^{(k)}_{i}}\) is a reduced form of \(\mathbf {f}^{(k)}_{i}\) that only contains the rows whose indices are in \(S^{(k)}_{i}\).
The identifiability of SEMs
Our main goal is to infer the adjacency matrices B(1) and B(2) from SMEs as in Eq. (10), and identify the difference between them (ΔB=B(1)−B(2)) in the meanwhile. Without any knowledge about the GRNs, no restriction is imposed on the structures specified by the adjacency matrices, that is to say, GRNs modeled with SEMs are considered as general directed networks that can possibly be DAGs or DCGs.
As mentioned before, we make some standard assumptions that are used by most popular GRN inference algorithms to ensure model identifiability. For example, the error terms \(\mathbf {e}^{(k)}_{i}\) are assumed as independent and identical normal distributions, and the diagonal entries of B(k) are all assumed to be zero so that there is no self-loop in GRNs.
While DAGs are always identified under the above assumptions, the identifiability of DCGs need further studies because of the challenge in model equivalence [11]. To make meaningful inference, it is important to have as small a set of equivalent models as possible [12]. Logsdon et al. [12] investigated this issue for DCGs in detail in their "Recovery" Theorem. According to their discussion, under the assumption that each gene is directly regulated by a unique nonempty set of cis-eQTLs, there will exist multiple equivalent DCGs, and the perturbation topology can completely change among equivalent DCGs. Furthermore, as in the Lemma of the "Recovery" Theorem, if we know which gene each cis-eQTL feeds into, then the cardinality of the equivalence class is reduced to one, that is, a unique DCG can be inferred. Therefore, we make the assumption that the the loci of the q cis-eQTLs have been determined by an existing eQTL method in advance, but the size of each regulatory effect is still unknown. In this way, the perturbation topology is determined, and a unique DCG can be the identified.
Now that the identifiability of SEMs are guaranteed for both DAGs and DCGs with appropriate assumptions, the pair-wise GRNs can be inferred by estimating B(1) and B(2) column by column by solving Eq. (11).
Methods
Joint inference model based on SEMs
By defining \(\mathbf {W}_{i}^{(k)} = \left [\mathbf {Y}^{(k)}_{-i}, \mathbf {X}^{(k)}_{S^{(k)}_{i}}\right ]\), \(\boldsymbol {\beta }^{(k)}_{i} = \left [\mathbf {b}^{(k)}_{i}, \mathbf {f}^{(k)}_{i,S^{(k)}_{i}} \right ]^{T}\), Eq. (11) can be rewritten as a linear type model
Therefore, we can first solve Eq. (12) by adopting appropriate regularized linear regression method and then extract \(\mathbf {b}^{(k)}_{i}\) from \(\boldsymbol {\beta }^{(k)}_{i}\).
As is known, a gene is usually regulated by a small number of genes, meaning that most entries in β(k) are equal to zero [23–26]. In addition, pair-wise GRNs under different conditions are biologically considered to be similar, that is to say, most entries in Δβ=β(1)−β(2) are also equal to zero [27]. In order to satisfy the sparsity of both the separate GRNs and the differential GRN, we penalize both β(k) and Δβ with l1-norm, which would yield the following optimization problem [19]:
where the l1-norms \(\lambda _{1}\left (\left \| \boldsymbol {\beta }^{(1)}_{i} \right \|_{1} + \left \| \boldsymbol {\beta }^{(2)}_{i} \right \|_{1}\right)\) and \(\lambda _{2}\left \| \boldsymbol {\beta }^{(1)}_{i} -\boldsymbol {\beta }^{(2)}_{i} \right \|_{1}\) are introduced to fulfill the sparsity of corresponding parameters, λ1>0 and λ2>0 are tuning parameters used to control the sparsity levels.
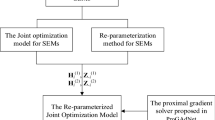
Inspired by the optimization model in Eq. (13), we re-parameterize the pair-wise re-parameterized SEMs as in Eq. (12) to an integrated model as follows,
where \(\mathbf {y}_{i}=\mathbf {y}_{i}^{(1)}+\mathbf {y}_{i}^{(2)}\), \(\mathbf {W}_{i} = \left [\mathbf {W}_{i}^{(1)},\mathbf {W}_{i}^{(2)}\right ]\), \(\boldsymbol {\beta }_{i} = \left [\boldsymbol {\beta }^{(1)}_{i},\boldsymbol {\beta }^{(2)}_{i}\right ]^{T}\) and \(\mathbf {e}_{i}=\mathbf {e}_{i}^{(1)}+\mathbf {e}_{i}^{(2)}\). By denoting the dimension of \(S_{i}^{(k)}\) as qi, the dimension of \(\boldsymbol {\beta }^{(k)}_{i}\) can be easily expressed as pi=p−1+qi. Therefore, yi and ei are n×1 vectors, Wi is an n×2pi design matrix and βi is a 2pi×1 vector containing all unknown parameters to be estimated. Then, the optimization problem in Eq. (13) can be transferred to
In the subsequent section, we infer Eq. (15) in a Bayesian framework by developing a novel appropriate prior to fulfill the required sparsity and estimating the parameters with a Gibbs sampler.
The BFDSEM algorithm
In this section, we develop the BFDSEM algorithm via a novel hierarchical prior for Eq. (14) to solve the optimization problem as in Eq. (15). Referring to the Bayesian fused Lasso [35], the prior for βi is defined as
Then the hierarchical prior can be represented as
The hyper parameters, ψ1,j and ψ2,k, are equivalent to the tuning parameters that adjust the sparsity of βi and Δβi. We consider the class of Gamma prior on them, namely Gamma(a,b), where a and b can be pre-specified appropriate values so that the hyper priors for ψ1,j and ψ2,k are essentially noninformative. It should be noted that here we employ adaptive tuning parameters for each penalized term in line with the adaptive Lasso [33] to improve the accuracy and robustness of estimation.
From Eq. (17), we see that \(\boldsymbol {\beta }_{i}|\sigma ^{2},\tau _{1}^{2},\cdots,\tau _{2p_{i}}^{2},\omega _{1}^{2},\cdots,\omega _{p_{i}}^{2}\) is in line with multivariate normal distribution, according to Eq. (16), it is deduced from
where
Therefore, \(\boldsymbol {\beta }_{i}|\sigma ^{2},\tau _{1}^{2},\cdots,\tau _{2p_{i}}^{2},\omega _{1}^{2},\cdots,\omega _{p_{i}}^{2}\) is multivariate normal distributed with mean vector 0 and covariance matrix \(\sigma ^{2}\Sigma _{\boldsymbol {\beta }_{i}}\) with
The hierarchical prior in Eqs. (16) and (17) implement the optimization problem as described in Eq. (15). We assign σ2 an Inverse-Gamma prior with hyper parameters ν0/2 and η0/2, the hyper parameters can be pre-specified appropriate values. With the likelihood
the full conditional posteriors of the hierarchical model can be given by:
Then a Gibbs sampler is used to draw samples iteratively from the above posteriors, and yields posterior estimates of βi, the uncertainty can also be characterized in a natural way through the credible intervals. The convergence of the Gibbs sampler is monitored by the potential scale reduction factor \(\widehat {R}\) as introduced in [37] and the convergence condition is set to \(\widehat {R}<1.1\). Once the Gibbs sampler converges, we continue to draw samples for several iterations and average the converged samples of βi as the estimations for βi. Vats [38] and Kyung et al. [34] have proved geometric ergodicity of the Gibbs samplers for the Bayesian fused lasso. Following the conclusion in [38], under the condition of n>3, no conditions on pi are required to fulfil the geometric ergodicity. Thus, the convergence of the Gibbs sampler is expected to be quite speed regardless of the dimension pi.
With the samples for all βi drawn from the Gibbs sampler, the posterior mean estimate and corresponding credible interval of \((\mathbf {B}^{(1)}_{i}\), \(\mathbf {B}^{(2)}_{i})\) can also be obtained. After applying the Gibbs Sampler on all the p models for i=1,⋯,p, the adjacency matrices of two GRNs B(1) and B(2) as well as the difference between them ΔB can be easily figured out.
Different from the frequency framework, a Bayesian hierarchical model with penalized prior can shrinkage the regression coefficients but does not produce exactly zero estimates. Several strategies have been proposed to go from a posterior distribution to a sparse point estimate [39–41]. Considering the computing complexity, here we adopt the simplest strategy suggested in [42–44] to preset a threshold value t. In the adjacency matrices B(1) and B(2), only the entries whose absolute value are larger than t are retained, all other entries are set to zero. Then the differential GRN can be obtained by computing ΔB=B(1)−B(2). Obviously, there is a trade off between power of detection (PD) and discovery rate (FDR), the smaller t is, more edges would be detected in the GRNs, which results in better PD but worse FDR; and reversely, a larger t yields worse PD but better FDR. As discussed in [42], the value of the threshold t is chosen subjectively. Referring to the threshold value in [42] (t=0.1) and [44] (t=0.05,0.1,0.2), we set t=0.2 for the following computer simulations.
Results
Computer Simulations
In this section, we run simulations on synthetic data by applying our proposed BFDSEM algorithm and two state-of-the-art joint differential analysis algorithms: FSSEM and ReDNet, and then compare the performance in terms of PD and FDR for (B(1),B(2)) and ΔB. Since the algorithms may have different performance in DAGs and DCGs, it is commonplace to run simulations on synthetic DAGs and DCGs, respectively.
Following the setup in [13, 20], both DAGs and DCGs under two different conditions are simulated. The simulated data have similar numeric data type and range with corresponding standardized experimental data, so the simulation studies could reflect the performance of the algorithms to some extent. The number of genes p varies from 10 to 30 or 50, the sample size n varies from 50 to 250. In the following simulations, the number of cis-eQTLs q is set as q=2p, meaning that each gene has two contributing cis-eQTLs. The average number of edges per node ne which determines the degree of sparsity varies from 1 to 3 or 4.
In detail, an adjacency matrix of a DAG or a DCG A(1) is first generated for the GRN under condition 1, then the corresponding adjacency matrix A(2) is generated by randomly changing nd entries of A(1), where nd is approximately equal to 10% of the nonzero entries, and the number of changes from 1 to 0 and from 0 to 1 are equal (denoted by nc). The network matrix of GRN under condition 1 B(1) is generated from A(1) by replacing its nonzero entries with random values generated from a uniform distribution over \((-1, -0.5)\bigcup (0.5, 1)\). Next, the corresponding network matrix under condition 2 B(2) is generated from A(2) and B(1) by steps as follows: For all \(A_{ij}^{(2)}=0\), we set \(B_{ij}^{(2)}=0\); for all \(A_{ij}^{(2)}=A_{ij}^{(1)}\), we randomly select nc entries and keep them unchanged, other entries are set as \(B_{ij}^{(2)}=B_{ij}^{(1)}\); for all \(A_{ij}^{(2)}=1\) but \(A_{ij}^{(1)}=0\), we generate \(B_{ij}^{(2)}\) from a uniformly distribution over interval \((-1, -0.5)\bigcup (0.5, 1)\). The genotypes of the q cis-eQTLs are simulated from an F2 cross. Values 1 and 3 were assigned to two homozygous genotypes, respectively, and value 2 to the heterozygous genotype. Then each entry in X(1) and X(2) are generated by sampling from {1, 2, 3} with corresponding probabilities {0.25, 0.5, 0.25}. The regulatory effects of corresponding cis-eQTLs are assumed to be 1, so F(1) and F(2) are simulated by randomly permuting the rows of matrix (Ip,Ip)T, where Ip denotes a p-dimensional identify matrix. In the following simulations, we assume F(1)=F(2). Each error term in E(1) and E(2) is independently sampled from a normal distribution with zero mean and variance σ2. Then, the gene expression matrices Y(1) and Y(2) can be obtained by computing Y(k)=(X(k)F(k)+E(k))(I−B(k))−1,k=1,2.
For each setup of the following simulated networks, 20 replicates are simulated, then the PD and FDR are calculated by averaging the results of all replicates in same setups. The variable selection threshold t is defined as 0.2.
We depict the results of DAGs and DCGs with p=30,ne=1,σ2=0.01 in Figs. 1 and 2, respectively. First, let us see the results of DAGs in Fig. 1. The PD and FDR of (B(1),B(2)) are shown in Fig. 1a and b. The three algorithms show similar performance in PD, which nearly reaches 1 for all sample sizes. As for the FDR, BFDSEM has similar results with FSSEM, which are better than ReDNet. The PD and FDR of ΔB are depicted in Fig. 1c and d. BFDSEM yields slightly better PD than ReDNet, and more better PD than FSSEM. It offers slightly worse FDR than FSSEM when sample size is ≤100, and much better FDR than ReDNet across all sample sizes. Next to see the results of DCGs in Fig. 2. The PD and FDR of (B(1),B(2)) can be observed in Fig. 2a and b. BFDSEM offers similar or very slightly worse PD and FDR than FSSEM, and provides visual better PD and FDR than ReDNet. The PD and FDR of ΔB are depicted in Fig. 2c and d. BFDSEM and FSSEM perform neck and neck PD and FDR, which are obviously better than ReDNet.

Performance of BFDSEM, FSSEM and ReDNet for DAGs. The number of genes p=30, the average number of edges per node ne=1, the noise variance σ2=0.01, and the sample sizes n1=n2 vary from 50 to 250

Performance of BFDSEM, FSSEM and ReDNet for DCGs. The number of genes p=30, the average number of edges per node ne=1, the noise variance σ2=0.01, and the sample sizes n1=n2 vary from 50 to 250
All of the simulation results of DAGs and DCGs under different setups (ne and σ2) can be found in Additional files 2, 3, 4, 5: Figure S1-S4. As a whole, BFDSEM generally outperforms ReDNet for all simulation setups. Compared to FSSEM, BFDSEM has similar or slightly better performance for synthetic data sets with σ2=0.01. When σ2=0.1, BFDSEM still exhibits similar or better PD for both (B(1),B(2)) and ΔB, but offers worse FDR when sample size is relatively smaller, especially for ΔB.
Finally, simulations on DAGs with p=50,ne=1,σ2=0.01 are run to show how does the value of threshold t affect the performance of BFDSEM. The simulation results for (B(1),B(2)) and ΔB with t ranging in {0.08,0.1,0.15,0.2} and n varies from 80 to 500 are depicted in Fig. 3. As shown in Fig. 3a and c, for all values of t, the PD of both (B(1),B(2)) and ΔB are similar and all equal to or slightly lower than 1. From Fig. 3b and d, we see that the FDR of (B(1),B(2)) and ΔB still achieve almost perfect results for t=0.15 or 0.2. Nevertheless, when t=0.08 or 0.1, the FDR of both (B(1),B(2)) and ΔB increase invisibly, especially for ΔB with small sample sizes.

Performance of BFDSEM for DAGs with different Bayesian variable selection threshold t. The number of genes p=50, the average number of edges per node ne=1, the noise variance σ2=0.01, the sample sizes n1=n2 vary from 80 to 500, and the variable selection threshold t ranges in {0.08, 0.1, 0.15, 0.2}
Real data analysis
We perform differential analysis on a real data set from 42 tumors and their adjacent normal tissues of non-smoking female patients with lung adenocarcinomas. The gene expression levels and genotypes of single nucleotide polymorphisms (SNPs) in this data set were reported in the gene expression omnibus data base GSE33356 by Lu et al. [45]. We preprocessed the raw data in GSE33356 following [20] with R package affy [62] and MatrixEQTL [63], resulting in 1,455 genes with at least one cis-eQTLs at an FDR = 0.01.
To perform more reliable inference, we further selected a smaller subset of the 1,455 genes with the GIANT database. The GIANT database which can be accessed in (http://hb.flatironinstitute.org) contains 144 tissue- and cell lineage-specific GRNs from an integration of data sets covering thousands of experiments contained in more than 14,000 distinct publications. We downloaded the lung network with Top Edges (lung_top.gz) from the GINAT database, the posterior probabilities of each edge can be found in the downloaded network. The edges whose posterior probabilities are less than 0.8 were deleted from the GIANT lung network. Then the 1455 genes with corresponding cis-eQTLs were further filtered with the GIANT lung network, and finally, 15 genes were identified to have interactions with at least one another gene with posterior probability ≥0.80 in the GIANT lung network. The details about these 15 lung genes are described in Additional file 6: Table S1.
Now we can apply BFDSEM on the filtered lung data set containing expression levels of 15 genes and genotypes of corresponding cis-eQTLs under two different conditions (in 42 normal tissues and 42 tumors) to make differential analysis.
First, BFDSEM was applied to quantify the uncertainty of the posterior Gibbs sampler by credible intervals. The posterior mean estimates and corresponding 95% equal-tailed credible intervals for B(1), B(2) and ΔB were estimated and computed, and each result of the first column is depicted in Fig. 4(a)(b)(c), respectively, denoting the regulatory effects of all the 15 genes on the first gene PPP4R2. For comparison, the point estimates of FSSEM and ReDNet are also depicted. Moreover, in Additional files 7, 8, 9: Figure S5-S7 give the results of 100 samples for each estimated edge.

Interval estimate of BFDSEM and point estimates of FSSEM and ReDNet for the first column of B(1), B(2) and ΔB. Including posterior mean estimates and corresponding 95% equal-tailed credible intervals of BFDSEM, and point estimates of FSSEM and ReDNet for the subset of human lung data set
Then we adopt BFDSEM to reconstruct the differential GRN. By directly applying BFDSEM to the original data set with 15 lung genes in 42 tumors and 42 normal tissues, 41 edges were detected. To evaluate the significance of the identified edges, we re-sampled from the original data sets with replacement to obtain 100 bootstraps, each bootstrap also has 42 tumor samples and 42 normal samples. Then BFDSEM is applied to the 100 bootstraps separately, and only the edges that were detected for more than 80 times were retained in the final GRNs. Finally, BFDSEM yielded a GRN with 18 edges for normal lung tissues B(1) and a GRN with 17 edges for lung tumors B(2). We compared the resulted normal GRN with the GIANT reference network inferred from a large number of samples, and found that 13 of the 18 edges were also in the corresponding GIANT lung network with relatively high confidence, which showed that the GRN inferred by the BFDSEM from only a small number of samples is in accordance with the GIANT lung network in some degree.
Since too small changes of the regulatory effects are often of little significance in biological, for a differential GRN identified by ΔB=B(1)−B(2), we only take the entries that satisfy the following condition: \(|B_{ij}^{(1)}-B_{ij}^{(2)}|>\textit {min}\left \{\left (B_{ij}^{(1)},B_{ij}^{(2)}\right)\right \}\)/5. This criteria was applied to all the 100 bootstraps, and the ultimate differential GRN was obtained by eliminating the edges that were detected for less than 80 times. The identified differential GRN with 7 genes and 5 edges is depicted in Fig. 5, in which the mainly related genes are: BTF3, RPS16, HSF1, RPS6, and MAPKAPK2.

The differential GRN of 15 lung genes identified by the BFDSEM algorithm. Including 7 genes and 5 edges, the other genes that were not involved in the differential GRN were omitted
Discussion
An SEM provides a systematic framework to integrate genetic perturbations with gene expression data to improve inference accuracy, and offers flexibility to model both DAGs and DCGs [13]. FSSEM and ReDNet are two state-of-the-art joint inference algorithms for differential analysis of two similar GRNs modeled with SEMs. The performance of these two joint inference algorithms have been proved much more efficient than naive approaches. The FSSEM algorithm in [20] modeled a penalized negative log-likelihood function and developed a proximal alternative linearize minimization algorithm to infer coefficients. The ReDNet algorithm in [21] re-parameterized the pair-wise SEMs as an integrated model regarding the averaged regulatory effects and differential regulatory effects as coefficients, and then penalized them to realize sparse learning. In this paper, we develop a novel algorithm named BFDSEM for joint inference of two similar GRNs modeled with SEMs. Different from FSSEM and ReDNet, BFDSEM is implemented based on re-parametrization and Bayesian penalized regression with a novel fused prior. First, the original pair-wise SEMs under different conditions are re-parameterized as an integrated linear model that incorporates all related data sources at the first stage; Next, considering the sparsity of the separate GRNs and the differential GRN, a penalized optimization model for the re-parameterized linear model is constructed and a corresponding penalized hierarchical prior is developed; Finally, the full conditional posteriors are deduced and a Gibbs sampler is conducted to draw samples iteratively from the posteriors, then the posterior credible interval and posterior mean estimation can be obtained from the samples.
Compared to FSSEM and ReDNet, the Gibbs sampler in BFDSEM is easy to implement, and not only provides point estimation via the posterior mean or median, but also quantifies the uncertainty via the credible interval automatically. The geometric ergodicity of Gibbs samplers for the Bayesian fused lasso have been proved in Vats [38] and Kyung et al. [34], which means fast convergence of the iterations. In addition, BFDSEM construct the penalized prior directly for the re-parameterized integrated linear model to achieve sparsity of the separate GRNs and differential GRN simultaneously. This approach is much simpler and faster than FSSEM, and can reach similar performance at the same time. ReDNet also re-parameterized the pair-wise SEMs as an integrated model, the adaptive Lasso was applied to achieve sparsity for the averaged GRN and differential GRN, rather than the separate GRNs, which may result in less accurate estimates.
Simulation studies have been run to compare the performance of BFDSEM with FSSEM and ReDNet, the results demonstrated that our BFDSEM algorithm has similar performance with FSSEM, and has better performance than ReDNet. The differential analysis of a real data set with 15 genes of 42 lung tumors and 42 normal tissues has been made to infer the underlying GRNs and differential GRN. The resulted normal GRN was demonstrated in good agreement with the GIANT reference network and the identified differential GRN contained 5 highly related genes. The 5 genes have been demonstrated to be related to lung cancer and some other kinds of cancers by experimental approaches in previous literatures. Specifically, BTF3 was confirmed aberrantly in various cancer tissues such as gastric cancer tissues [47, 48], prostate cancer tissues [49], colorectal cancer tissues[50] and pancreatic cancer cells [51]; RPS16 was found dysregulated in disc degeneration, which is one of the main causes of low back pain [52]; HSF1 influenced the expression of heat shock proteins as well as other activities like the induction of tumor suppressor genes, signal transduction pathway, and glucose metabolism. Its associations with gastric cancer [53], breast cancer and two of the studied SNPs correlated significantly with cancer development [54] have been proved; RPS6 was declared closely relevant to the non-small cell lung cancer (NSCLC) [55], the renal cell carcinoma [56] and some other cancers [57, 58]; MAPKAPK2 was demonstrated to contribute to tumor progression by promoting M2 macrophage polarization and tumor angiogenesis [59].
There are still some limitations of the BFDSEM algorithm: First, the selection of the Bayesian variable threshold t is somewhat arbitrary to some extent, an improper t may lead to less accurate results; Next, despite the apparent theoretical safeguard of geometric ergodicity, when p/n is large enough, it may be possible for the Gibbs samplers to converge at a slower rate [38, 60], thereby the uncertainty quantification may also be compromised; Moreover, the proposed re-parametrization method only supports pair-wise data sets with the same sample size. A natural direction for future research would be to investigate solutions for these limitations.
Conclusion
The differential analysis of pair-wise GRNs under different conditions is as important as the inference of single GRNs. In this paper, we develop a novel Bayesian fused differential analysis algorithm for GRNs modeled with SEMs, named BFDSEM, which provides valuable tool for joint inference of GRNs under two different conditions. To our knowledge, our BFDSEM algorithm is the first Bayesian inference method for joint analysis of GRNs modeled with SEMs.
Availability and Requirements
Project name: BFDSEM.Project home page: Not applicable.Operating system(s): Platform independent.Programming language: Matlab.Other requirements: None.License: None.Any restrictions to use by non-academics: None.
Availability of data and materials
The source code and the human lung data set used to make differential analysis are freely available upon request.
Abbreviations
- CNVs:
-
Copy number variants
- DAG:
-
Direct acyclic
- DCG:
-
Direct cyclic
- eQTL:
-
Expression quantitative trait loci
- FDR:
-
False discovery rate
- GRN:
-
Gene regulatory network
- GRN; GRN; PD:
-
Power of detection
- SEM:
-
Structural equation model
References
Shmulevich I, Dougherty ER, Zhang W. Gene perturbation and intervention in probabilistic boolean networks. Bioinformatics. 2002; 18(10):1319–31.
Luo W, Hankenson KD, Woolf PJ. Learning transcriptional regulatory networks from high throughput gene expression data using continuous three-way mutual information. BMC Bioinformatics. 2008; 9(1):1–15.
Zhang X, Zhao XM, et al.Inferring gene regulatory networks from gene expression data by path consistency algorithm based on conditional mutual information. Bioinformatics. 2011; 28(1):98–104.
Friedman J, Hastie T, Tibshirani R. Sparse inverse covariance estimation with the graphical lasso. Biostatistics. 2008; 9(3):432–41.
Oh JH, Deasy JO. Inference of radio-responsive gene regulatory networks using the graphical lasso algorithm. BMC Bioinformatics. 2014; 15 Suppl 7(7):S5.
Zou M, Conzen SD. A new dynamic bayesian network (DBN) approach for identifying gene regulatory networks from time course microarray data. Bioinformatics. 2005; 21(1):71–9.
Zhang Y, Deng Z, Jiang H, Jia P. Inferring gene regulatory networks from multiple data sources via a dynamic bayesian network with structural em In: Cohen-Boulakia S, Tannen V, editors. Data Integration in the Life Sciences. DILS 2007. Lecture Notes in Computer Science, vol 4544. Berlin, Heidelberg: Springer: 2007. p. 204–14.
Lu Y, Zhou Y, et al.A lasso regression model for the construction of microrna-target regulatory networks. Bioinformatics. 2011; 27(17):2406.
Omranian N, Eloundou-Mbebi J, et al.Gene regulatory network inference using fused lasso on multiple data sets. Sci Rep. 2016; 6(1):20533.
Xions MM, Li J, Fang X. Identification of genetic networks. Genetics. 2004; 166(2):1037–52.
Liu B, de la Fuente A, Hoeschele I. Gene network inference via structural equation modeling in genetical genomics experiments. Genetics. 2008; 178(3):1763–76.
Logsdon BA, Mezey J. Gene expression network reconstruction by convex feature selection when incorporating genetic perturbations. Plos Comput Biol. 2010; 6(12):e1001014.
Cai X, Andrés BJ, Giannakis GB. Inference of gene regulatory networks with sparse structural equation models exploiting genetic perturbations. Plos Comput Biol. 2013; 9(5):1003068.
Lewis C, Yang J, et al.Disease-specific gene expression profiling in multiple models of lung disease. Am J Respir Crit Care Med. 2012; 177(4):376–87.
Da R, Young A, Montana G. Differential analysis of biological networks. BMC Bioinformatics. 2015; 16(1):1–13.
Guo W, Zhu L, et al.Understanding tissue-specificity with human tissue-specific regulatory networks. Sci China Inf Sci. 2016; 59(7):070105.
Mohan H, London P, et al.Node-based learning of multiple gaussian graphical models. J Mach Learn Res. 2014; 15(1):445–88.
Danaher P, Wang P, Witten DM. The joint graphical lasso for inverse covariance estimation across multiple classes. J Royal Stat Soc. 2014; 76(2):373–97.
Wang C, Gao F, et al.Efficient proximal gradient algorithm for inference of differential gene networks. BMC Bioinformatics. 2019; 20(1):224.
Zhou X, Cai X. Inference of differential gene regulatory networks based on gene expression and genetic perturbation data. Association for Uncertainty in Artificial Intelligence. 2018.
Ren M, Zhang D. Differential analysis of directed networks. Bioinformatics. 2019; btz529.
Chen C, Ren M, et al.A two-stage penalized least squares method for constructing large systems of structural equations. J Mach Learn Res. 2018; 19(1):40–73.
Thieffry D, Huerta AM, Pérez-Rueda E, Collado-Vides J. From specific gene regulation to genomic networks: a global analysis of transcriptional regulation in escherichia coli. Bioessays News Rev Mole Cell Develop Biol. 1998; 20(5):433.
Brazhnik P, De l. F. A., Mendes P. Gene networks: how to put the function in genomics. Trends Biotechnol. 2002; 20(11):467–72.
Gardner T, Di Bernardo D, Lorenz D, Collins J. Inferring genetic networks and identifying compound mode of action via expression profiling. Science. 2003; 301(5629):102–5.
Tegner J, Yeung MK, Hasty J, Collins JJ. Reverse engineering gene networks: integrating genetic perturbations with dynamical modeling. Proc Nat Acad Sci USA. 2003; 100(10):5944.
de la Fuente A. From ’differential expression’ to ’differential networking’ - identification of dysfunctional regulatory networks in diseases. Trends Genet Tig. 2010; 26(7):326.
Tibshirani R. Regression shrinkage and selection via the lasso. J Royal Stat Soc: Ser B(Methodol). 1996; 58(1):267–88.
Park T, Casella G. The bayesian lasso. J Am Stat Assoc. 2008; 103(482):681–6.
Fan J, Li R. Variable selection via nonconcave penalized likelihood and its oracle properties. J Am Stat Assoc. 2001; 96(456):1348–60.
Zou H, Hastie T. Regularization and variable selection via the elastic net. J Royal Stat Soc: Ser B (Stat Methodol). 2005; 67(2):301–20.
Tibshirani R, Saunders M, Rosset S, Zhu J, Knight K. Sparsity and smoothness via the fused lasso. J Royal Stat Soc: Ser B (Stat Methodol). 2005; 67(1):91–108.
Zou H. The adaptive lasso and its oracle properties. J Am Stat Assoc. 2006; 101(476):1418–29.
Kyung MJ, Gill J, Ghosh M, Casella G. Penalized regression, standard errors, and bayesian lassos. Bayesian Anal. 2010; 5(2):369–412.
Shimamura K, Ueki M, Kawano S, Konishi S. Bayesian generalized fused lasso modeling via NEG distribution. Communication in Statistics- Theory and Methods. 2016.
Casella G. Empirical bayes gibbs sampling. Biostatistics. 2001; 2(4):485–500.
Gelman A, Carlin J, Stern H, Rubin D. Bayesian Data Analysis, 3rd. London: Chapman and Hall; 2003.
Vats Dootika. Geometric ergodicity of Gibbs samplers in Bayesian penalized regression models. J Am Stat Assoc. 2017; 11(2):4033–64.
Bondell HD, Reich BJ. Consistent high-dimensional Bayesian variable selection via penalized credible regions. J Am Stat Assoc. 2012; 107(500):1610–24.
Hahn PR, Carvalho CM. Decoupling shrinkage and selection in Bayesian linear models: A posterior summary perspective. J Am Stat Assoc. 2015; 110(509):435–48.
Zhang D, Chen B, et al.Bayesian Variable Selection and Estimation Based on Global-Local Shrinkage Priors. Sankhya A. 2018; 80(2):215–46.
Hoti F, Sillanp\(\ddot {\textup {a}}\ddot {\textup {a}}\) B, et al.Bayesian mapping of genotype × expression interactions in quantitative and qualitative traits. Heredity. 2006; 97(1):4–18.
Yi N, Xu S. Bayesian LASSO for quantitative trait loci mapping. Genetics. 2008; 179(2):1045–55.
Dong Z, Song T, Yuan C. Inference of Gene Regulatory Networks from Genetic Perturbations with Linear Regression Model. Plos One. 2013; 8(12):e83263.
Lu TP, Lai LC, et al.Integrated analyses of copy number variations and gene expression in lung adenocarcinoma. PLoS One. 2011; 6(9):e24829.
Greene CS, Krishnan A, et al.Understanding multicellular function and disease with human tissue-specific networks. Nature Genet. 2015; 47(6):569–76.
Zhang D, Chen B, et al.Basic Transcription Factor 3 is required for proliferation and Epithelial-Mesenchymal transition via regulation of FOXM1 and JAK2/STAT3 signaling in Gastric Cancer. Oncol Res. 2017; 25(9):1453–62.
Liu Q, Zhou J, et al.Basic transcription factor 3 is involved in gastric cancer development and progression. World J Gastroenterol. 2013; 19(28):4495–503.
Symes AJ, Eilertsen M, et al.Quantitative analysis of BTF3, HINT1, NDRG1 and ODC1 protein over-expression in human prostate cancer tissue. PLoS One. 2013; 8(12):e84295.
Wang C, Frånbergh-Karlson H, et al.Clinicopathological significance of BTF3 expression in colorectal cancer. Tumour Biol. 2013; 34(4):2141–6.
Kusumawidjaja G, Kayed H, et al.Basic transcription factor 3 (BTF3) regulates transcription of tumor-associated genes in pancreatic cancer cells. Canc Biol Ther. 2007; 6(3):367–76.
Yang Z, Chen X, et al.Dysregulated COL3A1 and RPL8, RPS16, and RPS23 in Disc Degeneration revealed by bioinformatics methods. Spine. 2015; 40(13):e745–51.
Kim SJ, Lee S, et al.Heat Shock Factor 1 Predicts Poor Prognosis of Gastric Cancer. Yonsei Med J. 2018; 59(9):1041–8.
Almotwaa S, Elrobh M, et al.Genetic polymorphism and expression of HSF1 gene is significantly associated with breast cancer in Saudi females. PLoS One. 2018; 13(3):e0193095.
Chen B, Tan Z, et al.Hyperphosphorylation of ribosomal protein S6 predicts unfavorable clinical survival in non-small cell lung cancer. J Experiment Clin Canc Res. 2015; 34(1):126.
Knoll M, Macher-Goeppinger S, et al.The ribosomal protein S6 in renal cell carcinoma: functional relevance and potential as biomarker. Oncotarget. 2016; 7(1):418–32.
Li G, Shan C, et al.Tanshinone IIA inhibits HIF-1 α and VEGF expression in breast cancer cells via mTOR/p70S6K/RPS6/4E-BP1 signaling pathway. PLoS One. 2015; 10(2):e0117440.
Grasso S, Tristante E, et al.Resistance to Selumetinib (AZD6244) in colorectal cancer cell lines is mediated by p70S6K and RPS6 activation. Neoplasis. 2014; 16(10):845–60.
Suarez-Lopez L, Sriram G, et al.MK2 contributes to tumor progression by promoting M2 macrophage polarization and tumor angiogenesis. Proc Nat Acad Sci USA. 2018; 115(18):E4236–44.
Rajaratnam B, Sparks D. MCMC-based inference in the era of big data: A fundamental analysis of the convergence complexity of high-dimensional chains. Statistics. 2015.
Holbrook JD, Parker JS, et al.Deep sequencing of gastric carcinoma reveals somatic mutations relevant to personalized medicine. J Transl Med. 2011; 9:119.
Gautier L, Cope L, et al.affy-analysis of affymetrix genechip data at the probe level. Bioinformatics. 2004; 20(3):307–15.
Shabalin AA. Matrix eqtl: ultra fast eqtl analysis via large matrix operations. Bioinformatics. 2012; 28(10):1353–8.
Acknowledgements
Not Applicable.
Funding
We would like to thank the National Natural Science Foundation of China (Grant Nos. 61502198, 61572226, 61472161, 61876069) for providing financial supports for this study and publication charges. The funding bodies did not play any role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
YL and DL conceived and deduced the algorithm. TL and YZ designed the simulation experiments and performed the real data analysis. YL wrote the manuscript. DL, TL and YZ help proofreading and revising the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not Applicable
Consent for publication
Not Applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1
Software. Software package that implements the BFDSEM algorithm.
Additional file 2
Figure A1. Performance of BFDSEM, FSSEM and ReDNet for DAGs with different setups. The number of genes p=10, the average number of edges per node ne=1,3 or 4, the noise variance σ2=0.01 or 0.1, and the sample sizes n1=n2 vary from 50 to 250.
Additional file 3
Figure A2. Performance of BFDSEM, FSSEM and ReDNet for DAGs with different setups. The number of genes p=30, the average number of edges per node ne=1,3 or 4, the noise variance σ2=0.01 or 0.1, and the sample sizes n1=n2 vary from 50 to 250.
Additional file 4
Figure A3. Performance of BFDSEM, FSSEM and ReDNet for DCGs with different setups. The number of genes p=10, the average number of edges per node ne=1,3 or 4, the noise variance σ2=0.01 or 0.1, and the sample sizes n1=n2 vary from 50 to 250.
Additional file 5
Figure A4. Performance of BFDSEM, FSSEM and ReDNet for DCGs with different setups. The number of genes p=30, the average number of edges per node ne=1,3 or 4, the noise variance σ2=0.01 or 0.1, and the sample sizes n1=n2 vary from 50 to 250.
Additional file 6
Table A1. A table including the Entrez ID, gene names, corresponding eQTL ID, aliases and brief description of the 15 filtered lung genes.
Additional file 7
Figure A5. The coefficient estimates of BFDSEM, FSSEM and ReDNet for the normal GRN of human lung. Depict the estimate of all the 225 edges in the normal GRN, including 100 samples for each edge drawn from the Gibbs sampler of BFDSEM (×), and point estimates of FSSEM (∘) and ReDNet (△).
Additional file 8
Figure A6. The coefficient estimates of BFDSEM, FSSEM and ReDNet for the tumor GRN of human lung. Depict the estimate of all the 225 edges in tumor GRN, including 100 samples for each edge drawn from the Gibbs sampler of BFDSEM (×), and point estimates of FSSEM (∘) and ReDNet (△).
Additional file 9
Figure A7. The coefficient estimates of BFDSEM, FSSEM and ReDNet for the differential GRN of human lung. Depict the estimate of all the 225 edges in the differential GRN, including 100 samples for each edge drawn from the Gibbs sampler of BFDSEM (×), and point estimates of FSSEM (∘) and ReDNet (△).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Li, Y., Liu, D., Li, T. et al. Bayesian differential analysis of gene regulatory networks exploiting genetic perturbations. BMC Bioinformatics 21, 12 (2020). https://doi.org/10.1186/s12859-019-3314-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-019-3314-3