
Abstract
Background
Selection for sound conformation has been widely used as a primary approach to reduce lameness and leg weakness in pigs. Identification of genomic regions that affect conformation traits would help to improve selection accuracy for these lowly to moderately heritable traits. Our objective was to identify genetic factors that underlie leg and back conformation traits in three Danish pig breeds by performing a genome-wide association study followed by meta-analyses.
Methods
Data on four conformation traits (front leg, back, hind leg and overall conformation) for three Danish pig breeds (23,898 Landrace, 24,130 Yorkshire and 16,524 Duroc pigs) were used for association analyses. Estimated effects of single nucleotide polymorphisms (SNPs) from single-trait association analyses were combined in two meta-analyses: (1) a within-breed meta-analysis for multiple traits to examine if there are pleiotropic genetic variants within a breed; and (2) an across-breed meta-analysis for a single trait to examine if the same quantitative trait loci (QTL) segregate across breeds. SNP annotation was implemented through Sus scrofa Build 10.2 on Ensembl to search for candidate genes.
Results
Among the 14, 12 and 13 QTL that were detected in the single-trait association analyses for the three breeds, the most significant SNPs explained 2, 2.3 and 11.4% of genetic variance for back quality in Landrace, overall conformation in Yorkshire and back quality in Duroc, respectively. Several candidate genes for these QTL were also identified, i.e. LRPPRC, WRAP73, VRTN and PPARD likely control conformation traits through the regulation of bone and muscle development, and IGF2BP2, GH1, CCND2 and MSH2 can have an influence through growth-related processes. Meta-analyses not only confirmed many significant SNPs from single-trait analyses with higher significance levels, but also detected several additional associated SNPs and suggested QTL with possible pleiotropic effects.
Conclusions
Our results imply that conformation traits are complex and may be partly controlled by genes that are involved in bone and skeleton development, muscle and fat metabolism, and growth processes. A reliable list of QTL and candidate genes was provided that can be used in fine-mapping and marker assisted selection to improve conformation traits in pigs.
Similar content being viewed by others

Background
Lameness and leg weakness are issues of concern in pig production due to economic and welfare aspects. Leg weakness has been reported as the second most common reason for involuntary culling in pigs for many years, and accounted for 8.6 to 15% of the sows being removed from commercials herds in Nordic countries [1, 2]. Several studies have reported favorable genetic correlations between good legs and litter size and sow stayability [3–5]. Thus, breeding for reduced leg weakness is expected to induce a favorable correlated response on sow reproduction and longevity, and thereby to improve these traits. In fact, conformation traits have been included in breeding goals in almost all Nordic countries, with the aim of reducing lameness [6]. The evaluation of conformation traits has often been carried out subjectively by scoring gait and movement, leg and feet visual observations, and knee and pastern postures [5, 7]. In the literature, heritability estimates for leg conformation traits range from 0.01 to 0.37 [7–10]. These low to moderate heritabilities suggest that faster genetic progress could be achieved by incorporating genetic marker information in the selection process rather than using a traditional pedigree-based selection scheme [11].
Reliability of genomic prediction can be increased by including single nucleotide polymorphisms (SNPs), that are significant in genome-wide association studies (GWAS), in the SNP chips used for routine genomic prediction [12]. Thus, once quantitative trait loci (QTL) associated with conformation traits have been identified, they can be used to improve the reliability of genomic estimated breeding values (GEBV) [13]. In addition, these QTL can be used in fine-mapping studies to identify the causal genetic factors and thus help to understand the biological processes that underlie conformational development in pigs. Information about which genetic factors are involved in the conformation and locomotion of an animal and how much they affect these traits may contribute in setting up a standard scoring system with more objective criteria for uniform evaluation.
Few studies have focused on the mapping of genes for leg conformation traits in pigs. Thus, the objectives of this study were (1) to identify the QTL that are associated with conformation traits in three Danish pig breeds (Landrace, Yorkshire and Duroc) by performing a GWAS; and (2) to examine if the identified genetic variants are associated with multiple conformation traits within one breed and if the same QTL are segregating across breeds by performing meta-analyses. The biological functions of the genes that were closest to the most significant SNP within the detected QTL were also examined to unravel the genetic background of conformation traits.
Methods
Animals and scoring of traits
Data on conformation traits from the three Danish breeds, Landrace, Yorkshire and Duroc, which were analyzed in this study, were provided by the Danish pig breeding company DanAvl, Axeltorv, Copenhagen, Denmark (http://www.danavl.dk/). In Denmark, purebred pigs in nucleus herds are performance-tested. Conformation traits are evaluated by trained technicians when the pigs are around five months of age and weigh approximately 100 kg. The data used here were recorded from 2002 to 2015 and included four conformation traits: front leg quality (FRONT), back quality (BACK), hind leg quality (HIND) and overall conformation trait (CONF). The first three traits (FRONT, BACK and HIND) were scored using a three-point scale from 1 to 3, with 3 corresponding to the best conformation. For CONF, a five-point scale from 1 to 5 was used to score the animals, with scores of 1 corresponding to animals that have serious legs or back problems, 3 to average animals, and 5 to animals with excellent conformation. The number of observations in each breed and the means and standard deviation of each trait are in Table 1. Due to the very low frequency of the extreme score categories 1 and 5, the few observations in these categories were merged into the adjacent categories for the association analyses.
Corrected phenotypes
Corrected phenotype, rather than raw phenotype, was used as the dependent variable in the association analysis. Fixed effects used for routine genetic evaluation in DanAvl were included in the following model:
where \({\mathbf{y}}\) is a vector of conformation scores, \({\mathbf{b}}\) is a vector of fixed effects including sex, and the combination of herd, year and month at performance testing; body weight at testing performance as covariate; \({\mathbf{u}}\) is a vector of additive genetic values of the animals; \({\mathbf{e}}\) is a vector of the residual effects; \({\mathbf{X}}\) and \({\mathbf{Z}}\) are incidence matrices that associate \({\mathbf{b}}\) and \({\mathbf{u}}\) with \({\mathbf{y}}\). The vectors of random effects \({\mathbf{a}}\) and \({\mathbf{e}}\) were assumed to be normally distributed, i.e. \({\mathbf{u }} \sim N \left( {0,{\upsigma }_{a}^{2} {\mathbf{A}}} \right)\) and \({\mathbf{e}} \sim N\left( {0,{\upsigma }_{e}^{2} {\mathbf{I}}} \right)\) where \(\sigma_{a}^{2}\) is additive genetic variance, \({\mathbf{A}}\) is additive relationship matrix derived from pedigree records, \(\sigma_{e}^{2}\) is residual variance and \({\mathbf{I}}\) is the identity matrix. The combination of herd, year and month at performance testing also accounts for the effect of the technician who performed the measures, since all records within each level of this combination were recorded by the same person. The analyses were carried out by using the REML method with an R interface to the DMU software package [14]. Heritabilities and genetic correlations between traits were estimated by using a bi-variate linear mixed model based on Model 1. Corrected phenotypes were obtained as the sum of the estimated breeding value and the residual value for each animal from Model 1 and were later used as dependent variables in the association model (Model 2).
Genotyping and SNP data
Genotyping was carried out using three types of SNP chip: Illumina PorcineSNP60 BeadChip (Illumina, San Diego, CA, USA), and two GeneSeek® custom SNP arrays (Neogen Corporation, Lansing, MI, USA) namely Genomic Profiler (GGP) Porcine LD array featuring over 8500 SNPs (8.5 K) or GGP Porcine HD array, featuring over 70,000 SNPs (70 K). SNPs were quality-controlled within each breed using the following criteria: SNPs with a call-rate lower than 80% across all samples genotyped with each chip or SNPs with a minor allele frequency (MAF) lower than 0.01 were excluded; SNPs that deviated strongly from the Hardy–Weinberg equilibrium (P < 10−7) and SNPs that were not mapped in the porcine reference genome build Sscrofa10.2 (http://www.ensembl.org/Sus scrofa/Info/Index) were also excluded. Missing genotypes for the remaining SNPs on the 60 K chip were imputed using Beagle version 3.3.2 [15]. After quality control and imputation, 37,080 SNPs, 36,080 SNPs and 32,376 SNPs on the 18 porcine autosomes were retained for the Landrace, Yorkshire and Duroc breeds, respectively. A total of 44,390 different SNPs were available for the three breeds and were later used in the across-breed meta-analysis. The genotype of an animal was removed if the call frequency was less than 90%. The numbers of animals genotyped by different SNP chips and used for the association analysis in each breed are in Table 1.
Association analyses
Single-trait association analysis
All steps in the single-trait association analyses were carried out using available options in the software Genome-wide complex trait analysis (GCTA) [16].
Estimation of the genomic relationship matrix (GRM)
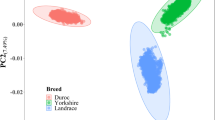
The GRM between individuals within each breed was used to perform principal component analysis and single-trait association analyses. The method that was used to estimate GRM between individuals using SNP data is described in Yang et al. [17]. In this approach, genotype dosages (0, 1, 2) and allele frequency at each SNP were used to calculate the “relationship score” between individual \(i\) and \(j\) for each SNP. The average “relationship score” across all SNPs was then used as the relationship between individuals \(i\) and \(j\).
Principal component analysis (PCA)
Principal component analysis was performed to assess the population structure within each breed. The top ten eigenvectors with the largest eigenvalues were subsequently included as covariates in the association analysis model to account for the confounding effect of population structure [18]. Including these ten eigenvectors resulted in acceptable genomic inflation factors (lambda-λ) (see Additional file 1: Figure S1, Additional file 2: Figure S2 and Additional file 3: Figure S3). Lambda is one of the standard quality-control measures used for GWAS and reflects the level of inflation of Chi square values when compared with their expectation under the null hypothesis of no association between the trait and SNPs [19].
SNP model for single-trait association analysis
Association analyses were carried out using a mixed-linear model with the leaving-one-chromosome-out approach described by Yang et al. [20]. Mixed-linear based association analysis treats the genotype at a single SNP as a fixed effect and the additive polygenic effect as a random effect, and will be called the SNP model (Model 2):
where \({\mathbf{y}}_{c}\) is a vector of corrected phenotypes obtained from Model 1; 1 is a vector of 1s; \(\mu\) is the general mean; \({\mathbf{X}}\) is a matrix containing the 10 eigenvectors that were derived from PCA and included as covariates, and \({\mathbf{b}}\) is a vector of associated effects; \({\mathbf{S}}\) is the design matrix that contains the allele contents at the fitted SNP, i.e. counts (0, 1, 2) of the second allele, and \(\alpha\) is the allele substitution effect; \({\mathbf{u}}\) is a vector of additive polygenic effect, and \({\mathbf{Z}}\) is an incidence matrix associating \({\mathbf{u}}\) with \({\mathbf{y}}\). Vectors of random effects \({\mathbf{u}}\) and \({\mathbf{e}}\) were assumed to be normally distributed, i.e. \({\mathbf{u}} \sim N\left( {0,\sigma_{g}^{2} {\mathbf{G}}} \right)\) and \({\mathbf{e}} \sim N\left( {0,\sigma_{e}^{2} {\mathbf{I}}} \right)\), where \(\sigma_{g}^{2}\) is polygenic variance, \({\mathbf{G}}\) is the GRM, \(\sigma_{e}^{2}\) is residual variance and \({\mathbf{I}}\) is the identity matrix. In the “leaving-one-chromosome-out” approach association analysis is performed for a candidate SNP using a \({\mathbf{G}}\) that is computed without including the chromosome on which the candidate SNP is located. For instance, an association analysis between a SNP on chromosome 1 used a \({\mathbf{G}}\) that was computed only from SNPs on chromosomes 2 to 18. This approach avoids “double-fitting” of the candidate SNP into the model (both as a fixed effect and a random effect) which can reduce power, as demonstrated by Listgarten et al. [21].
Multiple testing correction
The Bonferroni correction was applied to correct for multiple testing. Since the number of SNPs was relatively similar for the three breeds, the Bonferroni correction threshold was calculated for all three breeds based on the number of SNPs for the Landrace breed, i.e. 0.05/36,080 = 1.38 × 10−6. Thus, the 5% genome-wide significance level used to avoid type I errors was 1.38 × 10−6 for all three breeds.
Meta-analyses
Within-breed multi-trait meta-analysis
A multiple trait meta-analysis was performed within each breed using the approximate multi-trait test statistic described by Bolormaa et al. [22]. Effects of a SNP across all traits were calculated and combined with the genomic correlation matrix between traits to perform a multi-trait Chi square test with the number of degrees of freedom equal to the number of traits. The formula to calculate the multi-trait statistic for each SNP was as follows:
where \({\mathbf{t}}_{{\mathbf{i}}}\) is a vector of signed t-value of SNPi across traits (t-value = SNP effect/standard error of SNP effect), \({\mathbf{V}}^{ - 1}\) is the inverse matrix of the genomic correlation matrix between traits calculated from these t-values. The significance threshold from the single-trait association analyses (i.e. P value <1.38 × 10−6) was applied for these within-breed multi-trait analyses.
Across-breed meta-analysis
Meta-analyses across the three breeds for a trait was carried out using the METAL software developed by Willer et al. [23]. The direction of the effect and the P values from each study i were converted into z-scores. These z-scores for each SNP were weighted by the sample size of each study and combined in a weighted sum across breeds. The test statistic follows the standard normal distribution and was calculated as follows:
where \(P_{i}\) is the P value for study i; \(\Delta _{i}\) is the direction of effect for study i; and \(N_{i}\) is the sample size for study i. A Bonferroni correction was applied with a significance threshold of 0.05/44,390 = 1.13 × 10−6 for all four traits.
QTL characterization
QTL region
Since the software used does not report model convergence, SNPs with an allele substitution effect estimate that fell outside the range of ±2SD of the corrected phenotypes were filtered out to avoid their probably unrealistic large effect due to inappropriate convergence in the parameter estimation. Also, only SNPs that passed the Bonferroni correction threshold were selected to mark the QTL region. A QTL region was defined by extending the position of the most significant SNP (top SNP) on either side until all SNPs within that region had a \(- log_{10} \left( {P\text{-}value} \right)\) higher than the \(- log_{10} \left( {P\text{-}value} \right)\) of the top SNP minus 3 units.
Size of the QTL effect
The size of the effect of a QTL, which was defined as the contribution of the most significant SNP within that QTL to the phenotypic variance of the trait, was calculated as \(\left( {2pq\beta^{2} /\sigma_{P}^{2} } \right)\), in which \(p\) and \(q\) are the allele frequencies, \(\beta\) is the estimated SNP effect, and \(\sigma_{P}^{2}\) is the phenotypic variance of the trait. The corresponding contribution to the genetic variance for that SNP was calculated as a proportion of the genetic variance \(\sigma_{a}^{2}\).
Candidate genes underlying the QTL
Genes that harbored the most significant SNPs within each QTL region from single-trait analyses and across-breeds meta-analyses were searched based on the pig genome assembly, Sscrofa10.2 (http://www.ensembl.org/Susscrofa/Info/Index).
Results
Descriptive statistics
Means and standard deviations for each trait in the Landrace, Yorkshire and Duroc breeds are in Table 1 (see “Methods” section). Means for CONF were slightly above the middle score 3 and ranged from 3.09 to 3.31, with the standard deviations ranged from 0.60 to 0.70 across the three breeds. Means for FRONT, BACK and HIND were all significantly greater than the middle score 2. The trait BACK had the highest means (from 2.79 to 2.91) and low standard deviations (from 0.27 to 0.41) which indicate that most animals obtained the highest score i.e. 3 for this trait. For FRONT and HIND, means ranged from 2.29 to 2.36 and from 2.32 to 2.51, with standard deviations ranging from 0.45 to 0.48 and from 0.46 to 0.50, respectively.
Heritability estimates in each breed are presented on the diagonals in Table 2. In general, estimated heritabilities were low for all traits, ranging from 0.02 to 0.13. The trait CONF had a slightly higher heritability than the other traits in all three breeds.
Genetic correlations were estimated using the linear mixed model (Model 1) and genomic correlations were estimated from signed t-values of all SNPs between traits in each breed; they are presented on the lower and upper diagonals, respectively in Table 2. The traits FRONT, HIND and CONF were highly correlated with the genetic correlations ranging from 0.72 to 0.97. The trait BACK seems to be genetically more different from the other three traits, especially in the Landrace and Yorkshire breeds, for which the estimated genetic correlations ranged from 0.23 to 0.66. When breeds were compared, genetic correlations between traits were higher in Duroc than in Landrace and Yorkshire. Regardless of the breed, genomic correlations between traits followed the same pattern as the genetic correlations but at lower magnitudes. For instance, genomic correlations of BACK with the other traits ranged from 0.14 to 0.59.
Single-trait association analyses
Genomic regions that were found to be associated with conformation traits for Landrace, Yorkshire and Duroc are in Table 3, together with the candidate genes and the top associated SNPs within each region. Visual overviews of the location of the QTL are in the Manhattan plots of Additional file 1: Figure S1, Additional file 2: Figure S2 and Additional file 3: Figure S3. QTL regions were identified on Sus scrofa chromosome (SSC) 1, 2, 3, 4, 5, 6, 7, 10, 12, 13 and 18. In general, the number of associated regions was larger for CONF than for the other traits.
In total, 14 regions were significantly associated with conformation traits in Landrace, of which five were associated with BACK, one with HIND, and eight with CONF, but none with FRONT (Table 3). The most significant SNP identified for the Landrace breed, rs80828473, was located at 36.2 Mb within the PPARD gene on SCC7. This SNP explained 0.2% of phenotypic variance (2% of genetic variance) of BACK. The second most significant SNP, rs81389032 (SSC6: 74.4 Mb), was not within any reported coding gene, but was located in a region between the genes ENSSCT00000032147 and ENSSCT00000003913.
For the Yorkshire breed, 12 regions were associated with the traits analyzed: one region with FRONT, one with BACK, three with HIND, and seven with CONF (Table 3). The most significant SNP was rs80783847 (SSC1: 199.4 Mb) and was located close to gene ENSSSCT00000005518; explained 0.2% of the phenotypic variance (2.3% of the genetic variance) of CONF. The next two most significant SNPs were located between the genes RIPPLY2 and SNAP91 (SNP on SSC1: 92.7 Mb), and between the genes CD79B and GH1 (SNP rs81440562 on SSC12: 15.0 Mb).
Among the 13 QTL regions identified for the Duroc breed, the region on SSC3 between 100.2 and 100.4 Mb showed the highest peak (Table 3). The most significant SNP in this region, rs81373717, contributed 0.9% of the phenotypic variance (11.4% of the genetic variance) of BACK. This SNP was not located within any coding gene but was between the genes EPAS1 and PRKCE. The next two most significant SNPs were rs81373756 (SSC3: 100.4 Mb) within PRKCE and rs81333163 (SSC6: 57.6 Mb) between A1BG and RPS5. They were both associated with CONF and each explained 0.5% of the phenotypic variance of this trait.
Some regions and genes showed significant association with more than one trait within a breed, suggesting the presence of a variant that may affect multiple conformation traits. For example, strong associations were found: (1) PPARD (SSC7: 36.2 Mb) was highly associated with both BACK and CONF in Landrace; (2) the QTL region located between RIPPLY2 (92.6 Mb) and SNAP91 (92.8 Mb) on SSC1 exhibited high association with both CONF and HIND in Yorkshire; and (3) the region that comprises PRKCE (SSC3: 100.5 Mb) showed high association with both CONF and BACK in Duroc. In the single-trait within-breed association analyses, the only region that overlapped between breeds was on SSC7 and it was associated with CONF in Yorkshire and Duroc.
Meta-analyses
Within-breed multi-trait meta-analysis
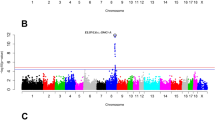
The numbers of significant SNPs identified in within-breed multi-trait meta-analyses for Landrace, Yorkshire and Duroc (206, 257 and 306 SNPs, respectively) were larger than in single-trait analyses (not shown). Their distribution on the genome was showed in the Manhattan plots in Additional file 4: Figure S4. Many of the SNPs that were found significantly associated with more than one trait in the single-trait analyses were confirmed in the multi-trait analyses, which suggests that the QTL containing these SNPs have pleiotropic effects. For instance, Fig. 1 shows the significance of the effects of SNPs on SSC6 and SSC3 from single-trait and multi-trait analyses in Duroc, which suggests that the corresponding QTL regions on these chromosomes affect multiple traits. Multi-trait meta-analysis can increase the power of QTL detection, since in general SNPs had lower P-values in the multi-trait analyses than in the single-trait analyses. Table 4 presents examples of such lower P-values for the most significant SNPs detected in the multi-trait analyses compared with the single-trait analyses.

The \(- \varvec{log}\left( {\varvec{P}\text{-}\varvec{value}} \right)\) of SNPs on SSC6 and SSC3 from single-trait and multi-trait meta-analyses in Duroc
Meta-analysis across breeds by trait
In total 36 regions were associated with the traits analyzed in the across-breed meta-analyses: three regions were associated with FRONT, eight with BACK, seven with HIND and 18 with CONF [Fig. 2 and Additional file 5: Table S1]. Among these 36 regions, several QTL regions that were detected in the single-trait within-breed analysis were confirmed and several additional regions with novel candidate genes were identified. For instance, the most significant SNP associated with CONF, rs81344309 (SSC6: 52.0 Mb), which is located between the two coding genes ENSSCG00000003243 and ZNF614, was not identified in the single-trait analyses. Similarly, the two second most significant SNPs are also new and are intergenic on SSC7: rs342640079 (34.9 Mb) between GRM4 and HMGA1 and rs80894106 (103.5 Mb) between VRTN and SYNDIG1L. The latter SNP is associated with CONF in both Yorkshire and Duroc in the single-trait analyses, but reached the higher level of significance in the across-breed meta-analysis.

Manhattan plots of across-breed meta-analyses for a FRONT, b BACK, c HIND and d CONF. x-axis represents the chromosomes and y-axis represents \({-}log_{10} \left( {P\text{-}value} \right)\). The red line indicates genome-wide significance threshold at P value <1.13 × 10−6
Discussion
Single-trait association analysis
The present GWAS detected several chromosomal regions that are associated with conformation traits in the three pig breeds, and in many cases, we were able to identify candidate genes within these regions. Results from GWAS in humans and other species suggest that conformation traits are complex and affected by various factors such as bone and cartilage development, muscle growth, fat accumulation and body weight gain [17, 24, 25]. Changes in these factors and how they interact with each other probably determine the skeletal structure and movement pattern of an individual. Several of the identified candidate genes e.g. LRPPRC, WRAP73, VRTN and PPARD are involved in bone and muscle development at different levels. Liu and McKeehan [26] suggested that LRPPRC has a role in the regulation of cytoskeleton network activity by analyzing its sequence and the proteins it interacts with. WRAP73 belongs to the WD repeat protein gene family, which includes the WDR8 gene that plays an essential role in the ossification process. Expression of WDR8 was observed in bone-forming cells and in bone and cartilage tissue during the early stage of ectopic ossification in mouse [27]. A polymorphism in the VRTN gene was found to be related to the number of vertebrae in domestic pigs with one allele resulting in an additional segment in the thoracic vertebrae compared with the wild type allele [28]. PPARD, a candidate gene for BACK and CONF in Landrace, was reported to be linked with muscle development and metabolism in pigs [29]. Expression of porcine PPARD inhibits the formation of myotube and increases adipocyte differentiation in mouse myoblasts [29]. In addition, several GWAS revealed the association of PPARD with limb bone length [30] and growth and fatness traits [31] in pigs. Similarly, HMGA1 which is located in a QTL region that we identified on SSC7 in Duroc, was reported to be associated with length of limb bone in pigs [30, 32] and height and length of hip axis in humans [24]. In vitro studies showed that addition of HMGA1 enhanced the proliferation of chondrocyte cells by regulating the expression of a chondrocyte-specific marker [33]. These findings suggest that VRTN, PPARD and HMGA1 are candidate genes for conformation traits in pigs by regulating bone and muscle development.
Growth-promoting factors, including insulin and IGF, are known to participate in bone and fat metabolism and in the growth process [34, 35]. In this study, several genes involved in the growth pathway were identified, such as IGF2BP2, GH1, CCND2 and MSH2. The protein IGF2BP2 plays an important role in controlling the action of IGF [36] which are associated with growth and fatness traits in pigs [31]. Similarly, GH1 is necessary for growth promotion and energy metabolism regulation [37]. Members of the candidate gene CCND2 for BACK in Landrace are essential for growth of pancreatic islets [38], which play a role in the regulation of the growth of an animal via its hormones. Another gene related to the growth pathway that was identified in this study, MSH2, regulates the activity of the melanocortin system which is involved in fat accumulation, feed intake and daily weight gain in pigs [39]. These results suggest that growth-related genes have a regulatory function on conformation traits in pigs but how they are associated with each other needs further study.
In this study, we identified several SNPs and chromosomal regions that were significantly associated with the traits analyzed. However, some of the top SNPs were located in intergenic regions between coding genes. Some of these genes, such as RIPPLY2 (SSC1: 92.6 Mb) and SYNDIG1L (SSC7: 103.5) are associated with vertebrae and rib development. RIPPLY2 is involved in somitogenesis during the embryo stage in mice, and knockout mice for this gene die during the perinatal period due to severe vertebrae and rib malformation [40]. In humans, it was shown that mutations in RIPPLY2 are associated with segment defects of the vertebrae [41]. In pigs, Verardo et al. [42] reported that SYNDIG1L was associated with number of teats while Duijvesteijn et al. [43] showed that number of teats and number of vertebras were controlled by several pleiotropic coding genes. These findings suggest that SYNDIG1L may have a role in the development of vertebrae in pigs. Many causal genetic factors have been previously reported to be located in the regulatory regions of genes which may explain why several candidate SNPs were identified at intergenic locations in this study. However, it may also indicate that the density of the SNP chips used was not sufficiently high. In that case, an association analysis with imputed whole-genome sequence might be able to further pinpoint the causal mutations [12].
The highly significantly associated gene with CONF in Duroc, PRKCE, is involved in several biological processes. PRKCE is a well-known key factor in cell proliferation and differentiation, muscle contraction, gene expression, cell growth and apoptosis, metabolism and diabetes, as reviewed by Akita [44] and Geraldes and King [45]. PRKCE plays an essential role in regulating lipogenesis by the interaction with GH [44] or IGF1 [46]. However, overexpression of PRKCE results in malignant tumors and diabetes [44, 46]. This relationship between growth-related factors and diabetes and bone metabolism could explain the association of PRKCE with conformation traits.
Not all candidate genes that were identified here have an obvious biological function on conformation traits. For example, some of the candidate genes are expressed in neuronal tissues and are related to the activity of the neuronal system, i.e. SLC14A2, PPM1G and NGFR [47–49]. The role of neuronal genes in the regulation of the metabolism, fat accumulation and body weight gain was investigated in humans and pigs [47, 50, 51]. Willer et al. [50] conducted a meta-analysis of 15 GWAS, which detected eight loci that are significantly associated with body mass index in humans. Another association study that combined human and pig data showed that several neuronal genes were associated with subcutaneous fat accumulation, which provides further support for a role of the nervous system on fat metabolism [47]. Another gene, SLC14A2 which is a member of the SLC super family, is mainly expressed in brain [52] and associated with fat thickness in humans and pigs [47]. Since the fat accumulation process probably affects the body structure and movement pattern of an animal, it would be interesting to investigate further how the nervous system influences conformation traits.
Within-breed multi-trait meta-analysis
In this study, the detection of QTL was enhanced in the multi-trait meta-analyses compared with the single-trait analyses. In other words, larger numbers of significant SNPs and higher significance levels of the top SNPs were observed in the meta-analyses. This phenomenon was also reported by Bolormaa et al. [22] for stature, fatness and reproduction related traits in beef cattle and more recently by Pausch et al. [53] for mammary gland morphology traits in dairy cattle. The ability to account for the relatedness between traits of multi-trait meta-analyses can explain the enhanced power of this approach here, where the four traits studied were highly genetically correlated [54]. The most significant SNPs detected in the multi-trait analyses for all three breeds reached higher significance level compared with the single-trait analysis, but they were not located within any annotated genes. Our results confirmed that multi-trait analyses can enhance the power to detect new associated SNPs, pleiotropic QTL and SNPs that were associated with only one of the correlated traits [55].
Across-breed meta-analysis
Phenotype and genotype data from all three breeds were available, and thus a GWAS analysis with pooled data could have been done instead of the across-breed meta-analysis. However, these breeds have been separated for many generations and have undergone strong artificial selection and genetic drift, which means that different sets of QTL may be segregating in each breed. Moreover, GWAS relies on linkage disequilibrium (LD) between markers and causal variants. The marker-QTL linkage phase can differ by breed and, thus, a joint analysis will have less power to detect such QTL. The genome-wide and local pattern of LD and the persistence of LD phase have been investigated in these three breeds [56], which showed that the persistence of LD phase was higher between Landrace and Yorkshire than between these two breeds and Duroc.
The results of the meta-analysis confirmed the associations of a number of candidate genes that had been detected in the single-breed analyses, such as PRKCE, SLC14A2, HMGA1, VRTN and SYNDIG1L. The greater significance level of the top SNPs was expected due to the improved power of detection for common SNPs in the meta-analyses compared with single-breed analyses. This advantage of meta-analyses has been reported in pigs [32], cattle [57, 58], and human [24, 50]. In this study, meta-analyses revealed several new QTL regions and candidate genes associated with the traits studied among which some are linked with bone, skeletal or muscle development, including SOS2, TRIM24 and ELMO1. Mutations in SOS2 are associated with the Noonan syndrome in humans, i.e. patients with this syndrome have short stature, weak muscles and malformed skeleton [59]. They are often diagnosed with GH deficiency and respond well to GH therapy [60], which suggests that SOS2 is associated with conformation traits via growth-regulated processes. TRIM24 belongs to the superfamily of tripartite motif-containing proteins which have a role in the immune system. A member of this protein family, TRIM76, was reported to be highly expressed in porcine skeletal muscle and significantly associated with carcass traits such as ham percentage and intramuscular fat [61]. The results of our meta-analysis suggested that ELMO1, which is associated with the development and progress of diabetes nephropathy in humans, is a candidate gene for CONF [62]. Another interesting region associated with CONF was detected at 25 Mb on SSC12 where the Antp homeobox (HOX) gene family is located. Four HOX gene clusters (HOXA, HOXB, HOXC and HOXD) are known to be associated with the formation and development of vertebrae [63]. The combined expression of these genes defines somite identities in mammalian embryos, which direct the differentiation and the development of the vertebrae according to their location [63]. In this study, HOXB5 and HOXB13 were the closest genes to the top SNP in the associated region. An association between HOXB genes and number of lumbar and thoracolumbar vertebrae was also reported in pigs [64].
The diversity in the candidate genes and their biological functions found in this study confirmed the complex pattern of the genetic mechanisms that underlie conformation traits in pigs. Bone and skeleton development, muscle and fat metabolism and growth processes probably interact together to determine the general conformation and movement of a pig. However, these interactions are still unclear and should be investigated further.
Conclusions
Conformation traits are complex and appear to be controlled by genes that are involved in different biological processes, including bone and skeleton development, muscle and fat metabolism and body growth. Our results suggested the association of the LRPPRC, WRAP73, VRTN, PPARD, IGF2BP2, GH1, CCND2 and MSH2 genes with conformation traits in pigs. We show that meta-analysis is a powerful QTL detection approach since we were able to detect possible QTL with pleiotropic effects in the multi-trait meta-analyses, and novel relevant candidate genes such as SOS2, TRIM24 and ELMO1 in the across-breed meta-analyses. Our findings are reliable and can be used in fine-mapping to confirm the effects of the genes identified, as well as in marker-assisted selection to improve the conformation in pigs.
References
Heinonen M, Peltoniemi O, Valros A. Impact of lameness and claw lesions in sows on welfare, health and production. Livest Sci. 2013;156:2–9.
Bonde M, Rousing T, Badsberg JH, Sørensen JT. Associations between lying-down behaviour problems and body condition, limb disorders and skin lesions of lactating sows housed in farrowing crates in commercial sow herds. Livest Prod Sci. 2004;87:179–87.
Le TH, Madsen P, Lundeheim N, Nilsson K, Norberg E. Genetic association between leg conformation in young pigs and sow longevity. J Anim Breed Genet. 2016;133:283–90.
Hoge MD, Bates RO. Developmental factors that influence sow longevity. J Anim Sci. 2011;89:1238–45.
Nikkilä MT, Stalder KJ, Mote BE, Rothshild MF, Gunsett FC, et al. Genetic associations for gilt growth, compositional, and structural soundness traits with sow longevity and lifetime reproductive performance. J Anim Sci. 2013;91:1570–9.
Rydhmer L. Swine breeding programmes in the Nordic countries. In: National Swine Improvement Federation. 2005. http://www.nsif.com/conferences/2005/pdf%255CBreedingNordicCountries.pdf.
Le HT, Nilsson K, Norberg E, Lundeheim N. Genetic association between leg conformation in young pigs and sow reproduction. Livest Sci. 2015;178:9–17.
Knauer MT, Cassady JP, Newcom DW, See MT. Phenotypic and genetic correlations between gilt estrus, puberty, growth, composition, and structural conformation traits with first-litter reproductive measures. J Anim Sci. 2011;89:935–42.
Nikkilä MT, Stalder KJ, Mote BE, Rothschild MF, Gunsett FC, Johnson AK, et al. Genetic parameters for growth, body composition, and structural soundness traits in commercial gilts. J Anim Sci. 2013;91:2034–46.
Aasmundstad T, Olsen D, Sehested E, Vangen O. The genetic relationships between conformation assessment of gilts and sow production and longevity. Livest Sci. 2014;167:33–40.
Calus MPL, Meuwissen THE, De Roos APW, Veerkamp RF. Accuracy of genomic selection using different methods to define haplotypes. Genetics. 2008;178:553–61.
Brøndum RF, Su G, Janss L, Sahana G, Guldbrandtsen B, Boichard D, et al. Quantitative trait loci markers derived from whole genome sequence data increases the reliability of genomic prediction. J Dairy Sci. 2015;98:4107–16.
Hidalgo AM. Exploiting genomic information on purebred and crossbred pigs. Doctoral Thesis. Wageningen University; 2015.
Madsen P, Jensen J, Labouriau R, Christensen OF, Sahana G. DMU—a package for analyzing multivariate mixed models in quantitative genetics and genomics. In: Proceedings of the 10th world congress on genetics applied to livestock production: 18–22 August 2014; Vancouver, p. 40–2.
Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2008;84:210–23.
Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82.
Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–9.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9.
Yang J, Weedon MN, Purcell S, Lettre G, Estrada K, Willer CJ, et al. Genomic inflation factors under polygenic inheritance. Eur J Hum Genet. 2011;19:807–12.
Yang J, Zaitlen NA, Goddard ME, Visscher PM, Price AL. Advantages and pitfalls in the application of mixed-model association methods. Nat Genet. 2014;46:100–6.
Listgarten J, Lippert C, Kadie CM, Davidson RI, Eskin E, Heckerman D. Improved linear mixed models for genome-wide association studies. Nat Methods. 2013;9:525–6.
Bolormaa S, Pryce JE, Reverter A, Zhang Y, Barendse W, Kemper K, et al. A multi-trait, meta-analysis for detecting pleiotropic polymorphisms for stature, fatness and reproduction in beef cattle. PLoS Genet. 2014;10:e1004198.
Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1.
Soranzo N, Rivadeneira F, Chinappen-Horsley U, Malkina I, Richards JB, Hammond N, et al. Meta-analysis of genome-wide scans for human adult stature identifies novel loci and associations with measures of skeletal frame size. PLoS Genet. 2009;5:e1000445.
Wu X, Fang M, Liu L, Wang S, Liu J, Ding X, et al. Genome wide association studies for body conformation traits in the Chinese Holstein cattle population. BMC Genomics. 2013;14:897.
Liu L, McKeehan WL. Sequence analysis of LRPPRC and its SEC1 domain interaction partners suggests roles in cytoskeletal organization, vesicular trafficking, nucleocytosolic shuttling and chromosome activity. Genomics. 2002;79:124–36.
Koshizuka Y, Ikegawa S, Sano M, Nakamura K, Nakamura Y. Isolation, characterization, and mapping of the mouse and human WDR8 genes, members of a novel WD-repeat gene family. Genomics. 2001;72:252–9.
Mikawa S, Sato S, Nii M, Morozumi T, Yoshioka G, Imaeda N, et al. Identification of a second gene associated with variation in vertebral number in domestic pigs. BMC Genet. 2011;12:5.
Yu YH, Wu SC, Cheng WT, Mersmann HJ, Ding ST. Ectopic expression of porcine peroxisome proliferator-activated receptor delta regulates adipogenesis in mouse myoblasts. J Anim Sci. 2008;86:64–72.
Zhang L-C, Li N, Liu X, Liang J, Yan H, Zhao K-B, et al. A genome-wide association study of limb bone length using a Large White × Minzhu intercross population. Genet Sel Evol. 2014;46:56.
Qiao R, Gao J, Zhang Z, Li L, Xie X, Fan Y, et al. Genome-wide association analyses reveal significant loci and strong candidate genes for growth and fatness traits in two pig populations. Genet Sel Evol. 2015;47:17.
Guo Y, Hou L, Zhang X, Huang M, Mao H, Chen H, et al. A meta analysis of genome-wide association studies for limb bone lengths in four pig populations. BMC Genet. 2015;16:95.
Richter A, Hauschild G, Murua Escobar H, Nolte I, Bullerdiek J. Application of high-mobility-group-A proteins increases the proliferative activity of chondrocytes in vitro. Tissue Eng Part A. 2009;15:473–7.
Kang H, Sung J, Jung HM, Woo KM, Hong SD, Roh S. Insulin-like growth factor 2 promotes osteogenic cell differentiation in the parthenogenetic murine embryonic stem cells. Tissue Eng Part A. 2012;18:331–41.
Bilezikian JP, Raisz LG, Rodan GA. Principles of bone biology. 2nd ed. San Diego: AcademicPress; 2002.
Clemmons DR. Insulin-like growth factor binding proteins and their role in controlling IGF actions. Cytokine Growth Factor Rev. 1997;8:45–62.
Goodman HM. Growth hormone and metabolism. In: Schreibman MP, Scanes CG, Pang PKT, editors. The endocrinology of growth, development, and metabolism in veterbrates. London: Academic Press Inc; 1993. p. 93–115.
Kushner JA, Ciemerych MA, Sicinska E, Wartschow M, Teta M, Long SY, et al. Cyclins D2 and D1 are essential for postnatal pancreatic β-cell growth. Mol Cell Biol. 2005;25:3752–62.
Switonski M, Mankowska M, Salamon S. Family of melanocortin receptor (MCR) genes in mammals-mutations, polymorphisms and phenotypic effects. J Appl Genet. 2013;54:461–72.
Chan T, Kondow A, Hosoya A, Hitachi K, Yukita A, Okabayashi K, et al. Ripply2 is essential for precise somite formation during mouse early development. FEBS Lett. 2007;581:2691–6.
McInerney-Leo AM, Sparrow DB, Harris JE, Gardiner BB, Marshall MS, O’Reilly VC, et al. Compound heterozygous mutations in RIPPLY2 associated with vertebral segmentation defects. Hum Mol Genet. 2015;24:123–42.
Verardo LL, Silva FF, Lopes MS, Madsen O, Bastiaansen JWM, Knol EF, et al. Revealing new candidate genes for reproductive traits in pigs: combining Bayesian GWAS and functional pathways. Genet Sel Evol. 2016;48:9.
Duijvesteijn N, Veltmaat JM, Knol EF, Harlizius B. High-resolution association mapping of number of teats in pigs reveals regions controlling vertebral development. BMC Genomics. 2014;15:542.
Akita Y. Protein kinase C-ε (PKC-ε) its unique structure and function. J Biochem. 2002;132:847–52.
Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res. 2010;106:1319–31.
Allen TR, Krueger KD, Hunter WJ III, Agrawal DK. Evidence that insulin-like growth factor-1 requires protein kinase C-epsilon, PI3-kinase and mitogen-activated protein kinase pathways to protect human vascular smooth muscle cells from apoptosis. Immunol Cell Biol. 2005;83:651–67.
Lee KT, Byun MJ, Kang KS, Park EW, Lee SH, Cho S, et al. Neuronal genes for subcutaneous fat thickness in human and pig are identified by local genomic sequencing and combined SNP association study. PLoS One. 2011;6:e16356.
Foster WH, Langenbacher A, Gao C, Chen J, Wang Y. Nuclear phosphatase PPM1G in cellular survival and neural development. Dev Dyn. 2013;242:1101–9.
Hefti F, Mash DC. Localization of nerve growth factor receptors in the normal human brain and in Alzheimer’s disease. Neurobiol Aging. 1989;10:75–87.
Willer CJ, Speliotes EK, Loos RJF, Li S, Lindgren CM, Heid IM, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25–34.
Fontanesi L, Schiavo G, Galimberti G, Calò DG, Scotti E, Martelli PL, et al. A genome wide association study for backfat thickness in Italian Large White pigs highlights new regions affecting fat deposition including neuronal genes. BMC Genomics. 2012;13:583.
Dahlin A, Royall J, Hohmann JG, Wang J. Expression profiling of the solute carrier gene family in the mouse brain. J Pharmacol Exp Ther. 2009;329:558–70.
Pausch H, Emmerling R, Schwarzenbacher H, Fries R. A multi-trait meta-analysis with imputed sequence variants reveals twelve QTL for mammary gland morphology in Fleckvieh cattle. Genet Sel Evol. 2016;48:14.
Zhou X, Stephens M. Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat Methods. 2014;11:407–9.
Stephens M. A unified framework for association analysis with multiple related phenotypes. PLoS One. 2013;8:e65245.
Wang L, Sørensen P, Janss L, Ostersen T, Edwards D. Genome-wide and local pattern of linkage disequilibrium and persistence of phase for 3 Danish pig breeds. BMC Genet. 2013;14:115.
Mao X, Sahana G, De Koning DJ, Guldbrandtsen B. Genome-wide association studies of growth traits in three dairy cattle breeds using whole-genome sequence data. J Anim Sci. 2016;94:1426–37.
Raven LA, Cocks BG, Hayes BJ. Multibreed genome wide association can improve precision of mapping causative variants underlying milk production in dairy cattle. BMC Genomics. 2014;15:62.
Allanson JE, Hall JG, Van Allen MI. Noonan phenotype associated with neurofibromatosis. Am J Med Genet. 1985;21:457–62.
Noonan JA, Kappelgaard AM. The efficacy and safety of growth hormone therapy in children with Noonan syndrome: a review of the evidence. Horm Res Paediatr. 2015;83:157–66.
Xu X, Xu X, Yin Q, Sun L, Liu B, Wang Y. The molecular characterization and associations of porcine cardiomyopathy asssociated 5 (CMYA5) gene with carcass trait and meat quality. Mol Biol Rep. 2011;38:2085–90.
Shimazaki A, Kawamura Y, Kanazawa A, Sekine A, Saito S, Tsunoda T, et al. Genetic variations in the gene encoding ELMO1 are associated with susceptibility to diabetic nephropathy. Diabetes. 2005;54:1171–8.
Kessel M, Gruss P. Homeotic transformations of murine vertebrae and concomitant alteration of Hox codes induced by retinoic acid. Cell. 1991;67:89–104.
Rohrer GA, Nonneman DJ, Wiedmann RT, Schneider JF. A study of vertebra number in pigs confirms the association of vertnin and reveals additional QTL. BMC Genet. 2015;16:129.
Authors’ contributions
BN extracted the data from the database. GS and OFC conceived and supervised the research project. THL performed the statistical analyses and wrote the manuscript. All authors contributed to the final manuscript. All authors read and approved the final manuscript.
Acknowledgements
Thu Hong Le benefited from a joint grant from the European Commission within the framework of the Erasmus Mundus joint doctorate “EGS-ABG”. We are grateful to EGS ABG for funding and DanAvl for providing the data. Xiaowei Mao and Xiaoping Wu are acknowledged for helpful discussions during the data analyses.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional files
12711_2017_289_MOESM1_ESM.docx
Additional file 1: Figure S1. Manhattan plot of GWAS in Landrace pigs for (a) FRONT, (b) BACK, (c) HIND and (d) CONF. The data provided represent the Manhattan plot of single-trait association analyses in Landrace pigs for four traits studied.
12711_2017_289_MOESM2_ESM.docx
Additional file 2: Figure S2. Manhattan plot of GWAS in Yorkshire pigs for (a) FRONT, (b) BACK, (c) HIND and (d) CONF. The data provided represent the Manhattan plot of single-trait association analyses in Yorkshire pigs for four traits studied.
12711_2017_289_MOESM3_ESM.docx
Additional file 3: Figure S3. Manhattan plot of GWAS in Duroc pigs for (a) FRONT, (b) BACK, (c) HIND and (d) CONF. The data provided represent the Manhattan plot of single-trait association analyses in Duroc pigs for four traits studied.
12711_2017_289_MOESM4_ESM.docx
Additional file 4: Figure S4. Manhattan plot of within-breed multi-trait meta-analyses in (a) Landrace, (b) Yorkshire and (c) Duroc. The data provided represent the Manhattan plot of within-breed multi-trait meta-analyses in three breeds studied.
12711_2017_289_MOESM5_ESM.docx
Additional file 5: Table S1. QTL regions and the most significant SNP within each region in across-breed meta-analyses. The data provided represent the QTL regions and the information of the most significant SNP within each region in across-breed meta-analyses.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Le, T.H., Christensen, O.F., Nielsen, B. et al. Genome-wide association study for conformation traits in three Danish pig breeds. Genet Sel Evol 49, 12 (2017). https://doi.org/10.1186/s12711-017-0289-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12711-017-0289-2