
Abstract
Background
Genomic prediction requires estimation of variances of effects of single nucleotide polymorphisms (SNPs), which is computationally demanding, and uses these variances for prediction. We have developed models with separate estimation of SNP variances, which can be applied infrequently, and genomic prediction, which can be applied routinely.
Methods
SNP variances were estimated with Bayes Stochastic Search Variable Selection (BSSVS) and BayesC. Genome-enhanced breeding values (GEBV) were estimated with RR-BLUP (ridge regression best linear unbiased prediction), using either variances obtained from BSSVS (BLUP-SSVS) or BayesC (BLUP-C), or assuming equal variances for each SNP. Datasets used to estimate SNP variances comprised (1) all animals, (2) 50% random animals (RAN50), (3) 50% best animals (TOP50), or (4) 50% worst animals (BOT50). Traits analysed were protein yield, udder depth, somatic cell score, interval between first and last insemination, direct longevity, and longevity including information from predictors.
Results
BLUP-SSVS and BLUP-C yielded similar GEBV as the equivalent Bayesian models that simultaneously estimated SNP variances. Reliabilities of these GEBV were consistently higher than from RR-BLUP, although only significantly for direct longevity. Across scenarios that used data subsets to estimate GEBV, observed reliabilities were generally higher for TOP50 than for RAN50, and much higher than for BOT50. Reliabilities of TOP50 were higher because the training data contained more ancestors of selection candidates. Using estimated SNP variances based on random or non-random subsets of the data, while using all data to estimate GEBV, did not affect reliabilities of the BLUP models. A convergence criterion of 10−8 instead of 10−10 for BLUP models yielded similar GEBV, while the required number of iterations decreased by 71 to 90%. Including a separate polygenic effect consistently improved reliabilities of the GEBV, but also substantially increased the required number of iterations to reach convergence with RR-BLUP. SNP variances converged faster for BayesC than for BSSVS.
Conclusions
Combining Bayesian variable selection models to re-estimate SNP variances and BLUP models that use those SNP variances, yields GEBV that are similar to those from full Bayesian models. Moreover, these combined models yield predictions with higher reliability and less bias than the commonly used RR-BLUP model.
Similar content being viewed by others

Background
Genomic prediction is currently used in many breeding schemes around the world. Initial challenges for genomic prediction were to overcome the so-called n < < p problem, because the number of SNP (single nucleotide polymorphism) effects (p) that needs to be estimated in the model is typically much larger than the number of individuals with records in the training dataset (n). Much research in the past few years has been conducted to develop and test different genomic prediction models [1]. With the costs of genotyping dropping continuously, and with the expected use of whole-genome sequence data in practical applications in the short term [2], the dimensions of datasets are growing rapidly. This also means that processing time and computer memory requirements of many genomic prediction models will rapidly increase.
Genomic prediction models can be roughly divided into two sets of models i.e. one that estimates the explained variance specific to each SNP or group of SNPs in the model, comprising most Bayesian genomic prediction models [1],[3],[4], and one that completely relies on prior assumptions to define the explained variance that may be common for all SNPs, such as the RR-BLUP (referring to Random Regression Best Linear Unbiased Prediction [5] or Ridge Regression BLUP [1]) and the GBLUP (genomic-BLUP [6]) models. Several studies have shown that using SNP-specific variances to give more weight to SNPs with large effects, may improve the accuracy of genomic prediction [4],[6],[7], although other studies, in particular those based on real data, have reported no differences in accuracies [1]. However, in traditional pedigree-based breeding value estimation models used in routine evaluations, variance components and breeding values are rarely estimated simultaneously, because this is computationally not feasible when using very large datasets [8]. Instead, variance components are usually estimated for a subset of the data, for which more stringent editing criteria are applied compared to data used for breeding value estimation. Because variance components are expected to be relatively consistent over time, they are re-estimated less frequently using REML or Bayesian models [9],[10]. However, for some species, breeding values are estimated much more frequently i.e. for dairy cattle [11] twice or four times per year for national genetic evaluations according to the ICAR guidelines [12] and for pig and poultry on a weekly or even daily basis. Compared to the models applied to estimate variance components, those applied to predict breeding values use different algorithms, for example preconditioned conjugate gradients (PCG) [13].
To reduce computational burden in genomic prediction models, while still being able to use SNP-specific variances, a similar strategy that estimates separately variance components at low frequency and breeding values at much higher frequency appears to be an interesting option. The objectives of this study were: (1) to describe genomic prediction models that involve separate steps to estimate SNP variances and to estimate GEBV (genome-enhanced breeding values) using BLUP, (2) to compare the performance of this two-step procedure with the equivalent Bayesian model that estimates SNP variances and breeding values simultaneously, and (3) to investigate the impact on the reliability of GEBV obtained using BLUP with SNP variances estimated on random or non-random subsets of the data. These objectives were investigated using dairy cattle data.
Methods
Bayesian models that include variance component estimation
Two Bayesian models were used to estimate SNP effects and SNP specific variances i.e. Bayesian Stochastic Search Variable Selection (BSSVS) [14],[15] and BayesC [16]. The general model was:
where y is a vector of phenotypic records, μ is the overall mean, 1 is a vector of 1s, Z is an incidence matrix that links records to individuals, u is a vector of the random polygenic effects of all individuals, X is a matrix that contains the scaled and centered genotypes (such that they have a distribution N(0,1) for each locus) of all individuals, α is a vector of the (random) allele substitution effects for all loci, and e a vector of the random residuals.
The difference between BSSVS and BayesC lies in the distributions from which the allele substitution effects are drawn. In both models, for each iteration of the implemented Gibbs sampler, a QTL (quantitative trait locus)-indicator I j is sampled for each locus j. In both models, the effect is sampled from a distribution with large effects if I j = 1. When I j = 0, the effect is either sampled from a distribution with small effects (BSSVS) or is set to 0 (BayesC).
For BSSVS, the estimate of α j is drawn from:
where is the sum of the products of the genotypes at locus j and λ is equal to , and
For BayesC, the estimate of α j is drawn from:
if I j = 1,
0 if I j = 0
where
For both models, has a prior distribution of:
where v is the degrees of freedom, set to 4.2 following [16], and the scale parameter is calculated as , where is the prior value of and is computed as for BSSVS, where is the total genetic variance and n is the number of loci, and as for BayesC [1]. The posterior value of is drawn from the following inverse-χ2 distribution for BSSVS:
where n is the total number of SNP loci in the data, is a vector with squares of the current estimates of the allele substitution effects of all loci, which is weighted by vector ω that contains values of 1 or 100 for each locus.
The posterior value of is drawn from the following inverse-χ2 distribution for BayesC:
For both models, the posterior distribution of the QTL-indicator for locus j (I j ) was (following the notation in [17]):
where , with y* containing the conditional phenotypes and f(r j |I j = δ), with δ being equal to either 0 or 1 and proportional to . For BSSVS, . For BayesC, and . Finally, the conditional posterior density of is an inverse-χ2 distribution:
where m is the number of animals with records, and e is a vector of the current residuals.
More details on the BSSVS model are in Calus et al. [14], Calus [18] and Verbyla et al. [15], and more details on the BayesC model are in Habier et al. [16]. Both BSSVS and BayesC were run using Gibbs sampling. For each BSSVS and BayesC analysis, two replicates that each consisted of a Gibbs chain of 60 000 iterations were run, discarding 10 000 iterations for burn-in. For BSSVS, parameter π was set to 0.999, based on our experience with this model. For BayesC, parameter π was set to 0.9 for BayesC, in line with estimates for this parameter in the literature [16].
BLUP models
In addition to the two Bayesian models, three BLUP models were used to predict GEBV of validation animals. The parameterization of all three BLUP models was similar to that of the Bayesian models, except that the BLUP models did not estimate SNP (specific) variances and they were solved using Gauss-Seidel instead of Gibbs sampling. Convergence criteria were computed across the mean, polygenic breeding values and SNP effects as the sum of squared differences between current and previous solutions, divided by the sum of squared current solutions [19]. The threshold used for convergence was 10−10[20]. To evaluate the impact of using a more relaxed convergence criterion on the reliability and the number of iterations required, a convergence criterion of 10−8 was also tested.
The first BLUP model, RR-BLUP, defined the SNP variance as the total genetic variance divided by the total number of SNP loci. The second BLUP model used SNP-specific variances that were computed using BSSVS. This model will hereafter be referred to as BLUP-SSVS. The third BLUP model used SNP-specific variances that were computed using BayesC. This model will hereafter be referred to as BLUP-C.
Variance components
Genetic variances were required to compute prior SNP-variances for BSSVS and BayesC, and to compute SNP-variances used in RR-BLUP. For RR-BLUP, the SNP-specific variance was set equal to 95% of the genetic variance divided by the number of SNPs. Note that it was divided by the number of SNPs because, after scaling and centering, all genotypes had a variance of 1. The variance of the polygenic effects was set as 5% of the genetic variance. Likewise, residual variances were required for the RR-BLUP model. The genetic and residual variances were estimated from the data with a pedigree-based model.
For both BLUP-SSVS and BLUP-C, the variance of the polygenic effect and the residual variance were directly obtained from the corresponding Bayesian models. The estimated SNP variances of the BSSVS model, to be used in the BLUP-SSVS model, were computed as:
where is the posterior probability of locus j, that is computed as the average of the QTL indicator I j across all iterations after the burn-in, and is the posterior mean of the SNP variance component. Likewise, the estimated SNP variances of the BayesC model, to be used in the BLUP-C model, were computed as:
Implementation of the models
As mentioned before, the BLUP models were implemented using Gauss-Seidel, while the Bayesian models were implemented using Gibbs sampling. The treatment of the SNP variances differed between models, in the sense that they were estimated in the Bayesian models and assumed known in the BLUP models. To compute the conditional values of the allele substitution effects within iterations, in both the BLUP and Bayesian models, the right-hand-side updating algorithm was used [18]. This algorithm is an extension of residual updating that uses the feature that each locus has only three genotypes, which drastically reduces the required number of computations. To avoid rounding errors due to residual updating, the residuals were recomputed every 100th iteration [20]. The order of the SNPs in which they were handled, was permuted every 10th iteration to speed up convergence in the BLUP models and to improve mixing in the Bayesian models solved with Gibbs sampling. This implies that the BLUP models also used a random seed, except that this was only used to permute the order in which the SNPs were evaluated. For each analysis of the BLUP and the Bayesian models, two replicates were performed using different seeds. Final estimates for each scenario were obtained as the average of the two replicates.
Data
The data comprised 5000 Holstein Friesian dairy bulls with genotypes and de-regressed estimated breeding values (EBV) obtained from the Dutch national evaluations for the six following traits: protein yield, udder depth (UD), somatic cell score (SCS), interval from first to last insemination (IFL), direct longevity (DLO), and longevity including information from the predictor traits UD, SCS and locomotion (LON). Reliabilities of the EBV were used to compute effective daughter contributions (EDC) [21], which were used as weights in the analyses, by dividing the residual variance for each bull by its EDC.
The genotypes were edited as part of a larger dataset. SNPs with a minor allele frequency below 0.025, a difference between observed and expected fraction of heterozygotes (based on Hardy-Weinberg disequilibrium) larger than 0.15, or a call rate higher than 90% were removed. Any missing individual genotype was imputed using DAGPHASE [22] and Beagle [23], to avoid missing genotypes in the final data. The training data consisted of 4245 to 4271 animals across the six traits. The validation population comprised all bulls with phenotypic information in the data born since January 1 2004 onwards, with a total number of 729.
Scenarios
In terms of animals used in the Bayesian models versus the BLUP models, four different scenarios were considered, as summarized in Table 1. In the first scenario, all animals in the training dataset were used in the Bayesian models and RR-BLUP. This scenario is termed `FULL¿ hereafter. In the second scenario, a random selection of 50% of the animals from the training dataset was used in the Bayesian models and RR-BLUP, and the BLUP-C and BLUP-SSVS used the SNP-wise variances from BayesC and BSSVS, with the corresponding reduced data. This scenario is termed `RAN50¿ hereafter. It reflects a situation for which SNP-variances are estimated on a reduced random subset of the data. In the third scenario, the 50% of the animals in the training dataset with the highest de-regressed EBV were used in the Bayesian models. This scenario is termed `TOP50¿ hereafter. It reflects a situation in which SNP-variances are estimated on a reduced non-random subset of the data. The fourth scenario is similar to the third, but uses the 50% animals with the lowest de-regressed EBV. This scenario is termed `BOT50¿ hereafter. It should be noted that both the TOP50 and BOT50 scenarios were defined for each trait separately and thus contained different animals for different traits. In all four scenarios, all training animals were used with the BLUP models BLUP-SSVS and BLUP-C.
Evaluation of reliability, bias and convergence
Reliability of GEBV was calculated as the squared correlation between de-regressed EBV and GEBV, divided by the average reliability of the initial EBV that were de-regressed (ranging from 0.82 to 0.96 across traits). It should be noted that the GEBV consisted of the sum of the SNP effects and the polygenic effect. Standard errors of the reliabilities were computed for both BLUP and Bayesian models using bootstrapping through the R-package ¿boot¿ [24]. The bootstrapping procedure involved computing reliabilities for 10 000 bootstrapping samples of the 729 validation animals. Standard errors were computed as the standard deviation of those 10 000 reliability estimates. For each scenario, this standard error was computed for each replicate and then averaged across the two replicates. Bias of the GEBV was assessed by comparing mean de-regressed EBV and mean GEBV across all validation animals, and by evaluating coefficients of the regression of de-regressed EBV on GEBV.
For the BLUP models, the number of iterations to reach convergence is reported as a measure of efficiency. For BSSVS and BayesC, the estimated value of the SNP variance component was plotted for each iteration in the Gibbs chain, for visual inspection of its convergence. In order to assess whether the length of the Gibbs chain was sufficient, the effective chain length of the 50 000 samples after burn-in was computed using the R package Coda [25].
Results
Reliability of GEBV
Estimated reliabilities of GEBV across traits, scenarios, and models are in Table 2. Results indicate that within scenarios, the BSSVS and BayesC models had very similar reliabilities, while the RR-BLUP model generally had slightly lower reliabilities than BSSVS and BayesC, although the difference was only significantly different from 0 (P < 0.05) for the trait DLO. Although not significant at P < 0.05, the P-value of the difference between the reliability using RR-BLUP and the reliability using the Bayesian models was less than 0.10 for protein and LON (results not shown). The models BLUP-SSVS and BLUP-C always used all training animals, but used SNP variances that were estimated with BSSVS and BayesC using different training datasets in the different scenarios. Reliabilities obtained with BLUP-SSVS and BLUP-C were always very similar to the reliabilities obtained with BSSVS and BayesC in the FULL scenario. Comparing the reliabilities of BSSVS and BayesC across different scenarios indicates that selecting the best animals (TOP50) as training animals yielded a slightly higher reliability for four out of six traits than selecting training animals at random, while selecting the worst animals (BOT50) yielded very low reliabilities for five out of six traits. In summary, these results indicate that using random (RAN50) or non-random subsets (TOP50 and BOT50) to estimate SNP variances does not affect the GEBV, as long as the training data used to predict the GEBV includes all animals.
The equivalence of the GEBV obtained in the FULL scenario, is illustrated by the correlation between GEBV obtained with those different models (Table 3). These correlations clearly indicate that BSSVS, BayesC, BLUP-SSVS and BLUP-C gave very similar GEBV (correlations > 0.99) for all traits and scenarios. The GEBV obtained with RR-BLUP that assumes that each SNP explains the same amount of variance, tended to be slightly different from the GEBV obtained with the other models, with correlations ranging from 0.94 to 0.99.
Bias of GEBV
For all scenarios, models and traits, de-regressed EBV were compared to the GEBV, to investigate potential bias in the GEBV. The difference between mean of the de-regressed EBV and mean of the GEBV gives an indication of the bias in the level of the GEBV. Those differences show that within scenarios, biases of the Bayesian models and RR-BLUP were very similar (Table 4). Compared to the FULL scenario, for BSSVS, BayesC and RR-BLUP, the bias with the RAN50 scenario was somewhat higher for all traits except DLO and LON, and even more so for the TOP50 and BOT50 scenarios. In all scenarios, the bias in the level of predictions with models BLUP-SSVS and BLUP-C was similar to the bias observed with the Bayesian models and RR-BLUP in the FULL scenario.
Slopes of the regression of de-regressed EBV on the GEBV indicate bias in the scale of the GEBV, i.e. values greater (lower) than 1.0 indicate underestimation (overestimation) of the variance of the GEBV. For the FULL scenario, regression coefficients deviated most from 1.0 for RR-BLUP and were substantially lower than 1.0 for all traits (Table 5). The other models yielded regression coefficients closer to 1.0, but also had values substantially lower than 1.0 for the traits IFL, DLO and LON. Compared to the FULL scenario, the GEBV obtained with BSSVS and BayesC always showed a greater bias for the BOT50 scenario except for protein, and for some traits also for the RAN50 scenario. However the TOP50 scenario tended to yield the least biased GEBV, even compared to the FULL scenario, except for SCS and UD.
Estimated SNP-specific variances
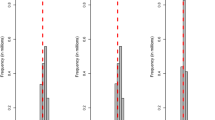
Distributions of estimated SNP-specific variances of the Bayesian models were studied since the most important difference between the Bayesian models and the RR-BLUP model lies in the SNP variance used to estimate the SNP effects. The distributions of the two independent replicates were very similar, and therefore only the distribution for the first replicates is shown in Figure 1. These results show that the maximum SNP variances were substantially larger for the BSSVS model compared to the BayesC model.

SNP-specific variances estimated with BSSVS and BayesC. Estimates were obtained for all six traits in the FULL scenario, for the first replicate. The largest SNP variances across traits were equal to 0.52, 0.0165, 0.0270, 0.0086, 76.95 and 76.15 for BSSVS and 0.059, 0.0041, 0.0033, 0.0020, 8.09, and 7.90 for BayesC.
Convergence of the Bayesian models
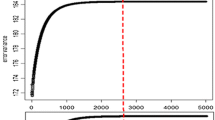
The pattern of the SNP variance component across iterations, appeared to be quite stable after the 10 000 iterations of burn-in, both for BSSVS and BayesC, as illustrated in Figure 2 for the first replicate of each trait in the FULL scenario. In fact, the patterns suggest that using 5000 iterations for burn-in would be sufficient for all traits and both models, while for some traits as little as 2000 iterations appears to be sufficient for burn-in.

SNP variance components across iterations, estimated with BSSVS and BayesC. Estimates were obtained for all six traits in the FULL scenario; values are shown only for the first replicate. The effective chain length of the last 50 000 samples is given for each figure.
Effective chain lengths ranged from 57.9 to 211.4 for BSSVS and from 179.6 to 522.2 for BayesC (Figure 2). In nearly all cases, the effective chain length was roughly twice as large for BayesC compared to BSSVS, suggesting that the SNP variance component reaches convergence faster in BayesC than in BSSVS. Considering that the effective chain length should be at least 50, this suggests that across traits and models, anywhere between ~5000 and 50 000 iterations are required after burn-in.
Convergence of the BLUP model
The number of iterations required for the different BLUP models until convergence, averaged across both replicates, is in Table 6. In general, the required number of iterations was rather similar for RR-BLUP, BLUP-SSVS and BLUP-C. The only clear difference was observed between RR-BLUP on the one hand, and BLUP-SSVS and BLUP-C on the other hand for the traits UD and SCS in the TOP50 scenario and for IFL in the BOT 50 scenario. In those cases, RR-BLUP required 4000 to 5000 iterations compared to 13 000 to 16 000 for BLUP-SSVS and BLUP-C.
When using a convergence criteria of 10−8 instead of 10−10 for the FULL scenario, GEBV had very similar reliabilities (Table 7), and were indeed virtually the same, i.e. they had a correlation higher than 0.999 (results not shown). The required number of iterations, however, was only 1832 to 4928 (Table 7), and thereby decreased by 71 to 90% compared to the more stringent convergence criterion of 10−10.
Discussion
The objectives of this study were to develop and describe genomic prediction models with a separate step to estimate SNP specific variances with a Bayesian model and a subsequent step to predict GEBV using a BLUP model. Such a system has the advantage that SNP variances can be estimated less frequently, while genomic evaluations can be performed at higher frequency with a BLUP model. BLUP models have the advantage that monitoring of convergence is straightforward and convergence is obtained within a limited number of iterations, as our results confirmed, while it was expected that the results of the BLUP models were similar to those of the Bayesian models. Our results confirmed that the BLUP models, using SNP-specific variances, yielded GEBV that were very similar to those with the Bayesian models, even if the SNP-specific variances were estimated from a non-random subset of the data. Other studies have drawn similar conclusions when SNP variances were estimated with Lasso [26] or BayesB [7] and later used in a GBLUP type of model, or when a non-linear weighting was directly incorporated in the GBLUP model [6], although those studies did not investigate the sensitivity of the models to estimating SNP variances from non-random subsets of the data. Another advantage of using pre-computed SNP variances from the data rather than using variances that are a priori distributed across the SNPs, is that the SNP variances used are not very dependent on assumptions that need to be made in RR-BLUP, where the variance for all SNPs is assumed equal and simply computed as the total genetic variance divided by the number of SNPs. Using estimated SNP variances instead, allows the variances to differ between SNPs, and even to adapt, for instance, to linkage disequilibrium between SNPs, which may affect the variance associated to them. Our results suggest that the assumptions of, e.g., RR-BLUP may result in less accurate and more biased predictions.
The GEBV obtained using BSSVS and BayesC were very similar, despite the observation that the distributions of SNP effects differed considerably between the two models (Figure 1). It should be noted that the differences in SNP-specific variances between the two Bayesian models were mainly due to differences in priors. Initially, π was set equal to 0.999 and 0.99 for BSSVS and BayesC, respectively. The value of 0.99 for BayesC was chosen to obtain the same prior SNP variance component for both models. However, the reliabilities obtained for BayesC with this initial value for π were substantially lower than those for BSSVS (results now shown). Therefore, we decided to use a π value of 0.90 for BayesC, which is closer to empirical estimates for BayesC reported in the literature [16]. With this value of π for BayesC, results of BSSVS and BayesC were very similar.
Use of subsets of data to estimate SNP variances
Although BSSVS and BayesC consistently outperformed RR-BLUP, the difference in observed reliabilities was not significantly different from 0 for nearly all cases, despite the relatively large number of validation animals used, i.e. 724. Also, the scale of the GEBV obtained with BLUP-SSVS and BLUP-C was consistently less biased than the scale of the GEBV obtained with RR-BLUP. In fact, when SNP variances were estimated with a subset of the data (RAN50, TOP50 and BOT50), both BLUP-SSVS and BLUP-C, which used all training data, were in most cases able to reduce the bias observed in the level of the GEBV (Table 4) and overcome the bias observed in the scale of the GEBV with the Bayesian models (Table 5). These results are in line with those of other studies that suggest that bias in GEBV due to genomic pre-selection can be overcome by including all information of selected and unselected animals when estimating GEBV [27],[28]. However, based on our results using all information does not seem necessary when estimating the SNP variances. Thus, it is concluded that genomic prediction models that use pre-computed SNP variances are efficient and can generate GEBV with improved properties, in terms of reliability and bias, compared to the commonly used RR-BLUP model.
One clear trend in the results was that GEBV predicted using the 50% of the animals in the training data with the highest de-regressed EBV (TOP50) resulted for most traits (Protein, SCS, DLO, and LON) in a higher reliability than using a random 50% of the animals as training data (RAN50). For all traits except IFL, selecting the 50% animals with the lowest de-regressed EBV resulted in a very low reliability. The explanation for these results is that the validation animals in our study are not just a random subset of animals, but selection candidates, i.e. their sires most likely have an above average breeding value and are therefore likely to be included in the training data in the TOP50 scenario. This explanation can be investigated by simply counting per scenario the number of selection candidates that have sires, paternal or maternal grandsires with de-regressed EBV in the training data. The results are in Table 8 and show that the number of male ancestors in the reference population were substantially larger for the TOP50 animals than for the RAN50 animals for all traits, except for IFL, in agreement with the observation that IFL had a substantially lower reliability with the TOP50 scenario compared to the RAN50 scenario. Thus, using only the TOP50 training animals results in a set of training data that are highly related to the selection candidates for most traits, which is known to result in higher reliabilities [5],[29],[30]. In fact, our results suggest that when resources are limited to compose a training dataset, the best approach may be to genotype only the animals with high EBV. Some simulation studies show that selecting only the top animals may lead to substantially biased [31] and inaccurate predictions [32]. In our study, reliabilities for four out of six traits were higher for the TOP50 scenario than for the RAN50 scenario. Only for IFL, was the reliability considerably lower for TOP50 than for BOT50, simply because the number of male ancestors for the validation animals was much greater for BOT50 than TOP50. At the same time, for four out of six traits, the bias was smaller for the TOP50 scenario than for the RAN50 scenario. Thus, in general, our results were better for TOP50 than for RAN50. The most likely reason for the discrepancy between our results and those in the aforementioned simulation studies [31],[32], is that the predicted animals were selection candidates in our study, and thereby more likely to be offspring of the top animals, while the predicted animals were generated through random mating in the studies of Jiménez-Montero et al. [31] and Bolignon et al. [32].
Computing efficiency
In our study, the effective chain length of the SNP variance component was evaluated as a measure of convergence of the Bayesian models. This clearly showed that the effective chain length increased almost continuously with the number of iterations [see Additional file 1: Figure S1]. However, it is unclear whether effective chain length is indeed an indicator of convergence, for instance at the level of estimated SNP-specific variances. One way to assess convergence of SNP-specific variances, is to evaluate the correlation between posterior means of variance estimates from two independent replicates, where a correlation close to 1 indicates that both independent chains have converged to very similar SNP-specific variances. We computed this correlation every 1000th iteration and compared it to the effective chain length of the SNP variance component achieved at that iteration after burn-in [see Additional file 2: Figure S2]. This shows, that with the BSSVS model, an effective chain length of 50 was sufficient to obtain similar SNP-specific variances between replicates for LON, DLO, and SCS (correlations ranged from 0.87 to 0.92). However for the traits Protein, UD and IFL, correlations between SNP variances ranged only from 0.47 to 0.54 when an effective chain length of ~50 was obtained. With BayesC, for all traits, the SNP-specific variances were very similar (correlations above 0.91) after an effective chain length of ~200, which was achieved for all traits within the 50 000 iterations performed after the burn-in [see Additional file 1: Figure S1]). This shows that the SNP-specific variances estimated with BayesC converged in considerably fewer iterations compared to BSSVS, as indicated by the observation that for a given number of iterations the effective chain length of the SNP variance component was roughly twice as large for BayesC than for BSSVS.
With both BLUP and Bayesian models, the order of the SNPs was permuted every 10th iteration. This strategy was initially implemented to improve mixing in the Gibbs chain for the Bayesian models. This strategy also helped to speed up convergence in the BLUP models (results not shown). Other reported strategies that speed up convergence are to order the SNPs based on decreasing minor allele frequency [33].
Our implementation of the BLUP models used Gauss Seidel. Legarra and Misztal [20] showed that Gauss Seidel was 4.6 times slower than PCG. Their comparison showed that PCG was more efficient because it required ~8 times fewer iterations, while one iteration took twice as long for PCG than one iteration of Gauss Seidel. It should be noted that we used right-hand-side updating [18] in the Gauss Seidel implementation, which is shown to be ~5 times faster than the residual updating algorithm used by Legarra and Misztal [20] when the training data contains ~5000 animals [18]. However, right-hand-side updating cannot be applied to the PCG algorithm.
The number of iterations required for RR-BLUP to reach convergence ranged from 4017 to 19 980 (Table 6). These numbers are much larger than for instance the 164 required iterations reported by Legarra and Misztal [20]. We expected that this difference may be due to the fact that our models included a polygenic effect that is at least partly confounded with the SNP effects. In such situations, the Gauss-Seidel algorithm may be inefficient. Since convergence was monitored at the level of the estimated SNP effects and polygenic breeding values, the GEBV may in fact have converged much faster. To investigate this, one additional replicate was run for all BLUP models for all traits and the FULL scenario, for 200 000 iterations. GEBV were stored every 1000 iterations, and their correlation with the final estimates after 200 000 iterations were computed, following a similar approach as [19]. These results showed that correlations with final estimates greater than 0.9999 and 0.999 were obtained within the first 1000 iterations for all traits with RRBLUP and BLUP-C, respectively. For BLUP-SSVS, correlations greater than 0.999 were obtained within 1000 iterations for four out of six traits. For UD and IFL, 4000 and 7000 iterations were required to obtain correlations above 0.99. This suggests that for most applications of the BLUP models included in our study, convergence at the level of the GEBV is expected to be reached within the first 1000 iterations. Thus, monitoring convergence at the level of the estimated SNP and polygenic effects may unnecessarily increase the total number of iterations. Whether this holds for a particular application can be investigated by computing correlations between GEBV after different numbers of iterations with ¿final¿ estimates, as outlined above.
To further test the hypothesis that the confounding between SNP and polygenic effects leads to poor convergence at the level of the estimated SNP and polygenic effects, the analyses with RR-BLUP in the FULL scenario were repeated without a polygenic effect in the model for all six traits, using a convergence criterion of 10−10. The results (Table 9) show that in this case, only 29 to 131 iterations were required, i.e. less than 1% of the iterations required for the RR-BLUP model that did include a polygenic effect. At the same time, however, the obtained reliabilities were 0.015 to 0.031 lower than those obtained with the RR-BLUP model that did include a polygenic effect (Table 9). This stresses that polygenic effects capture some additional variance in genomic prediction models [34], which results in slightly higher accuracy of GEBV, as also demonstrated in other studies [33],[35], although this may require much more iterations to reach convergence of the BLUP models, as shown in our study.
Frequency to re-estimate SNP variances
Within the proposed framework, an important question is how often the SNP variances should be estimated. Or in other words: how fast are estimated SNP effects expected to change in time? So far, there are no published reports based on real data that have investigated this issue. The answer probably depends on several factors, including selection intensity, effective size of the population, density of the SNP chip used, initial size of the training data, and whether or not the size and composition of the training data change over time. For instance, it has been shown that a strong increase in the size of the training dataset leads to a much wider range of estimated SNP effects, even in a model for which the variance allocated to each SNP was the same [33]. This indicates that increasing the size of the training dataset, will also change SNP variances because power to estimate these variances increases.
Similarly, an important question for traditional pedigree-based genetic evaluation models is how often variance components should be re-estimated. For traditional genetic evaluation models applied in dairy cattle, the Interbull recommendation is to estimate variance components as often as possible and definitely, at least, once per generation [12]. Since the additive genetic variance estimated with an animal model is expected to be the same as the sum of all SNP variances, SNP variances are expected to change more than variance components used in conventional pedigree-based animal models. This suggests that SNP variances should be estimated more frequently than overall variance components. Moreover, our results indicate that the results of the BLUP models are very robust against using non-random subsets of the data to estimate SNP variances. This suggests that re-estimating SNP variances once a year is expected to be more than sufficient.
Conclusions
Our results show that BLUP genomic prediction models can adopt the same characteristics and yield the same results as variable selection models, provided that they use SNP-specific variances that are estimated with the variable selection models. This permits a flexible genomic evaluation system, for which SNP variances are perhaps re-estimated once per year using a Bayesian model, while efficient BLUP models that permit easy evaluation of convergence during the analysis, can be applied to estimate GEBV at a much higher frequency.
To monitor convergence in the Bayesian models, computing the effective chain length of the SNP variance component appears to be a useful measure. For the two Bayesian models used here, the estimated SNP-specific variances converged in considerably fewer iterations with BayesC than with BSSVS.
Our results confirmed that in order to get unbiased GEBV, it is important that the training dataset covers the entire population and that it is not composed of a pre-selected group of animals. However, using a pre-selected group of animals to estimate the SNP-specific variances did not affect the resulting GEBV, provided that the training data used in the BLUP step covered the entire population. Genomic prediction models that use pre-computed SNP variances proved to be able to generate GEBV with better properties, in terms of reliability and bias, than the commonly used RR-BLUP model. Including a separate polygenic effect systematically improved the reliabilities of the GEBV but also substantially increased the number of iterations needed to reach convergence for the RR-BLUP model.
Additional files
References
de los Campos G, Hickey JM, Pong-Wong R, Daetwyler HD, Calus MPL: Whole-genome regression and prediction methods applied to plant and animal breeding. Genetics. 2013, 193: 327-345. 10.1534/genetics.112.143313.
Daetwyler HD, Capitan A, Pausch H, Stothard P, Van Binsbergen R, Brøndum RF, Liao X, Djari A, Rodriguez S, Grohs C, Jung S, Esquerré D, Bouchez O, Rossignol MN, Klopp C, Rocha D, Fritz S, Eggen A, Bowman P, Coote D, Chamberlain A, Vantassell CP, Hulsegge I, Goddard ME, Guldbrandtsen B, Lund MS, Veerkamp RF, Boichard DA, Fries R, Hayes BJ: Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat Rev Genet. 2014, 46: 858-865. 10.1038/ng.3034.
Gianola D, de los Campos G, Hill W, Manfredi E, Fernando R: Additive genetic variability and the Bayesian alphabet. Genetics. 2009, 183: 347-363. 10.1534/genetics.109.103952.
Meuwissen THE, Hayes BJ, Goddard ME: Prediction of total genetic value using genome-wide dense marker maps. Genetics. 2001, 157: 1819-1829.
Habier D, Fernando RL, Dekkers JCM: The impact of genetic relationship information on genome-assisted breeding values. Genetics. 2007, 177: 2389-2397.
VanRaden PM: Efficient methods to compute genomic predictions. J Dairy Sci. 2008, 91: 4414-4423. 10.3168/jds.2007-0980.
Zhang Z, Liu JF, Ding XD, Bijma P, de Koning DJ, Zhang Q: Best linear unbiased prediction of genomic breeding values using a trait-specific marker-derived relationship matrix. PLoS One. 2010, 5: e12648-10.1371/journal.pone.0012648.
Gilmour AR, Thompson R: Options for estimating variance components in large mixed models. Proc Adv Anim Breed Gen. 2003, 15: 206-209.
Searle SR, Casella G, McCulloch CE: Variance Components. 2009, Wiley, New York
Misztal I: Reliable computing in estimation of variance components. J Anim Breed Genet. 2008, 125: 363-370. 10.1111/j.1439-0388.2008.00774.x.
Interbull: National GES information.., [http://www.interbull.org/ib/nat_publication_links]
Guidelines Approved by the General Assembly Held in Cork, Ireland on June 2012. International Agreement of Recording Practices. 2012, ICAR, Rome
Stranden I, Lidauer M: Solving large mixed linear models using preconditioned conjugate gradient iteration. J Dairy Sci. 1999, 82: 2779-2787. 10.3168/jds.S0022-0302(99)75535-9.
Calus MPL, Meuwissen THE, De Roos APW, Veerkamp RF: Accuracy of genomic selection using different methods to define haplotypes. Genetics. 2008, 178: 553-561. 10.1534/genetics.107.080838.
Verbyla KL, Hayes BJ, Bowman PJ, Goddard ME: Accuracy of genomic selection using stochastic search variable selection in Australian Holstein Friesian dairy cattle. Genet Res. 2009, 91: 307-311. 10.1017/S0016672309990243.
Habier D, Fernando RL, Kizilkaya K, Garrick DJ: Extension of the Bayesian alphabet for genomic selection. BMC Bioinformatics. 2011, 12: 186-10.1186/1471-2105-12-186.
Jia Y, Jannink JL: Multiple-trait genomic selection methods increase genetic value prediction accuracy. Genetics. 2012, 192: 1513-1522. 10.1534/genetics.112.144246.
Calus MPL: Right-hand-side updating for fast computing of genomic breeding values. Genet Sel Evol. 2014, 46: 24-10.1186/1297-9686-46-24.
Lidauer M, Strandén I, Mäntysaari E, Pösö J, Kettunen A: Solving large test-day models by iteration on data and preconditioned conjugate gradient. J Dairy Sci. 1999, 82: 2788-2796. 10.3168/jds.S0022-0302(99)75536-0.
Legarra A, Misztal I: Computing strategies in genome-wide selection. J Dairy Sci. 2008, 91: 360-366. 10.3168/jds.2007-0403.
Fikse WF, Banos G: Weighting factors of sire daughter information in international genetic evaluations. J Dairy Sci. 2001, 84: 1759-1767. 10.3168/jds.S0022-0302(01)74611-5.
Druet T, Georges M: A hidden Markov model combining linkage and linkage disequilibrium information for haplotype reconstruction and quantitative trait locus fine mapping. Genetics. 2010, 184: 789-798. 10.1534/genetics.109.108431.
Browning SR, Browning BL: Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007, 81: 1084-1097. 10.1086/521987.
Canty A, Ripley B: Boot: Bootstrap R (S-Plus) Functions. R Package Version 1.2-34. 2009
Plummer M, Best N, Cowles K, Vines K: CODA: convergence diagnosis and output analysis for MCMC. R News. 2006, 6: 7-11.
Legarra A, Robert-Granie C, Croiseau P, Guillaume F, Fritz S: Improved Lasso for genomic selection. Genet Res. 2011, 93: 77-87. 10.1017/S0016672310000534.
Patry C, Ducrocq V: Accounting for genomic pre-selection in national BLUP evaluations in dairy cattle. Genet Sel Evol. 2011, 43: 30-10.1186/1297-9686-43-30.
Patry C, Ducrocq V: Evidence of biases in genetic evaluations due to genomic preselection in dairy cattle. J Dairy Sci. 2011, 94: 1011-1020. 10.3168/jds.2010-3804.
Pszczola M, Strabel T, Mulder HA, Calus MPL: Reliability of direct genomic values for animals with different relationships within and to the reference population. J Dairy Sci. 2012, 95: 389-400. 10.3168/jds.2011-4338.
Pérez-Cabal MA, Vazquez AI, Gianola D, Rosa GJM, Weigel KA: Accuracy of genome enabled prediction in a dairy cattle population using different cross-validation layouts. Front Genet. 2012, 3: 27-10.3389/fgene.2012.00027.
Jiménez-Montero JA, Gonzalez-Recio O, Alenda R: Genotyping strategies for genomic selection in dairy cattle. Animal. 2012, 6: 1216-1224. 10.1017/S1751731112000341.
Boligon AA, Long N, Albuquerque LG, Weigel KA, Gianola D, Rosa GJM: Comparison of selective genotyping strategies for prediction of breeding values in a population undergoing selection. J Anim Sci. 2012, 90: 4716-4722. 10.2527/jas.2012-4857.
Liu Z, Seefried FR, Reinhardt F, Rensing S, Thaller G, Reents R: Impacts of both reference population size and inclusion of a residual polygenic effect on the accuracy of genomic prediction. Genet Sel Evol. 2011, 43: 19-10.1186/1297-9686-43-19.
Jensen J, Su G, Madsen P: Partitioning additive genetic variance into genomic and remaining polygenic components for complex traits in dairy cattle. BMC Genet. 2012, 13: 44-10.1186/1471-2156-13-44.
Calus MPL, Veerkamp RF: Accuracy of breeding values when using and ignoring the polygenic effect in genomic breeding value estimation with a marker density of one SNP per cM. J Anim Breed Genet. 2007, 124: 362-368. 10.1111/j.1439-0388.2007.00691.x.
Acknowledgements
The authors acknowledge CRV BV (Arnhem, the Netherlands) for financial support and for providing the data, and also acknowledge financial support from the Dutch Ministry of Economic Affairs, Agriculture, and Innovation (Public-private partnership ¿Breed4Food¿ code KB-12-006.03-004-ASG-LR).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MPLC implemented the models, performed the analyses and wrote large parts of the initial manuscript. CS edited the data, helped to describe the dataset and interpret the results. RFV participated in discussions on the results. All authors read and approved the final manuscript.
Electronic supplementary material
12711_2014_52_MOESM1_ESM.docx
Additional file 1: Figure S1.: Effective chain length of the SNP variance component for various numbers of iterations after the burn-in. Average results across two replicates are shown for the FULL scenario and models BSSVS and BayesC. (DOCX 159 KB)
12711_2014_52_MOESM2_ESM.docx
Additional file 2: Figure S2.: The relationship between correlations between estimated SNP variances of two independent replicates and the effective chain length of the SNP variance component. Results are shown for the FULL scenario and models BSSVS and BayesC. Effective chain lengths are averaged across the two replicates. (DOCX 141 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
Cite this article
Calus, M.P., Schrooten, C. & Veerkamp, R.F. Genomic prediction of breeding values using previously estimated SNP variances. Genet Sel Evol 46, 52 (2014). https://doi.org/10.1186/s12711-014-0052-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12711-014-0052-x