
Abstract
The volume-sensitive outwardly rectifying or volume-regulated anion channel, VSOR/VRAC, which was discovered in 1988, is expressed in most vertebrate cell types, and is essentially involved in cell volume regulation after swelling and in the induction of cell death. This series of review articles describes what is already known and what remains to be uncovered about the functional and molecular properties as well as the physiological and pathophysiological roles of VSOR/VRAC. This Part 2 review article describes, from the physiological and pathophysiological standpoints, first the pivotal roles of VSOR/VRAC in the release of autocrine/paracrine organic signal molecules, such as glutamate, ATP, glutathione, cGAMP, and itaconate, as well as second the swelling-independent and -dependent activation mechanisms of VSOR/VRAC. Since the pore size of VSOR/VRAC has now well been evaluated by electrophysiological and 3D-structural methods, the signal-releasing activity of VSOR/VRAC is here discussed by comparing the molecular sizes of these organic signals to the channel pore size. Swelling-independent activation mechanisms include a physicochemical one caused by the reduction of intracellular ionic strength and a biochemical one caused by oxidation due to stimulation by receptor agonists or apoptosis inducers. Because some organic substances released via VSOR/VRAC upon cell swelling can trigger or augment VSOR/VRAC activation in an autocrine fashion, swelling-dependent activation mechanisms are to be divided into two phases: the first phase induced by cell swelling per se and the second phase caused by receptor stimulation by released organic signals.
Similar content being viewed by others
Introduction
Mammalian anion channels are known to be classified into six major groups: ligand-gated receptor-coupled, voltage-gated ClC-type, cyclic AMP/PKA-activated cystic fibrosis transmembrane conductance regulator (CFTR), Ca2+-activated TMEM16/ANO, acid-activated ASOR/PAC, and swelling-activated anion channels (see Review [1]). The last one, called the volume-activated anion channel (VAAC), is involved in cell volume regulation and consists of two types, the intermediate-conductance outwardly rectifying anion channel, which was termed the volume-sensitive outwardly rectifying anion channel (VSOR) or volume regulated anion channel (VRAC) (see Review [2]), and the large-conductance ohmic maxi-anion channel (Maxi-Cl) (see Review [3]).
The intermediate conductance one, here called the volume-sensitive outwardly rectifying/volume-regulatory anion channel (VSOR/VRAC), was first functionally found in 1988 independently by two groups ([4, 5]). Soon thereafter, its phenotypical properties were well-characterized [6,7,8], but the molecular nature remained unknown for over a quarter of a century. In recent years, the pore-forming core component and the swelling-sensing subcomponents of VSOR/VRAC were identified as LRRC8 members in 2014 [9, 10] and TRPM7 in 2021 [11], respectively, as summarized in the Part 1 article [12].
VSOR/VRAC is known to be activated not only by cell swelling but also by some other physicochemical and biochemical stimuli even in the absence of cell swelling. In addition, accumulating evidence showed that VSOR/VRAC plays not only the volume-regulatory role but also other roles including mediation of organic signal release, induction of apoptotic, necrotic, and pyroptotic cell death, and acquirement of anti-cancer drug resistance. Thus, here in this Part 2 article, from physiological and pathophysiological standpoints, I review how VSOR/VRAC is involved in the cellular release of autocrine/paracrine organic signals, and how it is activated, in a swelling-dependent and -independent manner, together with pointing out what remains to be elucidated in future studies. The VSOR/VRAC roles in cell death induction and acquisition of anti-cancer drug resistance will be reviewed in the next Part 3 article.
Mediation of release of organic substances and signals via VSOR/VRAC
Since organic solutes produced within cells are intracellularly accumulated, and their extracellular concentrations are negligibly low under ordinal conditions, they are readily driven out of cells by such chemical potential (concentration) gradients when some diffusional channel-mediated routes are available. For the release of negatively charged organic solutes, the intracellular negative potential is to be added to the driving force. For example for ATP4−, the electrochemical potential gradient reaches the order of 1010 when the intracellular electrical potential is around − 60 mV [13].
After the establishment of VSOR/VRAC role in cell volume regulation, its mediation of the cellular release of small organic substances was found to be another important role of VSOR/VRAC in the early 1990s, as summarized by Strange et al. [6, 14]. Thereafter, this VSOR/VRAC role was supported by the functional and structural evaluation of the pore size of VSOR/VRAC. Recent studies have elucidated the physiological/pathophysiological significance of VSOR/VRAC-mediated release of organic substances as paracrine/autocrine signals.
Swelling-activated release of intracellular organic solutes and the pore size of VSOR/VRAC
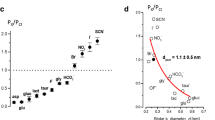
Swelling-activated Na+-independent (that is, Na+-driven cotransport-independent) release of intracellular glutamate, aspartate, and taurine from mammalian cells was, for the first time, observed in rat astrocytes in 1990 by Kimelberg et al. [15]. In 1991, hypotonicity-induced, Na+-independent taurine release was found to be sensitive to a Cl− channel blocker DIDS in rabbit lymphocytes [16]. Then, similar Na+-independent release of glutamate, taurine, and glycine from canine kidney MDCK cells was shown to be linearly dependent on these concentrations in 1992 [17], suggesting the involvement of diffusional channel-mediated, but not saturable carrier/transporter-mediated, transport which is activated by cell swelling. The swelling-activated, VSOR/VRAC-mediated currents conveyed by negatively charged organic substances were, in fact, recorded under voltage-clamp and bi-ionic conditions for gluconate in human epithelial Intestine 407 cells under the whole-cell configuration by Kubo and Okada [18] and for aspartate, glutamate, and taurine, which is a zwitter ion and electrically neutral at physiological pH but becomes negatively charged at alkaline pH, in MDCK cells in single-channel recording modes by Banderali and Roy [19] also in 1992. These patch-clamp studies evaluated their permeability coefficient: Paspartate/PCl ~ 0.2 [17], Pgluconate/PCl ~ 0.1 [18], Pglutamate/PCl ~ 0.2 [19], and Ptaurine/PCl ~ 0.75 [19]. Because VSOR/VRAC is a low-field anion channel, as described in the Part 1 article [12], it is reasonable that the sequence of these permeability coefficients (chloride > taurine > aspartate ~ glutamate > gluconate) is in fairly good accordance with the sequence for their effective diameters listed in Table 1. Also, it must be noteworthy that all these organic substances have much smaller diameters [13, 14, 20] than a pore diameter of VSOR/VRAC (7–13 Å) functionally estimated by three different and unrelated techniques by Nilius and Droogmans [21], by Droogmans et al. [22], and by Ternovsky et al. [23] (Table 2A).
Taking the capability of VSOR/VRAC to serve as the pathway for the swelling-induced release of intracellular organic substances into consideration, this volume-sensitive anion channel has sometimes been also called the volume-sensitive organic osmolyte and anion channel (VSOAC) [6, 14]. However, it must be noted that not only VSOR/VRAC but also Maxi-Cl and CFTR can provide the pathways for the release of organic solutes such as glutamate, ATP, and GSH, as pointed out in our previous article [1].
In agreement with the fact that LRRC8A represents an indispensable core molecule of VSOR/VRAC [9, 10], LRRC8A was found to be a prerequisite to the hypotonicity-induced release of organic osmolytes mediated by VSOR/VRAC. Gene knockout of LRRC8A abolished the release of taurine from HCT116 cells [10] and HEK293 cells [24, 25], that of glutamate from HEK293 cells [25] and rat astrocytes [26], and that of aspartate, lysine, serine, GABA, and myo-inositol from HEK293 cells [25]. Gene knockout experiments also showed that LRRC8D is essential for swelling-induced taurine release from HEK293 cells [24]. LRRC8A/8D heteromers can transport relatively large organic anions such as glutamate− and aspartate− as well as uncharged organic osmolytes, taurine, myo-inositol, and GABA, and even positively charged lysine+ [25]. In contrast, LRRC8A/8C and LRRC8A/8E heteromers conduct Cl− and aspartate− but are much less permeable to GABA and myo-inositol [25]. Thus, it is concluded that LRRC8D makes VSOR/VRAC more permeable to larger organic anions and uncharged or cationic organic osmolytes. These results are in good agreement with the 3D structures analyzed by cryo-electron microscopy (cryo-EM). The narrowest constriction part (at the selectivity filter) of the pore of LRRC8D homohexamers (at F143) [27] is much larger than that of LRRC8A homohexamers (at R103) [28,29,30,31,32], LRRC8A/8C heterohexamers (at R103/L105) [33, 34], and LRRC8C-8A(IL125) homoheptamers (at L105) [35] but is comparable to that of LRRC8C homoheptamers (at L105) [33], as listed in Table 2B. How large the pores of LRRC8A/8D and LRRC8A/8E heteromers awaits cryo-EM studies in the immediate future.
VSOR/VRAC-mediated transport of organic signaling molecules
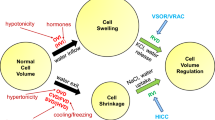
In addition to volume-regulatory roles, VSOR/VRAC plays roles in the transmission of paracrine/autocrine signaling by transporting numbers of negatively charged organic substances, such as glutamate–, aspartate–, ATP4–, glutathione (GSH–), itaconate2–, and 2′3′-cyclic-GMP-AMP (cGAMP2–). Moreover, glutamate and ATP released via VSOR/VRAC were shown to activate VSOR/VRAC in a positive feedback fashion through stimulation of their receptors (Fig. 1).

Autocrine/paracrine signaling roles of glutamate and ATP released via VSOR/VRAC and Maxi-Cl upon cell swelling in VSOR/VRAC activation. First ((1): brown arrows), such released glutamate and ATP activate, in an autocrine fashion, VSOR/VRAC via stimulation of GPCRs (mGluR and P2YR) in a cell in response to osmotic swelling. Second ((2): violet arrows), glutamate and ATP then activate, in a paracrine fashion, VSOR/VRAC via stimulation of GPCRs in another neighboring cell even in the absence of swelling. These glutamate and ATP may also trigger (black arrows), in a paracrine fashion, induction of inflammation in the surrounding cells/tissues from which BK and S1P are thereafter released. Third ((3): blue arrows), BK and S1P then activate VSOR/VRAC via stimulation of their receptors (B2R and S1PR1). (See text for details.)
VSOR/VRAC-mediated glutamate release
Kimelberg et al. [15] found that the swelling-induced release of glutamate, aspartate, and taurine from rat astrocytes in culture is sensitive to many known anion transport blockers. Also, Roy and Malo [17] observed that large losses of amino acids, such as glutamate, taurine, and glycine, take place during cell volume regulation upon a hypotonic challenge to canine MDCK cells through diffusional routes rather than transporters/carriers. Then, Banderali and Roy [19], for the first time, showed that an outwardly rectifying anion channel provides the diffusional pathway for the swelling-induced release of some intracellular organic substances including glutamate. Since some of these organic substances are known to play signaling roles as excitatory amino acids between neurons and between glial and neuronal cells in the brain under physiological/pathological situations, the roles of VSOR/VRAC in glutamate release mainly from glial cells were thereafter studied.
Electrophysiological and pharmacological evidence was provided for the VSOR/VRAC role in glutamate release from mouse astrocytes induced by hypoosmotic and ischemic stress [36]. Accordingly, DCPIB, which is a relatively most VSOR/VRAC-specific blocker among available anion channel blockers (see Reviews [1, 12]), was shown to inhibit osmotic swelling-induced glutamate release from rat primary astrocytes [37] and human retinal glial Müller MIO-M1 cells [38] as well as the hypotonicity-induced release of aspartate, which is a non-metabolized analog of glutamate, from rat astrocytes [37, 39]. However, DCPIB was unexpectedly found to inhibit not only glutamate release mediated by VSOR/VRAC but also that mediated by connexin hemichannels as well as glutamate uptake via glutamate transporter GLT1 in rat glial cells [40].
Molecular evidence for VSOR/VRAC-mediated glutamate/aspartate release from osmotically swollen astrocytes was recently provided by the effects of reduced expression of LRRC8A. First, siRNA-mediated LRRC8A knockdown brought about 70% and 90% inhibition of the release of glutamate and aspartate from rat astrocytes induced by a moderate hypotonic (70% osmolarity) challenge [41, 42] and by a severe hypotonic (30% osmolarity) challenge [42], respectively. Second, astrocyte-specific Lrrc8a knockout caused 93% and 70% inhibition of the release of glutamate and aspartate from swollen mouse astrocytes challenged by an intracellular hypertonic (135% osmolarity) solution [26] and an extracellularly hypotonic (70% osmolarity) solution [43], respectively. Third, aspartate release from primary rat astrocytes challenged by hypotonicity (70% osmolarity) was suppressed to ~ 60% and ~ 25% by double knockdown of Lrrc8c plus Lrrc8e and by that of Lrrc8c plus Lrrc8d, respectively [44]. However, it must be noted that swelling-induced glutamate release from astrocytes is mediated not only by VSOR/VRAC but also by Maxi-Cl channels [36], the latter of which represents another type of VAACs [1]. That is why gene deletion of LRRC8 members failed to cause the complete abolition of swelling-induced glutamate release from astrocytes. Since the core molecule of Maxi-Cl was recently identified as SLCO2A1 [45], we need hereafter to answer the question as to what degree VSOR/VRAC and Maxi-Cl contribute to glutamate release induced by cell swelling each other in the cell types in question under the given conditions.
Extracellular application of ATP was found to stimulate the release of aspartate [41, 46] and glutamate [46, 47] from astrocytes by activating VSOR/VRAC through stimulation of purinergic P2Y receptors (P2YRs) [47, 48] under isotonic conditions. Extracellular ATP-induced aspartate release was shown to be, as a matter of course, inhibited by siRNA-mediated Lrrc8a knockdown in rat astrocytes [41]. Extracellular application of glutamate was also shown to induce VSOR/VRAC activation under isotonic conditions through activation of muscarinic glutamate receptor (mGluR) in mouse astrocytes [49]. Thus, glutamate released via both types of VAACs contributes to the activation of VSOR/VRAC via mGluRs in the cell itself and in a nearby cell in autocrine and paracrine fashions (Fig. 1: right side (1) and (2)), respectively. Since both glutamate and ATP are known to be released from swollen cells via both types of VAACs, Maxi-Cl and VSOR/VRAC, hereafter we need to answer the question as to what extent swelling-induced glutamate release is caused by cell swelling per se and by glutamate and ATP secondary released in the particular cell types under the given conditions.
Exposure to extracellular bradykinin (BK), which is a proinflammatory nine-amino acid peptide, was shown to trigger VSOR/VRAC activation [50, 51] via bradykinin B2 receptor (B2R) and to stimulate glutamate release [50] without exhibiting cell swelling. BK is generated from kininogens by the action of kallikrein, represents an initial mediator of inflammation [52], and is known to be released from injured and inflammatory sites such as the central nervous system after brain trauma and stroke [53, 54]. Since major excitatory neurotransmitter glutamate exerts as signaling and causal factors for inflammation coupled to some disorders in the central system [55] and in the peripheral system [56, 57], glutamate released via VSOR/VRAC and Maxi-Cl may be involved in the induction of cell/tissue inflammation (Fig. 1: right side, black arrow) and therefrom secondarily causes BK release thereby inducing B2R-mediated VSOR/VRAC activation (Fig. 1: right side (3)) followed by glutamate release, in a positive feedback manner, from the cells stimulated by BK.
Extracellular application of sphingosine-1-phosphate (S1P) was shown to activate VSOR/VRAC via S1P receptor 1 (S1PR1) under isotonic conditions in murine RAW 264.7 macrophages [58]. A pleiotropic lipid mediator S1P plays a significant role in inflammation [59, 60] and is known to be released from the inflamed cells [61,62,63]. Therefore, VSOR/VRAC may be activated by S1P released from inflammatory cells/tissues (Fig. 1: left side (3)), which were caused by exposure to glutamate released via VSOR/VRAC and Maxi-Cl (Fig. 1: right side, black arrow), thereby causing VSOR/VRAC activation in the cells stimulated by S1P and therefrom glutamate release in a positive feedback manner.
VSOR/VRAC-mediated ATP release
ATP acts as a major messenger molecule for autocrine and paracrine signaling in the extracellular space [13, 64, 65], whereas it serves as an energy source in the cytosol. ATP is released not only by vesicular exocytosis but also by the transport via several non-vesicular pathways including anion channels [13]. In particular, large-conductance Maxi-Cl anion channels have been shown to serve as a major pathway for swelling- and ischemia-induced release of ATP4– from many cell types such as astrocytes and cardiomyocytes [3]. Swelling-induced ATP release was suggested to be mediated also via VSOR/VRAC largely based on pharmacological evidence in bovine aortic endothelial cells [66], mouse DRG neurons [67], and mouse RAW 264.7 macrophages [58]. In contrast, swelling-induced ATP release was not inhibited by a number of VSOR/VRAC blockers in bovine ocular ciliary epithelial cells [68], human epithelial Intestine 407 cells [69], rat cardiomyocytes [70], and mouse astrocytes [71]. ATP release induced by mechanical stimulation was found to be sensitive to VSOR/VRAC blockers but insensitive to Lrrc8a knockdown in rat astrocytes [72]. In contrast, gene knockdown of LRRC8A was shown to suppress ATP release induced by hypoosmotic stimulation in HEK293 cells [73], HeLa cells [74], and mouse microglial BV-2 cells [74] and that induced by application of S1P in mouse microglial BV-2 cells [74, 75] and in human breast cancer MCF7 and MDA-MB-231 cells [76]. Collectively, it appears that VSOR/VRAC can mediate ATP release in many, but not all, cell types. Notably, the functional pore diameter of VSOR/VRAC (Table 2A) is very close to the effective diameter of ATP4‒ and MgATP2‒ (Table 1). Thus, the ATP conductivity of VSOR/VRAC pores may be prone to be affected by alterations in the surrounding microenvironment at the plasma membrane, especially in the lipid microenvironment. Also, the LRRC8 heteromer composition of VSOR/VRAC may affect its ATP conductivity, because ATP release was found to be provoked by hypotonic stimulation in Xenopus oocytes when LRRC8A was co-expressed with LRRC8E or LRRC8C but not with LRRC8B or LRRC8D [77]. In light to these observations, we now need to examine to what degree each type of VAACs contributes to swelling-induced ATP release from the concerned cell types under the given conditions. Also, we hereafter need to answer the question as to what degree swelling-induced ATP release is induced by cell swelling per se and by glutamate and ATP secondary released upon cell swelling from the concerned cell types under the given conditions. Furthermore, from now on, we need to pay attention to what extent swelling-induced ATP release is affected by following two opposite autocrine actions in the particular cell types under the given conditions because extracellular ATP exerts two contradictory effects, an open-channel blocking action [78,79,80] and receptor-mediated swelling-independent activating action [48].
ATP released via both types of VAACs provokes the activation of VSOR/VRAC via P2YRs in a cell itself and also in a nearby cell in autocrine and paracrine fashions (Fig. 1: left side (1) and (2)), respectively. From now on, we thus need to answer the question as to what extent swelling-induced ATP release is caused by cell swelling per se and by ATP and glutamate secondary released in the particular cell types under the given conditions. VSOR/VRAC must be activated by BK and S1P released from inflamed cells caused by exposure to extracellular ATP (Fig. 1: left side, black arrow), which is one of the danger-associated molecular patterns (DAMPs) causing inflammation in a variety of tissues [81,82,83,84,85], thereby bringing about further VSOR/VRAC activation in the cells stimulated by BK and S1P (Fig. 1: right and left sides (3)) and therefrom ATP release in a positive feedback manner.
VSOR/VRAC-mediated transport of other important negatively charged organic substances
VSOR/VRAC has been shown to serve as conductive pathways also for other negatively charged organic substances, such as GSH, methylene succinic acid or itaconic acid (itaconate), and cGAMP, that are known to be important signaling molecules involved in anti-oxidation, anti-inflammation, and anti-viral defense, respectively.
The most abundant antioxidant GSH is involved in essential cell processes including antioxidant defense, drug detoxification, cell metabolism, and proliferation [86,87,88]. The release of GSH is a prerequisite to apoptosis induction [89,90,91]. The first evidence for VSOR/VRAC-mediated GSH release was reported in 2013 by Sabirov et al. [92]. The molecular size of GSH (Table 1) is smaller than the functional pore diameter of VSOR/VRAC (Table 2A). Hypotonicity-induced GSH release from rat thymocytes was largely abolished by a variety of VSOR/VRAC blockers including DCPIB. The VSOR/VRAC permeability to GSH is significant with PGSH/PCl of 0.10 for release from and 0.32 for entry to thymocytes. Subsequently, Friard et al. [93] showed that swelling-induced GSH release is sensitive not only to DCPIB but also to LRRC8A gene knockout in HEK293 cells and that the PGSH/PCl values can be evaluated as 0.08 in HEK293 cells and 0.11 in human kidney tubular epithelial HK2 cells. Even under isotonic conditions, VSOR/VRAC was found to be activated by exposure to TGFβ1, which is a pleiotropic growth factor inducing the epithelial-to-mesenchymal transition (EMT), thereby releasing GSH in HK2 cells [93].
Itaconate, a Krebs cycle-derived metabolite, is produced upon stimulation of Toll-like receptor (TLR) in myeloid cells and is accumulated upon prolonged inflammatory situations. The intracellular itaconate accumulation was shown to inhibit NLRP3 inflammasome activation [94,95,96]. The itaconate-induced inhibition of NLRP3 inflammasomes was observed to be greatly abolished by myeloid LRRC8A gene knockout [97], suggesting that VSOR/VRAC activity is involved in NLRP3 inflammasome activation, presumably by mediating itaconate efflux. The size of this anti-inflammatory signal, itaconate (C5H6O4), should be smaller than those of glutamate (C5H9NO4) and gluconate (C6H12O7). In fact, the unhydrated diameter of itaconate was calculated to be 6.6 Å as a geometric mean of three dimensions by RZ Sabirov (personal communication). Thus, itaconate is expected to be VSOR/VRAC-permeable. Confirming this inference, Wu et al. [97] showed that hypotonicity induces activation of whole-cell inward currents mediated by efflux of negatively charged itaconate filled in the pipette (intracellular) solution in LPS-primed macrophages and estimated the permeability coefficient of itaconate (Pitaconate/PCl) for VSOR/VRAC of around 0.2.
cGAMP is an immune-transmitting second messenger produced by cyclic-AMP-GMP synthase (cGAS) in response to cytosolic double-stranded DNAs (dsDNAs) and is an agonist for its receptor, stimulator of interferon genes (STING). cGAMP thereby serves as an important messenger for the cGAS-cGAMP-STING pathway which represents an essential innate immune signaling cascade responsible for the sensing of aberrant cytosolic dsDNA and then plays roles of anti-viral defense and anti-cancer immunity by eliciting interferons (IFNs) [98,99,100]. Since the size of cGAMP (Table 1) is a little smaller than the effective diameter of VSOR/VRAC pore (Table 2A) estimated by non-electrolyte partitioning [23], VSOR/VRAC channels may mediate cGAMP transport under appropriate conditions. Consistently, the cGAMP uptake/import induced by extracellular application of cGAMP was found to be inhibited by DCPIB in HEK293 cells and primary human umbilical vein endothelial (HUVEC) cells [101], in mouse lung fibroblast (MLF) cells [102, 103], and in murine bone marrow-derived macrophage (BMDM) cells [103]. Also, extracellular cGAMP treatment was found to activate the STING pathway due to VSOR/VRAC-mediated cGAMP import in human lymphoma U937, epithelial HEK293, and endothelial TIME cells incubated in a serum-free isotonic electrolyte solution containing glucose [101]. Gene knockout of LRRC8A suppressed the cGAMP import in HEK293, HUVEC, MLF, and BMDM cells [101,102,103] as well as in mouse CD4+ T cells [104]. Osmotic swelling-induced cGAMP export was electrophysiologically evidenced by the recording of inward currents conveyed by cGAMP2–, in a manner sensitive to LRRC8A gene knockdown, in human epithelial HeLa and HCT116 cells [101, 102]. Taken together, it is concluded that VSOR/VRAC is a cGAMP-transporting channel that can mediate bilateral transport of cGAMP. Supporting this conclusion, Zou et al. [102] demonstrated that cGAMP released via VSOR/VRAC channels from host cells infected with DNA viruses is transmitted to distant filter-separated bystander cells and then taken up via VSOR/VRAC channels, in a manner sensitive to LRRC8A gene knockout, by using a trans-well chamber assay in mouse embryonic fibroblasts (MEFs). Thus, it is evident that VSOR/VRAC mediates bilateral transport of cGAMP, especially in association with anti-viral defense immunity.
STING activation induced by extracellular cGAMP application was found to be suppressed by gene knockout not only of LRRC8A but also LRRC8C in U937 and TIME cells [101]. Similarly, LRRC8C gene knockout was observed to inhibit STING activation induced by extracellular cGAMP in CD4+ T cells [104]. Thus, VSOR/VRAC responsible for the cGAMP import is likely formed mainly with LRRC8A plus LRRC8C. In contrast, STING activation induced by extracellular cGAMP was suppressed by gene knockout of LRRC8A or LRRC8E in BMDM and MLF cells [102, 103]. Also, increased expression of interferon in response to infection with a DNA virus, HSV-1, was inhibited by gene knockout of LRRC8A or LRRC8E but not by triple knockout of LRRC8B, 8C, and 8D genes in MLF cells [102]. Thus, VSOR/VRAC channels formed mainly with LRRC8A plus LRRC8E and with LRRC8A plus LRRC8C play essential roles in anti-viral immunity [103] and in suppression of the cytotoxic T cell function [104, 105] by bilaterally transporting cGAMP presumably via the channels, respectively. However, further studies are required to precisely determine whether LRRC8 heteromer compositions of cGAMP-transporting VSOR/VRAC vary depending on cell types or cell functions.
Activation mechanisms of VSOR/VRAC
Activation of VSOR/VRAC was first found to be induced by cell swelling or volume expansion by Hazama and Okada [4] and Cahalan and Lewis [5] in 1988. Thereafter, even without visible cell swelling, VSOR/VRAC was shown to be activated by GTPγS by Doroshenko et al. [106] in 1991 and by a reduction of intracellular ionic strength (Γin) by Cannon et al. [107] and Nilius et al. [108] in 1998. These findings suggest that there are not only a swelling-dependent physiological activation mechanism but also some other swelling-independent activation mechanisms including G-protein-linked biochemical events and Γin-related physicochemical events.
Swelling-independent physicochemical activation of VSOR/VRAC
In intact cell systems, VSOR/VRAC activation in the absence of cell swelling was shown to be induced by a large reduction (down to around 30 to 60%) in Γin [107,108,109,110]. After the identification of LRRC8 members as the pore-forming core components [9, 10], similar low Γin-induced channel activation was found in the cell-free reconstitution system formed with purified LRRC8A and LRRC8C, 8D, or 8E in lipid droplet bilayers [111]. However, it is noted that the properties of channels reconstituted in droplet bilayers are different from native VSOR/VRAC, as follows. The reconstituted heteromeric LRRC8 channels are not activated by inflation (volume increase) of droplets but activated by a reduction of Γin in a manner independent of intracellular ATP and do not exhibit voltage-dependent inactivation kinetics. The homomeric LRRC8A channels reconstituted in liposomes were also found to be activated only in low Γin solutions [31]. The channel activity was observed even in the absence of ATP and in the presence of a high concentration of free Mg2+ on the intracellular side, in contrast to the phenotypical properties of native VSOR/VRAC existing in living cells (see Table 1 in Part 1 article [12]). Since the reduction in Γin should increase the surface potential on the peripheral surface of highly charged domains of channel-forming proteins, physicochemical/electrostatic repulsion or attraction would take place between any pairs of closely adjacent charged domains or proteins. Deneka et al. [28] suggested that the hydrophilic leucine-rich repeats (LRR) domains (LRRDs: see Fig. 2 in the Part 1 article [12]) are involved in the low Γin-induced activation of VSOR/VRAC, because many basic (negatively charged) and acidic (positively charged) residues exist on the molecular surface of cytoplasmic LRRD. In association with the activation of LRRC8A channels, such physicochemical conformational changes in LRRDs were recently observed [112] by using five synthetic nanobodies called sybodies (sbs): namely, LRRC8A channels expressed in Lrrc8-knockout (LRRC8−/−) HEK293 cells were found to be activated by sb4 and sb5 but inhibited by sb1, sb2, and sb3, the former two sbs and latter three sbs of which were shown to bind to the concave inside and convex outside, respectively, of the horseshoe-shaped LRRDs by cryo-EM. On the other hand, the involvement of unfolding of the N-terminal (NT) domain of LRRC8 in the VSOR/VRAC activation induced by a large reduction (down to 33%) in Γin was suggested by Liu et al. [32], mainly based on the molecular dynamics (MD) simulations of cryo-EM structure of LRRC8A. Thus, it can be concluded that VSOR/VRAC is physicochemically activated by the reduction in Γin through the conformational change in LRRC8 proteins in a manner independent of cell swelling or membrane expansion (Fig. 2A). However, the question as to which domains of LRRC8 proteins are conformationally affected by the Γin reduction to activate VSOR/VRAC remains to be precisely elucidated.

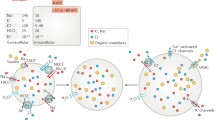
The activation mechanisms of VSOR/VRAC. A Swelling-independent activation physicochemically induced by the Γin reduction. B Swelling-independent activation biochemically induced by oxidation due to NOX-mediated ROS production in response to activation of GPCRs and death receptors. C First-phase swelling-induced ATP-dependent activation in association with swelling-triggered activation of TRPM7 which physically interacts with LRRC8A. Here, the ATP dependence is assumed to be granted by some ATP-bound ABC protein (here called ABCX) which is assumed to physically interact with VSOR/VRAC molecules, presumably at the convex outside of the LRRD of LRRC8A, but released therefrom upon osmotic swelling followed by an interaction with some cytoskeletal component. D Second-phase swelling-induced ROS-dependent activation due not only to NOX-mediated ROS production after GPCR stimulation induced by glutamate (Glu−) and ATP4− released as a result of first-phase activation of VSOR/VRAC but also to the elevation of cytosolic ROS level as a result of the loss of intracellular GSH caused by VSOR/VRAC-mediated GSH− release. (See text for details.)
It must, however, be pointed out that this physicochemical activation mechanism cannot principally account for the swelling-induced activation mechanism because such large extents of Γin reduction required for VSOR/VRAC activation are unlikely to occur under physiological conditions. Furthermore, cell swelling is known to activate VSOR/VRAC even under constant Γin conditions [113,114,115,116].
Swelling-independent oxidation-induced activation of VSOR/VRAC
In 2004, hydrogen peroxide (H2O2), one of the reactive oxygen species (ROS), was found to activate VSOR/VRAC currents under iso-osmotic conditions without leading to cell swelling independently by Shimizu et al. [117] in HeLa cells, Varela et al. [118] in HCT and HeLa cells, and Browe and Baumgarten [119] in cardiomyocytes. This fact was subsequently confirmed by many groups in a variety of cell types [50, 120,121,122,123,124,125,126,127,128,129,130,131,132,133].
Furthermore, Shimizu et al. [117], for the first time, demonstrated that both a mitochondrion-mediated apoptosis inducer staurosporine (STS) and a death receptor-mediated apoptosis inducer tumor necrosis factor-α (TNFα) rapidly activate VSOR/VRAC currents under isotonic conditions in association with significant production of ROS in HeLa cells. Similarly, an ER stress-mediated apoptosis inducer tunicamycin was later shown to increase ROS production and thereby activate VSOR/VRAC currents in rabbit chondrocytes under isotonic conditions [129]. On the other hand, Browe and Baumgarten [119] showed that isotonic VSOR/VRAC activation is induced by angiotensin II via G protein-coupled receptor (GPCR) angiotensin receptor type 1 (AT1), the activation of which is known to induce ROS generation in rabbit ventricular myocytes [134]. GPCR-mediated isotonic VSOR/VRAC activation was also shown to be associated with increased ROS production through B2R activation in mouse astrocytes [50, 51], endothelin-1 ETA receptor activation in rabbit atrial and ventricular myocytes [125], and S1PR1 activation in murine RAW 264.7 macrophages [58]. However, the exact molecular mechanism of GPCR-mediated VSOR/VRAC activation is still elusive.
A variety of other chemical stimuli have been shown to elicit ROS production thereby activating VSOR/VRAC currents under isotonic conditions, including a glucan zymosan in rat microglia [123], HIV protease inhibitors in rabbit ventricular myocytes and LL1 cardiomyocytes [125], a brief acid exposure in mouse nodose ganglia neurons [133], and sub-micromolar ouabain in cancer HT-29, KB, and HepG2 cells [135]. Zinc pyrithione (ZPT), which is known to stimulate ROS production [136, 137], was also found to induce VSOR/VRAC activation in the absence of cell swelling in HEK293 cells [138].
It appears that NADPH oxidase (NOX), which generates superoxide and other downstream ROS, is involved in swelling-independent VSOR/VRAC activation, under normal Γin conditions, in light of the following observations. First, NOX1 was demonstrated to physically interact not only with LRRC8A [139] but also with LRRC8C and 8D [140]. Second, a NOX inhibitor DPI abolished VSOR/VRAC currents induced by STS [117] and β-integrin stretch [119]. Third, a cell-permeable NOX blocker gp9/de-tat was shown to inhibit EGF-induced VSOR/VRAC activation in cardiac myocytes [141]. Fourth, an inhibitor of NOX assembling, 4-(2-aminoethyl)-benzene sulfonyl fluoride (AEBSF), markedly suppressed VSOR/VRAC currents triggered by β-integrin stretch [119]. Since it is known that activation of NOX requires the phosphorylation of its subunit p47phox [142] by PKC [143], VSOR/VRAC activation may also be induced by other chemical agonists for Gq-coupled receptors, for example, a P2YR agonist ATP [48, 144] and a mGluR agonist glutamate [49] in astrocytes through NOX-mediated ROS production presumably triggered by a local intracellular Ca2+ rise in the vicinity of Ca2+-permeable cation channels, called Ca2+ nanodomain [48, 49, 51]. Taken together, it is evident that VSOR/VRAC is biochemically activated by oxidation through NOX-mediated ROS production caused by activation of GPCRs and death receptors, as schematically depicted in Fig. 2B.
Then, the next question is how ROS activate VSOR/VRAC. One possibility is that ROS directly oxidize LRRC8 members thereby inducing some conformational changes in LRRC8. Using Xenopus oocytes overexpressed with fluorescently tagged LRRC8 proteins, Pusch and his collaborators found that LRRC8A/8E heteromeric channels were dramatically activated by oxidation [145] through the disulfide bond formation between two cysteines, C424 of LRR1 and C448 of LRR2, in the intracellular LRR regions of LRRC8E [146]. In contrast, they found that LRRC8A/8C and LRRC8A/8D heteromeric channels were rather inhibited by oxidant chloramine-T [145] and by the oxidation of the start methionine (M1) in LRRC8C [146]. In agreement with these observations, VSOR/VRAC currents were inhibited by oxidation in Jurkat T lymphocytes which exhibit a low expression of LRRC8E [145]. However, ROS were, in contrast, found to activate VSOR/VRAC currents in HeLa cells [117, 118] and KB cells [135], both of which express LRRC8D mRNA at much higher levels (around 13.5 and 11.1 times, respectively) than that of LRRC8E mRNA [147], Thus, it seems likely that sensitivity of LRRC8 members to ROS is different from each other depending on cell types and/or experimental conditions. Another possibility is that ROS indirectly lead to the opening of the VSOR/VRAC pore via some second messengers because ROS are known to stimulate a variety of intracellular mediators including several protein kinases and G-proteins [148], some of which have been suggested to regulate VSOR/VRAC activity (see Reviews [7, 8]). Intracellular ATP is expected to be essential for GPCR- and protein kinase-mediated VSOR/VRAC activation. However, it has yet to be determined how ROS activate VSOR/VRAC and whether intracellular ATP is required for ROS-induced VSOR/VRAC activation.
As described in the preceding section, TGFβ1 can activate VSOR/VRAC under isotonic conditions [93]. Also, swelling-independent activation of VSOR/VRAC was recently found to be induced by stimulation not only with TNFα but also with another cytokine IL-1β and some other heat-labile serum protein, in an additive fashion, under isotonic conditions in a manner sensitive to gene knockout of LRRC8A or LRRC8E [103]. This activation was shown to be dependent on the plasmalemmal expression of cGAS which exhibits a physical interaction with LRRC8A [103]. However, it is not known how TGFβ1 and IL-1β induce VSOR/VRAC activation as well as which heat-labile serum protein, other than TNFα and IL-1β, can activate VSOR/VRAC.
Swelling-induced activation of VSOR/VRAC
Osmotic swelling-induced VSOR/VRAC currents were reported to be only partially inhibited by NOX inhibitors; that is, around 35% suppression by AEBSF in human neutrophils [149] and around 40% inhibition by DPI in mouse astrocytes [49]. Furthermore, swelling- and ROS-induced activation mechanisms were elucidated to be independent of each other, since hypotonicity- and chloramine-T-induced VSOR/VRAC currents observed in Xenopus oocytes overexpressed with LRRC8A and 8E were additive [145]. Thus, it appears that a major component of swelling-induced VSOR/VRAC currents is independent of ROS.
First-phase ROS-independent, cytosolic ATP-dependent component of swelling-induced activation
As summarized in the Part 1 article [12], TRPM7, which was shown to exert as a mechano-sensitive swelling-activated cation channel by Numata et al. [150, 151], serves as the swelling-sensing subcomponent of VSOR/VRAC not only by enhancing LRRC8A mRNA expression via steady-state Ca2+ influx but also by exhibiting real-time functional coupling to VSOR/VRAC activity with showing a physical interaction to LRRC8A protein [11]. It is likely that the TRPM7-mediated Ca2+ influx is somehow implicated in the VSOR/VRAC activation caused by cell swelling or membrane expansion. Although a global rise of the intracellular free Ca2+ concentration ([Ca2+]i) is not required for swelling-induced VSOR/VRAC activation (see Review [7]), there remains a possibility of an involvement of localized Ca2+ rise therein [49, 152]. In any case, it is evident that hypotonicity-induced VSOR/VRAC is associated with conformational changes in pore-forming LRRC8 proteins, because this activation was observed to be coupled to the displacement of C-terminal LRRDs in HeLa cells and LRRC8−/− HEK293 cells transfected with fluorescence-labeled LRRC8A and LRRC8E by FRET studies [153].
Although cytosolic ATP dependence is one of the most important physiological properties of VSOR/VRAC, its exact molecular mechanism is still missing, as pointed out in the previous review articles [7, 12]. Non-hydrolytic requirement of intracellular ATP may suggest that some ATP-binding protein plays an essential role in the mechanism of VSOR/VRAC activation. In agreement with this inference, so far, four members of the ATP-binding cassette (ABC) transporter superfamily proteins, three of which are membrane-spanning and another is cytosolic proteins, have been reported to be involved in the regulation of VSOR/VRAC activity. The chronologically first one is P-glycoprotein (PGP), the MDR1 gene product, which was initially proposed as the molecular entity of VSOR/VRAC [154, 155]. Although its “PGP = VSOR/VRAC” hypothesis was later rejected [156], PGP was shown to upregulate the volume sensitivity of VSOR/VRAC channel [157]. Second, CFTR which has a structural similarity to PGP and exerts as the cAMP/PKA-dependent Cl− channel, was shown to downregulate VSOR/VRAC currents [158, 159] through the second nucleotide-binding domain (NBD2) [159]. The third one ABCF2, a cytosolic member of the ABC proteins, was shown to suppress VSOR/VRAC activity [160], as detailed below. The last one ABCG1, a cholesterol-exporting ATPase, was shown to enhance hypotonicity-induced ATP release mediated by VSOR/VRAC presumably through reduction of the cholesterol level within the plasma membrane [73]. Depletion of membrane cholesterol content was shown to enhance VSOR/VRAC activation induced by mild hypotonic stimulation [161,162,163]. Subsequently, cholesterol depletion-induced VSOR/VRAC activation was demonstrated to be mediated by F-actin [164], the expression of which was recently shown to be essential for VSOR/VRAC activity [165]. Since cholesterol depletion was reported to release several cytoskeletal proteins, such as actin, α-actinin, and ezrin from the cellular membrane fractions [166], there arises a possibility that an interaction between an ATP-binding protein and a cytoskeletal component released in response to osmotic cell swelling is involved in the swelling-induced activation mechanism of VSOR/VRAC. Taken together, swelling-induced activation of VSOR/VRAC is likely regulated by TRPM7 and some ABC proteins, which may interact with LRRC8 members as well as some cytoskeletal components in the native cell system, as schematically drawn in Fig. 2C.
Ando-Akatsuka et al. [160] found that an actin-binding and -crosslinking protein, α-actinin-4 (ACTN4), which is ubiquitously expressed in non-muscle cells [167] and participates in the cytoskeleton organization [168, 169], becomes associated with the plasma-membrane upon osmotic cell swelling. Next, by the protein overlay assays combined with proteomics approaches, a cytosolic member of the ABC transporter protein superfamily, ABCF2, was identified as the binding partner of ACTN4, and then the physical interaction (binding in the broad sense) between ACTN4 and ABCF2 was found to be prominently enhanced by hypotonic cell swelling. Furthermore, knockdown and overexpression of ABCF2 were shown to augment and suppress the VSOR/VRAC activity, respectively. Therefore, it was concluded that swelling-induced activation of VSOR/VRAC is accomplished by the protein–protein interaction between ACTN4 and ABCF2, thereby preventing ABCF2 from inhibiting VSOR/VRAC activity. Thus, it is likely that the cytosolic ATP-binding protein ABCF2 represents an endogenous blocking subcomponent of VSOR/VRAC. ABCF2 may also grant non-hydrolytic ATP dependence and free Mg2+ sensitivity to VSOR/VRAC, if only the form of ABCF2 bound to ATP, but not to Mg-ATP, can be released via VSOR/VRAC, thereby activating VSOR/VRAC upon osmotic swelling. Further investigations are warranted to prove this inference by testing the possibility that ABCF2 physically interacts with or directly binds to LRRC8 member proteins, especially to LRRDs which were shown to be required for swelling-induced VSOR/VRAC activation [170].
Second-phase ROS-dependent, GPCR-mediated component of swelling-induced activation
Hypotonic stimulation has been often found to bring about ROS production under certain conditions [118, 128, 129, 133, 171, 172]. However, this fact does not necessarily imply a direct action of osmotic swelling. As described in the preceding section, osmotic cell swelling often induces VSOR/VRAC-mediated release of GSH, glutamate, and ATP depending on cell type. Therefore, osmotic swelling may indirectly result in a rise of intracellular ROS level caused by GSH release mediated by VSOR/VRAC and by ROS production due to GPCR activation induced by glutamate and ATP. Consistently, hypotonicity-induced ROS production was shown to be mediated by NMDA receptors in rat astrocytes [173]. Thus, it is conceivable that swelling-induced VSOR/VRAC activity is enhanced by GPCR-mediated ROS production and VSOR/VRAC-mediated GSH− release in a manner of positive feedback control, as schematically depicted in Fig. 2D. However, it must be noted that this component is the secondary result of earlier ROS-independent swelling-induced VSOR/VRAC activation (Fig. 2C).
Swelling-induced VSOR/VRAC activity was observed to be upregulated by an increase in intracellular cAMP through adenylate cyclase (AC)-coupled Ca2+-sensing receptor, CaR, and arginine vasopressin type-2 receptor, V2R, both of which belong to the Gs-coupled receptor family, in response to elevation of extracellular Ca2+ [174] and arginine vasopressin [175], respectively. Stimulation of protein-tyrosine kinase (PTK)-coupled epidermal growth factor receptor, EGFR, was also shown to upregulate swelling-induced VSOR/VRAC activity [176]. The exact upregulating mechanisms of cAMP/AC- and PTK-mediated signaling pathways remain elusive.
Conclusions and perspectives
The volume-sensitive outwardly rectifying/volume-regulatory anion channel (VSOR/VRAC) activated by cell swelling transports inorganic halide anions (mainly Cl−), thereby regulating the cell volume after osmotic swelling. In addition, this channel was shown to serve as transporting pathways for many organic substances, the sizes of which are smaller than the VSOR/VRAC pore size. These organic substances include major extracellular messenger molecules for autocrine/paracrine signaling such as glutamate and ATP as well as anti-oxidant GSH, anti-inflammatory itaconate, and anti-viral defensing cGAMP. The activation mechanisms of VSOR/VRAC are classified into swelling-dependent and -independent ones. Reduction of intracellular ionic strength (Γin) physicochemically activates VSOR/VRAC due to the conformational changes in LRRC8 proteins in a manner independent of cell swelling. Also, VSOR/VRAC can be biochemically activated by oxidation even in the absence of cell swelling, because LRRC8 proteins are physically interacting with NOX which releases ROS, when some GPCRs and death receptors are activated. The mechanisms of swelling-induced activation are composed of two phases. The first phase is dependent on swelling-sensing TRPM7 which exhibits a physical interaction with the LRRC8A molecule and on the nonhydrolytic existence of intracellular free ATP. The second phase is dependent on GPCR activation triggered by glutamate and ATP which are released via VSOR/VRAC activated in the first phase and on the ROS production due to GPCR-mediated NOX activation and GSH release via VSOR/VRAC activated in the first phase. After the identification of the pore-forming core components of VSOR/VRAC as LRRC8 members, a large number of recent studies have elucidated the molecular processes of VSOR/VRAC-mediated release of organic substances and of VSOR/VRAC activation. However, still much remains unanswered, and many new questions have arisen, as pointed out in each section of this article and collectively listed in Supplementary Table as research subjects that remain to be studied in the near future.
Since VSOR/VRAC was recently demonstrated to be activated by inflammatory signals, BK and S1P, as well as by anti-inflammatory signals, itaconate and cGAMP, there arises a possibility that VSOR/VRAC activity plays some important and reciprocal roles in the inflammation processes. Further studies are warranted to investigate this possibility.
Availability of data and materials
The data underlying this article will be obtained via PubMed and Google Scholar or available from the author upon reasonable request.
Abbreviations
- CFTR:
-
Cystic fibrosis transmembrane conductance regulator
- VAAC:
-
Volume-activated anion channel
- VSOR:
-
Volume-sensitive outwardly rectifying anion channel
- VRAC:
-
Volume-regulated anion channel
- Maxi-Cl:
-
Large-conductance ohmic maxi-anion channel
- VSOR/VRAC:
-
The volume-sensitive outwardly rectifying/volume-regulatory anion channel
- VSOAC:
-
Volume-sensitive organic osmolyte and anion channel
- cryo-EM:
-
Cryo-electron microscopy
- GSH:
-
Glutathione
- cGAMP:
-
2′3′-Cyclic-GMP-AMP
- mGluR:
-
Muscarinic glutamate receptor
- P2YR:
-
Purinergic P2Y receptor
- BK:
-
Bradykinin
- B2R:
-
Bradykinin B2 receptor
- S1P:
-
Sphingosine-1-phosphate
- DAMP:
-
Danger-associated molecular pattern
- EMT:
-
Epithelial-to-mesenchymal transition
- TLR:
-
Toll-like receptor
- cGAS:
-
Cyclic-AMP-GMP synthase
- dsDNA:
-
Double-stranded DNA
- STING:
-
Stimulator of interferon genes
- IFN:
-
Interferon
- MLF:
-
Mouse lung fibroblast
- BMDM:
-
Bone marrow-derived macrophage
- MEF:
-
Mouse embryonic fibroblast
- Γin :
-
Intracellular ionic strength
- LRR:
-
Leucine-rich repeat
- NT:
-
N-terminal
- MD:
-
Molecular dynamics
- sbs:
-
Sybodies
- ROS:
-
Reactive oxygen species
- STS:
-
Staurosporine
- TNFα:
-
Tumor necrosis factor-α
- GPCR:
-
G protein-coupled receptor
- AT1 :
-
Angiotensin receptor type 1
- S1PR1:
-
Sphingosine-1-phosphate receptor 1
- ZPT:
-
Zinc pyrithione
- NOX:
-
NADPH oxidase
- AEBSF:
-
4-(2-Aminoethyl)-benzene sulfonyl fluoride
- ABC:
-
ATP-binding cassette
- PGP:
-
P-glycoprotein
- NBD2:
-
Second nucleotide-binding domain
- AC:
-
Adenylate cyclase
- CaR:
-
Ca2+-sensing receptor
- PTK:
-
Protein-tyrosine kinase
References
Okada Y, Okada T, Sato-Numata K, Islam MR, Ando-Akatsuka Y, Numata T, Kubo M, Shimizu T, Kurbannazarova RS, Marunaka M, Sabirov RZ (2019) Cell volume-activated and -correlated anion channels in mammalian cells: their biophysical, molecular and pharmacological properties. Pharmacol Rev 71:49–88. https://doi.org/10.1124/pr.118.015917
Pedersen SF, Okada Y, Nilius B (2016) Biophysics and physiology of the volume-regulated anion channel (VRAC)/volume-sensitive outwardly rectifying anion channel (VSOR). Pflügers Arch Eur J Physiol 468(3):371–383. https://doi.org/10.1007/s00424-015-1781-6
Sabirov RZ, Islam MR, Okada T, Merzlyak PG, Kurbannazarova RS, Tsiferova NA, Okada Y (2021) The ATP-releasing Maxi-Cl channel: its identity, molecular partners, and physiological/pathophysiological implications. Life 11:509. https://doi.org/10.3390/life11060509
Hazama A, Okada Y (1988) Ca2+ sensitivity of volume-regulatory K+ and Cl− channels in cultured human epithelial cells. J Physiol (London) 402:687–702. https://doi.org/10.1113/jphysiol.1988.sp017229
Cahalan MD, Lewis RS (1988) Role of potassium and chloride channels in volume regulation by T lymphocytes. Soc Gen Physiol Ser 43:281–301
Strange K, Emma F, Jackson PS (1996) Cellular and molecular physiology of volume-sensitive anion channels. Am J Physiol 270(3 Pt 1):C711–C730. https://doi.org/10.1152/ajpcell.1996.270.3.C711
Okada Y (1997) Volume expansion-sensing outward-rectifier Cl− channel: fresh start to the molecular identity and volume sensor. Am J Physiol 273(3 Pt 1):C755–C789. https://doi.org/10.1152/ajpcell.1997.273.3.C755
Nilius B, Eggermont J, Voets T, Buyse G, Manolopoulos V, Droogmans G (1997) Properties of volume-regulated anion channels in mammalian cells. Prog Biophys Mol Biol 68(1):69–119. https://doi.org/10.1016/s0079-6107(97)00021-7
Qiu Z, Dubin AE, Mathur J, Tu B, Reddy K, Miraglia LJ, Reinhardt J, Orth AP, Patapoutian A (2014) SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell 157(2):447–458. https://doi.org/10.1016/j.cell.2014.03.024
Voss FK, Ullrich F, Münch J, Lazarow K, Lutter D, Mah N, Andrade-Navarro MA, von Kries JP, Stauber T, Jentsch TJ (2014) Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 344(6184):634–638. https://doi.org/10.1126/science.1252826
Numata T, Sato-Numata K, Hermosura MC, Mori Y, Okada Y (2021) TRPM7 is an essential regulator for volume-sensitive outwardly rectifying anion channel. Commun Biol 4(1):599. https://doi.org/10.1038/s42003-021-02127-9
Okada Y (2024) Physiology of the volume-sensitive/regulatory anion channel VSOR/VRAC. Part 1: From its discovery and phenotype characterization to the molecular entity identification. J Physiol Sci 74:3. https://doi.org/10.1186/s12576-023-00897-x
Sabirov RZ, Okada Y (2005) ATP release via anion channels. Purinergic Signal 1:311–328. https://doi.org/10.1007/s11302-005-1557-0
Strange K, Jackson PS (1998) Swelling-activated organic osmolyte efflux: a new role for anion channels. Kidney Int 48(4):994–1003. https://doi.org/10.1038/ki.1995.381
Kimelberg HK, Goderie SK, Higman S, Pang S, Waniewski RA (1990) Swelling-induced release of glutamate, aspartate, and taurine from astrocyte cultures. J Neurosci 10(5):1583–1591. https://doi.org/10.1523/JNEUROSCI.10-05-01583.1990
Jesuś García J, Sánchez Olea R, Pasantes-Morales H (1991) Taurine release associated to volume regulation in rabbit lymphocytes. J Cell Biochem 45(2):207–212. https://doi.org/10.1002/jcb.240450212
Roy G, Malo C (1992) Activation of amino acid diffusion by a volume increase in cultured kidney (MDCK) cells. J Membr Biol 130(1):83–90. https://doi.org/10.1007/BF00233740
Kubo M, Okada Y (1992) Volume-regulatory Cl− channel currents in cultured human epithelial cells. J Physiol (London) 456:351–371. https://doi.org/10.1113/jphysiol.1992.sp019340
Banderali U, Roy G (1992) Anion channels for amino acids in MDCK cells. Am J Physiol 263(6 Pt 1):C1200–C1207. https://doi.org/10.1152/ajpcell.1992.263.6.C1200
Linsdell P, Hanrahan JW (1998) Adenosine triphosphate-dependent asymmetry of anion permeation in the cystic fibrosis transmembrane conductance regulator chloride channel. J Gen Physiol 111(4):601–614. https://doi.org/10.1085/jgp.111.4.601
Nilius B, Droogmans G (2003) Amazing chloride channels: an overview. Acta Physiol Scand 177(2):119–147. https://doi.org/10.1046/j.1365-201X.2003.01060.x
Droogmans G, Maertens C, Prenen J, Nilius B (1999) Sulphonic acid derivatives as probes of pore properties of volume-regulated anion channels in endothelial cells. Br J Pharmacol 28(1):35–40. https://doi.org/10.1038/sj.bjp.0702770
Ternovsky VI, Okada Y, Sabirov RZ (2004) Sizing the pore of the volume-sensitive anion channel by differential polymer partitioning. FEBS Lett 576(3):433–436. https://doi.org/10.1016/j.febslet.2004.09.051
Planells-Cases R, Lutter D, Guyader C, Gerhards NM, Ullrich F, Elger DA, Kucukosmanoglu A, Xu G, Voss FK, Reincke SM, Stauber T, Blomen VA, Vis DJ, Wessels LF, Brummelkamp TR, Borst P, Rottenberg S, Jentsch TJ (2015) Subunit composition of VRAC channels determines substrate specificity and cellular resistance to Pt-based anti-cancer drugs. EMBO J 34(24):2993–3008. https://doi.org/10.15252/embj.201592409
Lutter D, Ullrich F, Lueck JC, Kempa S, Jentsch TJ (2017) Selective transport of neurotransmitters and modulators by distinct volume-regulated LRRC8 anion channels. J Cell Sci 130(6):1122–1133. https://doi.org/10.1242/jcs.196253
Yang J, Vitery MDC, Chen J, Osei-Owusu J, Chu J, Qiu Z (2019) Glutamate-releasing SWELL1 channel in astrocytes modulates synaptic transmission and promotes brain damage in stroke. Neuron 102(4):813-827.e6. https://doi.org/10.1016/j.neuron.2019.03.029
Nakamura R, Numata T, Kasuya G, Yokoyama T, Nishizawa T, Kusakizako T, Kato T, Hagino T, Dohmae N, Inoue M, Watanabe K, Ichijo H, Kikkawa M, Shirouzu M, Jentsch TJ, Ishitani R, Okada Y, Nureki O (2020) Cryo-EM structure of the volume-regulated anion channel LRRC8D isoform. Commun Biol 3:240. https://doi.org/10.1038/s42003-020-0951-z
Deneka D, Sawicka M, Lam AKM, Paulino C, Dutzler R (2018) Structure of a volume-regulated anion channel of the LRRC8 family. Nature 558(7709):254–259. https://doi.org/10.1038/s41586-018-0134-y
Kasuya G, Nakane T, Yokoyama T, Jia Y, Inoue M, Watanabe K, Nakamura R, Nishizawa T, Kusakizako T, Tsutsumi A, Yanagisawa H, Dohmae N, Hattori M, Ichijo H, Yan Z, Kikkawa M, Shirouzu M, Ishitani R, Nureki O (2018) Cryo-EM structures of the human volume-regulated anion channel LRRC8. Nat Struct Mol Biol 25(9):797–804. https://doi.org/10.1038/s41594-018-0109-6
Kefauver JM, Saotome K, Dubin AE, Pallesen J, Cottrell CA, Cahalan SM, Qiu Z, Hong G, Crowley CS, Whitwam T, Lee W-H, Ward AB, Patapoutian A (2018) Structure of the human volume regulated anion channel. Elife 7:e38461. https://doi.org/10.7554/eLife.38461
Kern DM, Oh S, Hite RK, Brohawn SG (2019) Cryo-EM structures of the DCPIB-inhibited volume-regulated anion channel LRRC8A in lipid nanodiscs. Elife 8:e42636. https://doi.org/10.7554/eLife.42636
Liu H, Polovitskaya MM, Yang L, Li M, Li H, Han Z, Wu J, Zhang Q, Jentsch TJ, Liao J (2023) Structural insights into anion selectivity and activation mechanism of LRRC8 volume-regulated anion channels. Cell Rep 42(8):112926. https://doi.org/10.1016/j.celrep.2023.112926
Rutz S, Deneka D, Dittmann A, Sawicka M, Dutzler R (2023) Structure of a volume-regulated heteromeric LRRC8A/C channel. Nat Struct Mol Biol 30(1):52–61. https://doi.org/10.1038/s41594-022-00899-0
Kern DM, Bleier J, Mukherjee S, Hill JM, Kossiakoff AA, Isacoff EY, Brohawn SG (2023) Structural basis for assembly and lipid-mediated gating of LRRC8A: C volume-regulated anion channels. Nat Struct Mol Biol 30(6):841–852. https://doi.org/10.1038/s41594-023-00944-6
Takahashi H, Yamada T, Denton JS, Strange K, Karakas E (2023) Cryo-EM structures of an LRRC8 chimera with native functional properties reveal heptameric assembly. Elife 12:e82431. https://doi.org/10.7554/eLife.82431
Liu H-T, Tashmukhamedov BA, Inoue H, Okada Y, Sabirov RZ (2006) Roles of two types of anion channels in glutamate release from mouse astrocytes under ischemic or osmotic stress. Glia 54:343–357. https://doi.org/10.1002/glia.20400
Hyzinski-García MC, Vincent MY, Haskew-Layton RE, Dohare P, Keller RW Jr, Mongin AA (2011) Hypo-osmotic swelling modifies glutamate-glutamine cycle in the cerebral cortex and in astrocyte cultures. J Neurochem 118(1):140–152. https://doi.org/10.1111/j.1471-4159.2011.07289.x
Netti V, Pizzoni A, Pérez-Domínguez M, Ford P, Pasantes-Morales H, Ramos-Mandujano G, Capurro C (2018) Release of taurine and glutamate contributes to cell volume regulation in human retinal Müller cells: differences in modulation by calcium. J Neurophysiol 120(3):973–984. https://doi.org/10.1152/jn.00725.2017
Abdullaev IF, Rudkouskaya A, Schools GP, Kimelberg HK, Mongin AA (2006) Pharmacological comparison of swelling-activated excitatory amino acid release and Cl− currents in cultured rat astrocytes. J Physiol 572(Pt 3):677–689. https://doi.org/10.1113/jphysiol.2005.103820
Bowens NH, Dohare P, Kuo Y-H, Mongin AA (2013) DCPIB, the proposed selective blocker of volume-regulated anion channels, inhibits several glutamate transport pathways in glial cells. Mol Pharmacol 83(1):22–32. https://doi.org/10.1124/mol.112.080457
Hyzinski-García MC, Rudkouskaya A, Mongin AA (2014) LRRC8A protein is indispensable for swelling-activated and ATP-induced release of excitatory amino acids in rat astrocytes. J Physiol 92(22):4855–4862. https://doi.org/10.1113/jphysiol.2014.278887
Wilson CS, Bach MD, Ashkavand Z, Norman KR, Martino N, Adam AP, Mongin AA (2019) Metabolic constraints of swelling-activated glutamate release in astrocytes and their implication for ischemic tissue damage. J Neurochem 151(2):255–272. https://doi.org/10.1111/jnc.14711
Balkaya M, Dohare P, Chen S, Schober AL, Fidaleo AM, Nalwalk JW, Sah R, Mongin AA (2023) Conditional deletion of LRRC8A in the brain reduces stroke damage independently of swelling-activated glutamate release. iScience. 26(5):106669. https://doi.org/10.1016/j.isci.2023.106669
Schober AL, Wilson CS, Mongin AA (2017) Molecular composition and heterogeneity of the LRRC8-containing swelling-activated osmolyte channels in primary rat astrocytes. J Physiol 595(22):6939–6951. https://doi.org/10.1113/JP275053
Sabirov RZ, Merzlyak PG, Okada T, Islam MR, Uramoto H, Mori T, Makino Y, Matsuura H, Xie Y, Okada Y (2017) The organic anion transporter SLCO2A1 constitutes the core component of the Maxi-Cl channel. EMBO J 36(22):3309–3324. https://doi.org/10.15252/embj.201796685
Jeremic A, Jeftinija K, Stevanovic J, Glavaski A, Jeftinija S (2001) ATP stimulates calcium-dependent glutamate release from cultured astrocytes. J Neurochem 77(2):664–675. https://doi.org/10.1046/j.1471-4159.2001.00272.x
Rudkouskaya A, Chernoguz A, Haskew-Layton RE, Mongin AA (2008) Two conventional protein kinase C isoforms, alpha and beta I, are involved in the ATP-induced activation of volume-regulated anion channel and glutamate release in cultured astrocytes. J Neurochem 105(6):2260–2270. https://doi.org/10.1111/j.1471-4159.2008.05312.x
Akita T, Fedorovich SV, Okada Y (2011) Ca2+ nanodomain-mediated component of swelling-induced volume-sensitive outwardly rectifying anion current triggered by autocrine action of ATP in mouse astrocytes. Cell Physiol Biochem 28(6):1181–1190. https://doi.org/10.1159/000335867
Akita T, Okada Y (2014) Characteristics and roles of the volume-sensitive outwardly rectifying (VSOR) anion channel in the central nervous system. Neuroscience 275:211–231. https://doi.org/10.1016/j.neuroscience.2014.06.015
Liu H-T, Akita T, Shimizu T, Sabirov RZ, Okada Y (2009) Bradykinin-induced astrocyte-neuron signaling: glutamate release is mediated by ROS-activated volume-sensitive outwardly rectifying anion channels. J Physiol (London) 587:2197–2209. https://doi.org/10.1113/jphysiol.2008.165084
Akita T, Okada Y (2011) Regulation of bradykinin-induced activation of volume-sensitive outwardly rectifying anion channels by Ca2+ nanodomains in mouse astrocytes. J Physiol (London) 589:3909–3927. https://doi.org/10.1113/jphysiol.2011.208173
Bhoola KD, Figueroa CD, Worthy K (1992) Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol Rev 44(1):1–80
Ding-Zhou L, Margaill I, Palmier B, Pruneau D, Plotkine M, Marchand-Verrecchia C (2003) LF 16–0687 Ms, a bradykinin B2 receptor antagonist, reduces ischemic brain injury in a murine model of transient focal cerebral ischemia. Br J Pharmacol 139(8):1539–1547. https://doi.org/10.1038/sj.bjp.0705385
Leeb-Lundberg LM, Marceau F, Müller-Esterl W, Pettibone DJ, Zuraw BL (2005) International union of pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol Rev 57(1):27–77. https://doi.org/10.1124/pr.57.1.2
Cui W, Ning Y, Hong W, Wang J, Liu Z, Li MD (2019) Crosstalk between inflammation and glutamate system in depression: signaling pathway and molecular biomarkers for ketamine’s antidepressant effect. Mol Neurobiol 56(5):3484–3500. https://doi.org/10.1007/s12035-018-1306-3
Haas HS, Linecker A, Pfragner R, Sadjak A (2010) Peripheral glutamate signaling in head and neck areas. Head Neck 32(11):1554–1572. https://doi.org/10.1002/hed.21438
Wen ZH, Chang YC, Jean YH (2015) Excitatory amino acid glutamate: role in peripheral nociceptive transduction and inflammation in experimental and clinical osteoarthritis. Osteoarthritis Cartilage 23(11):2009–2016. https://doi.org/10.1016/j.joca.2015.03.017
Burow P, Klapperstück M, Markwardt F (2015) Activation of ATP secretion via volume-regulated anion channels by sphingosine-1-phosphate in RAW macrophages. Pflügers Arch 467(6):1215–1226. https://doi.org/10.1007/s00424-014-1561-8
Hait NC, Maiti A (2017) The role of sphingosine-1-phosphate and ceramide-1-phosphate in inflammation and cancer. Mediators Inflamm 2017:4806541. https://doi.org/10.1155/2017/4806541
Nagahashi M, Abe M, Sakimura K, Takabe K, Wakai T (2018) The role of sphingosine-1-phosphate in inflammation and cancer progression. Cancer Sci 109(12):3671–3678. https://doi.org/10.1111/cas.13802
Olivera A, Buckley NE, Spiegel S (1992) Sphingomyelinase and cell-permeable ceramide analogs stimulate cellular proliferation in quiescent Swiss 3T3 fibroblasts. J Biol Chem 267(36):26121–26127
Van Brocklyn JR, Lee MJ, Menzeleev R, Olivera A, Edsall L, Cuvillier O, Thomas DM, Coopman PJ, Thangada S, Liu CH, Hla T, Spiegel S (1998) Dual actions of sphingosine-1-phosphate: extracellular through the Gi-coupled receptor Edg-1 and intracellular to regulate proliferation and survival. J Cell Biol 142(1):229–240. https://doi.org/10.1083/jcb.142.1.229
Blaho VA, Hla T (2014) An update on the biology of sphingosine 1-phosphate receptors. J Lipid Res 55(8):1596–1608. https://doi.org/10.1194/jlr.R046300
Fields RD, Stevens B (2000) ATP: an extracellular signaling molecule between neurons and glia. Trends Neurosci 23(12):625–633. https://doi.org/10.1016/s0166-2236(00)01674-x
Burnstock G (2004) Introduction: P2 receptors. Curr Top Med Chem 4(8):793–803. https://doi.org/10.2174/1568026043451014
Hisadome K, Koyama T, Kimura C, Droogmans G, Ito Y, Oike M (2002) Volume-regulated anion channels serve as an auto/paracrine nucleotide release pathway in aortic endothelial cells. J Gen Physiol 119(6):511–520. https://doi.org/10.1085/jgp.20028540
Fields RD, Ni Y (2010) Nonsynaptic communication through ATP release from volume-activated anion channels in axons. Sci Signal. 3(142):ra73. https://doi.org/10.1126/scisignal.2001128
Mitchell CH, Carré DA, McGlinn AM, Stone RA, Civan MM (1998) A release mechanism for stored ATP in ocular ciliary epithelial cells. Proc Natl Acad Sci USA 95(12):7174–7178. https://doi.org/10.1073/pnas.95.12.7174
Hazama A, Shimizu T, Ando-Akatsuka Y, Hayashi S, Tanaka S, Maeno E, Okada Y (1999) Swelling-induced, CFTR-independent ATP release from a human epithelial cell line: lack of correlation with volume-sensitive Cl− channels. J Gen Physiol 114(4):525–533. https://doi.org/10.1085/jgp.114.4.525
Dutta AK, Sabirov RZ, Uramoto H, Okada Y (2004) Role of ATP-conductive anion channel in ATP release from neonatal rat cardiomyocytes in ischaemic or hypoxic conditions. J Physiol 559(Pt 3):799–812. https://doi.org/10.1113/jphysiol.2004.069245
Liu H-T, Toychiev AH, Takahashi N, Sabirov RZ, Okada Y (2008) Maxi-anion channel as a candidate pathway for osmosensitive ATP release from mouse astrocytes in primary culture. Cell Res 18(5):558–565. https://doi.org/10.1038/cr.2008.49
Fujii Y, Maekawa S, Morita M (2017) Astrocyte calcium waves propagate proximally by gap junction and distally by extracellular diffusion of ATP released from volume-regulated anion channels. Sci Rep 7(1):13115. https://doi.org/10.1038/s41598-017-13243-0
Dunn PJ, Salm EJ, Tomita S (2020) ABC transporters control ATP release through cholesterol-dependent volume-regulated anion channel activity. J Biol Chem 295(16):5192–5203. https://doi.org/10.1074/jbc.RA119.010699
Chu J, Yang J, Zhou Y, Chen J, Chen KH, Zhang C, Cheng HY, Koylass N, Liu JO, Guan Y, Qiu Z (2023) ATP-releasing SWELL1 channel in spinal microglia contributes to neuropathic pain. Sci Adv 9(13):eade9931. https://doi.org/10.1126/sciadv.ade9931
Zahiri D, Burow P, Großmann C, Müller CE, Klapperstück M (1868) Markwardt F (2021) Sphingosine-1-phosphate induces migration of microglial cells via activation of volume-sensitive anion channels, ATP secretion and activation of purinergic receptors. Biochim Biophys Acta Mol Cell Res 2:118915. https://doi.org/10.1016/j.bbamcr.2020.118915
Furuya K, Hirata H, Kobayashi T, Sokabe M (2021) Sphingosine-1-phosphate induces ATP release via volume-regulated anion channels in breast cell lines. Life (Basel) 11(8):851. https://doi.org/10.3390/life11080851
Gaitán-Peñas H, Gradogna A, Laparra-Cuervo L, Solsona C, Fernández-Dueñas V, Barrallo-Gimeno A, Ciruela F, Lakadamyali M, Pusch M, Estévez R (2016) Investigation of LRRC8-mediated volume-regulated anion currents in Xenopus oocytes. Biophys J 111(7):1429–1443. https://doi.org/10.1016/j.bpj.2016.08.030
Jackson PS, Strange K (1995) Characterization of the voltage-dependent properties of a volume-sensitive anion conductance. J Gen Physiol 105(5):661–676. https://doi.org/10.1085/jgp.105.5.661
Tsumura T, Oiki S, Ueda S, Okuma M, Okada Y (1996) Sensitivity of volume-sensitive Cl− conductance in human epithelial cells to extracellular nucleotides. Am J Physiol 271:C1872–C1878. https://doi.org/10.1152/ajpcell.1996.271.6.C1872
Sato-Numata K, Numata T, Inoue R, Okada Y (2016) Distinct pharmacological and molecular properties of the acid-sensitive outwardly rectifying (ASOR) anion channel from those of the volume-sensitive outwardly rectifying (VSOR) anion channel. Pflügers Arch Eur J Physiol 468:795–803. https://doi.org/10.1007/s00424-015-1786-1
Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S (2017) The P2X7 receptor in infection and inflammation. Immunity 47(1):15–31. https://doi.org/10.1016/j.immuni.2017.06.020
Le TT, Berg NK, Harting MT, Li X, Eltzschig HK, Yuan X (2019) Purinergic signaling in pulmonary inflammation. Front Immunol 10:1633. https://doi.org/10.3389/fimmu.2019.01633
Fodor P, White B, Khan R (2020) Inflammation—the role of ATP in pre-eclampsia. Microcirculation 27(1):e12585. https://doi.org/10.1111/micc.12585
Vuerich M, Mukherjee S, Robson SC, Longhi MS (2020) Control of gut inflammation by modulation of purinergic signaling. Front Immunol 11:1882. https://doi.org/10.3389/fimmu.2020.01882
Schädlich IS, Winzer R, Stabernack J, Tolosa E, Magnus T, Rissiek B (2023) The role of the ATP-adenosine axis in ischemic stroke. Semin Immunopathol 45(3):347–365. https://doi.org/10.1007/s00281-023-00987-3
Hammond CL, Lee TK, Ballatori N (2001) Novel roles for glutathione in gene expression, cell death, and membrane transport of organic solutes. J Hepatol 34(6):946–954. https://doi.org/10.1016/s0168-8278(01)00037-x
Wu G, Fang YZ, Yang S, Lupton JR, Turner ND (2004) Glutathione metabolism and its implications for health. J Nutr 134(3):489–492. https://doi.org/10.1093/jn/134.3.489
Orlowski M, Meister A (1970) The gamma-glutamyl cycle: a possible transport system for amino acids. Proc Natl Acad Sci USA 67(3):1248–1255. https://doi.org/10.1073/pnas.67.3.1248
Franco R, Cidlowski JA (2006) SLCO/OATP-like transport of glutathione in FasL-induced apoptosis: glutathione efflux is coupled to an organic anion exchange and is necessary for the progression of the execution phase of apoptosis. J Biol Chem 281(40):29542–32957. https://doi.org/10.1074/jbc.M602500200
Franco R, Panayiotidis MI, Cidlowski JA (2007) Glutathione depletion is necessary for apoptosis in lymphoid cells independent of reactive oxygen species formation. J Biol Chem 282(42):30452–33065. https://doi.org/10.1074/jbc.M703091200
Circu ML, Stringer S, Rhoads CA, Moyer MP, Aw TY (2009) The role of GSH efflux in staurosporine-induced apoptosis in colonic epithelial cells. Biochem Pharmacol 77(1):76–85. https://doi.org/10.1016/j.bcp.2008.09.011
Sabirov RZ, Kurbannazarova RS, Melanova NR, Okada Y (2013) Volume-sensitive anion channels mediate osmosensitive glutathione release from rat thymocytes. PLoS ONE 8(1):e55646. https://doi.org/10.1371/journal.pone.0055646
Friard J, Corinus A, Cougnon M, Tauc M, Pisani DF, Duranton C, Rubera I (2019) LRRC8/VRAC channels exhibit a noncanonical permeability to glutathione, which modulates epithelial-to-mesenchymal transition (EMT). Cell Death Dis 10(12):925. https://doi.org/10.1038/s41419-019-2167-z
Hooftman A, Angiari S, Hester S, Corcoran SE, Runtsch MC, Ling C, Ruzek MC, Slivka PF, McGettrick AF, Banahan K, Hughes MM, Irvine AD, Fischer R, O’Neill LA (2020) The immunomodulatory metabolite itaconate modifies NLRP3 and inhibits inflammasome activation. Cell Metab 32(3):468-478.e7. https://doi.org/10.1016/j.cmet.2020.07.016
Swain A, Bambouskova M, Kim H, Andhey PS, Duncan D, Auclair K, Chubukov V, Simons DM, Roddy TP, Stewart KM, Artyomov MN (2020) Comparative evaluation of itaconate and its derivatives reveals divergent inflammasome and type I interferon regulation in macrophages. Nat Metab 2(7):594–602. https://doi.org/10.1038/s42255-020-0210-0
Bambouskova M, Potuckova L, Paulenda T, Kerndl M, Mogilenko DA, Lizotte K, Swain A, Hayes S, Sheldon RD, Kim H, Kapadnis U, Ellis AE, Isaguirre C, Burdess S, Laha A, Amarasinghe GK, Chubukov V, Roddy TP, Diamond MS, Jones RG, Simons DM, Artyomov MN (2021) Itaconate confers tolerance to late NLRP3 inflammasome activation. Cell Rep 34(10):108756. https://doi.org/10.1016/j.celrep.2021.108756
Wu X, Yi X, Zhao B, Zhi Y, Xu Z, Cao Y, Cao X, Pang J, Yung KKL, Zhang S, Liu S, Zhou P (2023) The volume regulated anion channel VRAC regulates NLRP3 inflammasome by modulating itaconate efflux and mitochondria function. Pharmacol Res 198:107016. https://doi.org/10.1016/j.phrs.2023.107016
Motwani M, Pesiridis S, Fitzgerald KA (2019) DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet 20(11):657–674. https://doi.org/10.1038/s41576-019-0151-1
Ergun SL, Li L (2020) Structural insights into STING signaling. Trends Cell Biol 30(5):399–407. https://doi.org/10.1016/j.tcb.2020.01.010
Zhang X, Bai XC, Chen ZJ (2020) Structures and mechanisms in the cGAS-STING innate immunity pathway. Immunity 53(1):43–53. https://doi.org/10.1016/j.immuni.2020.05.013
Lahey LJ, Mardjuki RE, Wen X, Hess GT, Ritchie C, Carozza JA, Böhnert V, Maduke M, Bassik MC, Li L (2020) LRRC8A:C/E heteromeric channels are ubiquitous transporters of cGAMP. Mol Cell 80(4):578-591.e5. https://doi.org/10.1016/j.molcel.2020.10.021
Zhou C, Chen X, Planells-Cases R, Chu J, Wang L, Cao L, Li Z, López-Cayuqueo KI, Xie Y, Ye S, Wang X, Ullrich F, Ma S, Fang Y, Zhang X, Qian Z, Liang X, Cai SQ, Jiang Z, Zhou D, Leng Q, Xiao TS, Lan K, Yang J, Li H, Peng C, Qiu Z, Jentsch TJ, Xiao H (2020) Transfer of cGAMP into bystander cells via LRRC8 volume-regulated anion channels augments STING-mediated interferon responses and anti-viral immunity. Immunity 52(5):767-781.e6. https://doi.org/10.1016/j.immuni.2020.03.016
Chen X, Wang L, Cao L, Li T, Li Z, Sun Y, Ding J, Zhou C, Xie Y, Yue N, Nan J, Jia XM, Peng C, Li H, Yang J, Xiao H (2021) Regulation of anion channel LRRC8 volume-regulated anion channels in transport of 2′ 3′-cyclic GMP-AMP and cisplatin under steady state and inflammation. J Immunol 206(9):2061–2074. https://doi.org/10.4049/jimmunol.2000989
Concepcion AR, Wagner LE 2nd, Zhu J, Tao AY, Yang J, Khodadadi-Jamayran A, Wang YH, Liu M, Rose RE, Jones DR, Coetzee WA, Yule DI, Feske S (2022) The volume-regulated anion channel LRRC8C suppresses T cell function by regulating cyclic dinucleotide transport and STING-p53 signaling. Nat Immunol 23(2):287–302. https://doi.org/10.1038/s41590-021-01105-x
Chu J, Qiu Z (2022) An anion channel for cyclic dinucleotides in T cells. Nat Immunol 23(2):157–158. https://doi.org/10.1038/s41590-021-01118-6
Doroshenko P (1991) Second messengers mediating activation of chloride current by intracellular GTP gamma S in bovine chromaffin cells. J Physiol 436:725–738. https://doi.org/10.1113/jphysiol.1991.sp018576
Cannon CL, Basavappa S, Strange K (1998) Intracellular ionic strength regulates the volume sensitivity of a swelling-activated anion channel. Am J Physiol 275(2):C416–C422. https://doi.org/10.1152/ajpcell.1998.275.2.C416
Nilius B, Prenen J, Voets T, Eggermont J, Droogmans G (1998) Activation of volume-regulated chloride currents by reduction of intracellular ionic strength in bovine endothelial cells. J Physiol 506(Pt 2):353–361. https://doi.org/10.1111/j.1469-7793.1998.353bw.x
Voets T, Droogmans G, Raskin G, Eggermont J, Nilius B (1999) Reduced intracellular ionic strength as the initial trigger for activation of endothelial volume-regulated anion channels. Proc Natl Acad Sci USA 96(9):5298–5303. https://doi.org/10.1073/pnas.96.9.5298
Sabirov RZ, Prenen J, Tomita T, Droogmans G, Nilius B (2000) Reduction of ionic strength activates single volume-regulated anion channels (VRAC) in endothelial cells. Pflügers Arch 439(3):315–320. https://doi.org/10.1007/s004249900186
Syeda R, Qiu Z, Dubin AE, Murthy SE, Florendo MN, Mason DE, Mathur J, Cahalan SM, Peters EC, Montal M, Patapoutian A (2016) LRRC8 proteins form volume-regulated anion channels that sense ionic strength. Cell 164(3):499–511. https://doi.org/10.1016/j.cell.2015.12.031
Deneka D, Rutz S, Hutter CAJ, Seeger MA, Sawicka M, Dutzler R (2021) Allosteric modulation of LRRC8 channels by targeting their cytoplasmic domains. Nat Commun 12(1):5435. https://doi.org/10.1038/s41467-021-25742-w
Best L, Brown P (2009) Studies of the mechanism of activation of the volume-regulated anion channel in rat pancreatic beta-cells. J Membr Biol 230(2):83–91. https://doi.org/10.1007/s00232-009-9189-x
Hagiwara N, Masuda H, Shoda M, Irisawa H (1992) Stretch-activated anion currents of rabbit cardiac myocytes. J Physiol 456:285–302. https://doi.org/10.1113/jphysiol.1992.sp019337
Doroshenko P (1998) Pervanadate inhibits volume-sensitive chloride current in bovine chromaffin cells. Pflügers Arch 435(2):303–309. https://doi.org/10.1007/s004240050516
Zhang Y, Xie L, Gunasekar SK, Tong D, Mishra A, Gibson WJ, Wang C, Fidler T, Marthaler B, Klingelhutz A, Abel ED, Samuel I, Smith JK, Cao L, Sah R (2017) SWELL1 is a regulator of adipocyte size, insulin signalling and glucose homeostasis. Nat Cell Biol 19(5):504–517. https://doi.org/10.1038/ncb3514
Shimizu T, Numata T, Okada Y (2004) A role of reactive oxygen species in apoptotic activation of volume-sensitive Cl− channel. Proc Natl Acad Sci USA 101(17):6770–6773. https://doi.org/10.1073/pnas.0401604101
Varela D, Simon F, Riveros A, Jørgensen F, Stutzin A (2004) NAD(P)H oxidase-derived H2O2 signals chloride channel activation in cell volume regulation and cell proliferation. J Biol Chem 279(14):13301–13304. https://doi.org/10.1074/jbc.C400020200
Browe DM, Baumgarten CM (2004) Angiotensin II (AT1) receptors and NADPH oxidase regulate Cl− current elicited by beta1 integrin stretch in rabbit ventricular myocytes. J Gen Physiol 124(3):273–287. https://doi.org/10.1085/jgp.200409040
Wang X, Takahashi N, Uramoto H, Okada Y (2005) Chloride channel inhibition prevents ROS-dependent apoptosis induced by ischemia–reperfusion in mouse cardiomyocytes. Cell Physiol Biochem 16:147–154. https://doi.org/10.1159/000089840
Jiao J-D, Xu C-Q, Yue P, Dong D-L, Li Z, Du Z-M, Yang B-F (2006) Volume-sensitive outwardly rectifying chloride channels are involved in oxidative stress-induced apoptosis of mesangial cells. Biochem Biophys Res Commun 340(1):277–285. https://doi.org/10.1016/j.bbrc.2005.11.175
Varela D, Simon F, Olivero P, Armisén R, Leiva-Salcedo E, Jørgensen F, Sala F, Stutzin A (2007) Activation of H2O2-induced VSOR Cl− currents in HTC cells require phospholipase Cgamma1 phosphorylation and Ca2+ mobilization. Cell Physiol Biochem 20(6):773–780. https://doi.org/10.1159/000110437
Harrigan TJ, Abdullaev IF, Jourd’heuil D, Mongin AA (2008) Activation of microglia with zymosan promotes excitatory amino acid release via volume-regulated anion channels: the role of NADPH oxidases. J Neurochem 106(6):2449–24462. https://doi.org/10.1111/j.1471-4159.2008.05553.x
Deng W, Baki L, Baumgarten CM (2010) Endothelin signalling regulates volume-sensitive Cl− current via NADPH oxidase and mitochondrial reactive oxygen species. Cardiovasc Res 88(1):93–100. https://doi.org/10.1093/cvr/cvq125
Deng W, Baki L, Yin J, Zhou H, Baumgarten CM (2010) HIV protease inhibitors elicit volume-sensitive Cl− current in cardiac myocytes via mitochondrial ROS. J Mol Cell Cardiol 49(5):746–752. https://doi.org/10.1016/j.yjmcc.2010.08.013
l’Hoste S, Chargui A, Belfodil R, Corcelle E, Duranton C, Rubera I, Poujeol C, Mograbi B, Tauc M, Poujeol P (2009) CFTR mediates apoptotic volume decrease and cell death by controlling glutathione efflux and ROS production in cultured mice proximal tubules. Am J Physiol Renal Physiol 298(2):F435–F453. https://doi.org/10.1152/ajprenal.00286.2009
Matsuda JJ, Filali MS, Moreland JG, Miller FJ, Lamb FS (2010) Activation of swelling-activated chloride current by tumor necrosis factor-alpha requires ClC-3-dependent endosomal reactive oxygen production. J Biol Chem 285(30):22864–22873. https://doi.org/10.1074/jbc.M109.099838
Crutzen R, Shlyonsky V, Louchami K, Virreira M, Hupkens E, Boom A, Sener A, Malaisse WJ, Beauwens R (2012) Does NAD(P)H oxidase-derived H2O2 participate in hypotonicity-induced insulin release by activating VRAC in β-cells? Pflügers Arch 463(2):377–390. https://doi.org/10.1007/s00424-011-1047-x
Kumagai K, Imai S, Toyoda F, Okumura N, Isoya E, Matsuura H, Matsusue Y (2012) 17β-Oestradiol inhibits doxorubicin-induced apoptosis via block of the volume-sensitive Cl− current in rabbit articular chondrocytes. Br J Pharmacol 166(2):702–720. https://doi.org/10.1111/j.1476-5381.2011.01802.x
Holm JB, Grygorczyk R, Lambert IH (2013) Volume-sensitive release of organic osmolytes in the human lung epithelial cell line A549: role of the 5-lipoxygenase. Am J Physiol Cell Physiol 305(1):C48–C60. https://doi.org/10.1152/ajpcell.00412.2012
Shen M, Wang L, Wang B, Wang T, Yang G, Shen L, Wang T, Guo X, Liu Y, Xia Y, Jia L, Wang X (2014) Activation of volume-sensitive outwardly rectifying chloride channel by ROS contributes to ER stress and cardiac contractile dysfunction: involvement of CHOP through Wnt. Cell Death Dis 5(11):e1528. https://doi.org/10.1038/cddis.2014.479
Xia Y, Liu Y, Xia T, Li X, Huo C, Jia X, Wang L, Xu R, Wang N, Zhang M, Li H, Wang X (2016) Activation of volume-sensitive Cl− channel mediates autophagy-related cell death in myocardial ischaemia/reperfusion injury. Oncotarget 7(26):39345–39362. https://doi.org/10.18632/oncotarget.10050
Wang R, Lu Y, Gunasekar S, Zhang Y, Benson CJ, Chapleau MW, Sah R, Abboud FM (2017) The volume-regulated anion channel (LRRC8) in nodose neurons is sensitive to acidic pH. JCI Insight 2(5):e90632. https://doi.org/10.1172/jci.insight.90632
Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK (2002) Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circ Res 91(5):406–413. https://doi.org/10.1161/01.res.0000033523.08033.16
Fujii T, Shimizu T, Yamamoto S, Funayama K, Fujita K, Tabuchi Y, Ikari A, Takeshima H (1864) Sakai H (2018) Crosstalk between Na+, K+-ATPase and a volume-regulated anion channel in membrane microdomains of human cancer cells. Biochim Biophys Acta Mol Basis Dis 11:3792–3804. https://doi.org/10.1016/j.bbadis.2018.09.014
Carraway RE (1823) Dobner PR (2012) Zinc pyrithione induces ERK- and PKC-dependent necrosis distinct from TPEN-induced apoptosis in prostate cancer cells. Biochim Biophys Acta 2:544–557. https://doi.org/10.1016/j.bbamcr.2011.09.013
Mo J, Lin D, Wang J, Li P, Liu W (2018) Apoptosis in HepG2 cells induced by zinc pyrithione via mitochondrial dysfunction pathway: involvement of zinc accumulation and oxidative stress. Ecotoxicol Environ Saf 161:515–525. https://doi.org/10.1016/j.ecoenv.2018.06.026
Figueroa EE, Denton JS (2021) Zinc pyrithione activates the volume-regulated anion channel through an antioxidant-sensitive mechanism. Am J Physiol Cell Physiol 320(6):C1088–C1098. https://doi.org/10.1152/ajpcell.00070.2021
Choi H, Ettinger N, Rohrbough J, Dikalova A, Nguyen HN, Lamb FS (2016) LRRC8A channels support TNFα-induced superoxide production by Nox1 which is required for receptor endocytosis. Free Radic Biol Med 101:413–423. https://doi.org/10.1016/j.freeradbiomed.2016.11.003
Choi H, Rohrbough JC, Nguyen HN, Dikalova A, Lamb FS (2021) Oxidant-resistant LRRC8A/C anion channels support superoxide production by NADPH oxidase 1. J Physiol 599(12):3013–3036. https://doi.org/10.1113/JP281577
Browe DM, Baumgarten CM (2006) EGFR kinase regulates volume-sensitive chloride current elicited by integrin stretch via PI-3K and NADPH oxidase in ventricular myocytes. J Gen Physiol 127(3):237–251. https://doi.org/10.1085/jgp.200509366
Bedard K, Krause K-H (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87(1):245–313. https://doi.org/10.1152/physrev.00044.2005
Abramov AY, Jacobson J, Wientjes F, Hothersall J, Canevari L, Duchen MR (2005) Expression and modulation of an NADPH oxidase in mammalian astrocytes. J Neurosci 25(40):9176–9984. https://doi.org/10.1523/JNEUROSCI.1632-05.2005
Li G, Olson JE (2008) Purinergic activation of anion conductance and osmolyte efflux in cultured rat hippocampal neurons. Am J Physiol Cell Physiol 295(6):C1550–C1560. https://doi.org/10.1152/ajpcell.90605.2007
Gradogna A, Gavazzo P, Boccaccio A, Pusch M (2017) Subunit-dependent oxidative stress sensitivity of LRRC8 volume-regulated anion channels. J Physiol 595(21):6719–6733. https://doi.org/10.1113/JP274795
Bertelli S, Zuccolini P, Gavazzo P, Pusch M (2022) Molecular determinants underlying volume-regulated anion channel subunit-dependent oxidation sensitivity. J Physiol 600(17):3965–3982. https://doi.org/10.1113/JP283321
Okada T, Islam MR, Tsiferova NA, Okada Y, Sabirov RZ (2017) Specific and essential but not sufficient roles of LRRC8A in the activity of volume-sensitive outwardly rectifying anion channel (VSOR). Channels (Austin) 11:109–120. https://doi.org/10.1080/19336950.2016.1247133
Cai H, Griendling KK, Harrison DG (2003) The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci 24(9):471–478. https://doi.org/10.1016/S0165-6147(03)00233-5
Ahluwalia J (2008) Chloride channels activated by swell can regulate the NADPH oxidase generated membrane depolarisation in activated human neutrophils. Biochem Biophys Res Commun 365(2):328–333. https://doi.org/10.1016/j.bbrc.2007.10.176
Numata T, Shimizu T, Okada Y (2007) Direct mechano-stress sensitivity of TRPM7 channel. Cell Physiol Biochem 19(1–4):1–8. https://doi.org/10.1159/000099187
Numata T, Shimizu T, Okada Y (2007) TRPM7 is a stretch- and swelling-activated cation channel involved in volume regulation in human epithelial cells. Am J Physiol Cell Physiol 292(1):C460–C467. https://doi.org/10.1152/ajpcell.00367.2006
Liu Y, Zhang H, Men H, Du Y, Xiao Z, Zhang F, Huang D, Du X, Gamper N, Zhang H (2019) Volume-regulated Cl− current: contributions of distinct Cl− channels and localized Ca2+ signals. Am J Physiol Cell Physiol 317(3):C466–C480. https://doi.org/10.1152/ajpcell.00507.2018
König B, Hao Y, Schwartz S, Plested AJ, Stauber T (2019) A FRET sensor of C-terminal movement reveals VRAC activation by plasma membrane DAG signaling rather than ionic strength. Elife 8:e45421. https://doi.org/10.7554/eLife.45421
Valverde MA, Díaz M, Sepúlveda FV, Gill DR, Hyde SC, Higgins CF (1992) Volume-regulated chloride channels associated with the human multidrug-resistance P-glycoprotein. Nature 355(6363):830–833. https://doi.org/10.1038/355830a0
Gill DR, Hyde SC, Higgins CF, Valverde MA, Mintenig GM, Sepúlveda FV (1992) Separation of drug transport and chloride channel functions of the human multidrug resistance P-glycoprotein. Cell 71(1):23–32. https://doi.org/10.1016/0092-8674(92)90263-c
Tominaga M, Tominaga T, Miwa A, Okada Y (1995) Volume-sensitive chloride channel activity does not depend on endogenous P-glycoprotein. J Biol Chem 270:27887–27893. https://doi.org/10.1074/jbc.270.46.27887
Miwa A, Ueda K, Okada Y (1997) Protein kinase C-independent correlation between P-glycoprotein expression and volume sensitivity of Cl− channel. J Membrane Biol 157:63–69. https://doi.org/10.1007/s002329900216
Vennekens R, Trouet D, Vankeerberghen A, Voets T, Cuppens H, Eggermont J, Cassiman JJ, Droogmans G, Nilius B (1999) Inhibition of volume-regulated anion channels by expression of the cystic fibrosis transmembrane conductance regulator. J Physiol 515(Pt 1):75–85. https://doi.org/10.1111/j.1469-7793.1999.075ad.x
Ando-Akatsuka Y, Abdullaev IF, Lee EL, Okada Y, Sabirov RZ (2002) Down-regulation of volume-sensitive Cl− channels by CFTR is mediated by the second nucleotide-binding domain. Pflügers Arch Eur J Physiol 445:177–186. https://doi.org/10.1007/s00424-002-0920-z
Ando-Akatsuka Y, Shimizu T, Numata T, Okada Y (2012) Involvements of the ABC protein ABCF2 and α-actinin-4 in regulation of cell volume and anion channels in human epithelial cells. J Cell Physiol 227:3498–3510. https://doi.org/10.1002/jcp.24050
Levitan I, Christian AE, Tulenko TN, Rothblat GH (2000) Membrane cholesterol content modulates activation of volume-regulated anion current in bovine endothelial cells. J Gen Physiol 115(4):405–416. https://doi.org/10.1085/jgp.115.4.405
Romanenko VG, Rothblat GH, Levitan I (2004) Sensitivity of volume-regulated anion current to cholesterol structural analogues. J Gen Physiol 123(1):77–87. https://doi.org/10.1085/jgp.200308882
Byfield FJ, Hoffman BD, Romanenko VG, Fang Y, Crocker JC, Levitan I (2006) Evidence for the role of cell stiffness in modulation of volume-regulated anion channels. Acta Physiol (Oxf) 187(1–2):285–294. https://doi.org/10.1111/j.1748-1716.2006.01555.x
Klausen TK, Hougaard C, Hoffmann EK, Pedersen SF (2006) Cholesterol modulates the volume-regulated anion current in Ehrlich-Lettre ascites cells via effects on Rho and F-actin. Am J Physiol Cell Physiol 291(4):C757–C771. https://doi.org/10.1152/ajpcell.00029.2006
Shimizu T, Fujii T, Ohtake H, Tomii T, Takahashi R, Kawashima K, Sakai H (2020) Impaired actin filaments decrease cisplatin sensitivity via dysfunction of volume-sensitive Cl− channels in human epidermoid carcinoma cells. J Cell Physiol 235(12):9589–9600. https://doi.org/10.1002/jcp.29767
Harder T, Kellner R, Parton RG, Gruenberg J (1997) Specific release of membrane-bound annexin II and cortical cytoskeletal elements by sequestration of membrane cholesterol. Mol Biol Cell 8(3):533–545. https://doi.org/10.1091/mbc.8.3.533
Sjöblom B, Salmazo A, Djinović-Carugo K (2008) Alpha-actinin structure and regulation. Cell Mol Life Sci 65(17):2688–2701. https://doi.org/10.1007/s00018-008-8080-8
Honda K, Yamada T, Endo R, Ino Y, Gotoh M, Tsuda H, Yamada Y, Chiba H, Hirohashi S (1998) Actinin-4, a novel actin-bundling protein associated with cell motility and cancer invasion. J Cell Biol 140(6):1383–1393. https://doi.org/10.1083/jcb.140.6.1383
Jayadev R, Kuk CY, Low SH, Murata-Hori M (2012) Calcium sensitivity of α-actinin is required for equatorial actin assembly during cytokinesis. Cell Cycle 11(10):1929–1937. https://doi.org/10.4161/cc.20277
Platt CD, Chou J, Houlihan P, Badran YR, Kumar L, Bainter W, Poliani PL, Perez CJ, Dent SYR, Clapham DE, Benavides F, Geha RS (2017) Leucine-rich repeat containing 8A (LRRC8A)-dependent volume-regulated anion channel activity is dispensable for T-cell development and function. J Allergy Clin Immunol 140(6):1651-1659.e1. https://doi.org/10.1016/j.jaci.2016.12.974
Lambert IH (2003) Reactive oxygen species regulate swelling-induced taurine efflux in NIH3T3 mouse fibroblasts. J Membr Biol 192(1):19–32. https://doi.org/10.1007/s00232-002-1061-1
Ørtenblad N, Young JF, Oksbjerg N, Nielsen JH, Lambert IH (2003) Reactive oxygen species are important mediators of taurine release from skeletal muscle cells. Am J Physiol Cell Physiol 284(6):C1362–C1273. https://doi.org/10.1152/ajpcell.00287.2002
Schliess F, Foster N, Görg B, Reinehr R, Häussinger D (2004) Hypoosmotic swelling increases protein tyrosine nitration in cultured rat astrocytes. Glia 47(1):21–29. https://doi.org/10.1002/glia.20019
Shimizu T, Morishima S, Okada Y (2000) Ca2+-sensing receptor-mediated regulation of volume-sensitive Cl− channels in human epithelial cells. J Physiol (London) 528:457–472. https://doi.org/10.1111/j.1469-7793.2000.00457.x
Sato K, Numata T, Saito T, Ueta Y, Okada Y (2011) V2 receptor-mediated autocrine role of somatodendritic release of AVP in rat vasopressin neurons under hypo-osmotic conditions. Sci Signal 4(157):5. https://doi.org/10.1126/scisignal.2001279
Abdullaev IF, Sabirov RZ, Okada Y (2003) Upregulation of swelling-activated Cl− channel sensitivity to cell volume by activation of EGF receptors in murine mammary cells. J Physiol (London) 549:749–758. https://doi.org/10.1152/ajprenal.00313.2002
Poole CP, Owens FJ (2003) Introduction to nanotechnology. Wiley, Hoboken
Sabirov RZ, Okada Y (2004) Wide nanoscopic pore of maxi-anion channel suits its function as an ATP-conductive pathway. Biophys J 87(3):1672–1685. https://doi.org/10.1529/biophysj.104.043174
Okada Y, Sabirov RZ, Sato-Numata K, Numata T (2021) Cell death induction and protection by activation of ubiquitously expressed anion/cation channels. Part 1: roles of VSOR/VRAC in cell volume regulation, release of double-edged signals and apoptotic/necrotic cell death. Front Cell Develop Biol. 8:614040. https://doi.org/10.3389/fcell.2020.614040
Acknowledgements
The author thanks R.Z. Sabirov, T. Numata, K. Sato-Numata, Y. Ando-Akatsuka, and M. Kashio for their discussion and suggestions for this manuscript. Thanks are also due to all the previous and present coworkers for their nice collaboration, technical assistance, and secretarial help.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
YO conceptualized the study; prepared an original draft; reviewed and edited the manuscript; and read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This article does not contain any studies with human participants or animals performed by any authors.
Consent for publication
Consent for publication is not required for this article.
Competing interests
The author declares no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Okada, Y. Physiology of the volume-sensitive/regulatory anion channel VSOR/VRAC: part 2: its activation mechanisms and essential roles in organic signal release. J Physiol Sci 74, 34 (2024). https://doi.org/10.1186/s12576-024-00926-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12576-024-00926-3