
Abstract
The volume-sensitive outwardly rectifying or volume-regulated anion channel, VSOR/VRAC, which was discovered in 1988, is expressed in most vertebrate cell types and is essentially involved in cell volume regulation after swelling and in the induction of cell death. This series of review articles describes what is already known and what remains to be uncovered about the functional and molecular properties as well as the physiological and pathophysiological roles of VSOR/VRAC. This Part 1 review article describes, from the physiological standpoint, first its discovery and significance in cell volume regulation, second its phenotypical properties, and third its molecular identification. Although the pore-forming core molecules and the volume-sensing subcomponent of VSOR/VRAC were identified as LRRC8 members and TRPM7 in 2014 and 2021, respectively, it is stressed that the identification of the molecular entity of VSOR/VRAC is still not complete enough to explain the full set of phenotypical properties.
Similar content being viewed by others
Background
In animal cells, on which the rigid cell wall is lacking, cell volume regulation is essential for their survival and functions. Even under extracellular hypoosmotic or intracellular hyperosmotic conditions, the cells exhibit the regulatory volume decrease (RVD) shortly after osmotic swelling. In 1988, the RVD process was, for the first time, demonstrated to be attained by water efflux driven by KCl efflux due to parallel activation of K+ channels and Cl− channels by Hazama & Okada [1] and independently by Cahalan & Lewis [2]. Although the volume-regulatory K+ channel in epithelial cells was shown to be directly activated by an increase in the intracellular Ca2+ concentration and is thus classified into a well-known Ca2+-activated K+ channel, the Cl− channel was demonstrated to be not directly activated by cytosolic Ca2+ rise, unlike the Ca2+-activated Cl− channel (CaCC), and to be a new type of anion channel [1]. This swelling-activated anion channel is expressed in most vertebrate cell types studied to date and is nowadays called the volume-sensitive outwardly rectifying anion channel (VSOR: [3]) or the volume-regulated/regulatory anion channel (VRAC: [4]). The volume-sensitive/regulatory outwardly rectifying anion channel, here called VSOR/VRAC, was then discovered to play an essential role also in the induction of apoptotic cell death [5, 6] and cisplatin resistance in cancer cells [7]. Thereafter, in 2014, LRRC8A was identified as the prerequisite molecule of VSOR/VRAC [8, 9], and the heteromer of LRRC8A and the other member(s) of LRRC8 (LRRC8C/D/E) was shown to exert as the core component of VSOR/VRAC channels [9]. Furthermore, the volume expansion sensitivity of VSOR/VRAC was recently shown to be granted by mechano-sensitive Ca2+-permeable cation channel TRPM7 which serves as the essential subcomponent of VSOR/VRAC channels by physically interacting with LRRC8A [10]. However, from the standpoint of the phenotypical properties of VSOR/VRAC, there is still a missing subcomponent for the molecular entity of VSOR/VRAC [11]. As the 35th anniversary of the VSOR/VRAC discovery, this review article describes the roles in cell volume regulation, the phenotypical properties, and the molecular entities, including its pore-forming core component and volume-sensing subcomponent, of VSOR/VRAC with evaluating what is already elucidated and what remains unknown mainly from the physiological standpoint.
Introduction: cell volume regulation and discovery of VSOR/VRAC
All animal cells have appropriate cell volume, and cell volume regulation is essential for their survival (see Reviews: [12,13,14]; also see Books: [15, 16]).
Cell volume perturbation
Although body fluid osmolarity is kept constant (within 1–2%) by the thirst-antidiuretic hormone mechanisms, significant changes in local osmolarity are, even under physiological conditions, elicited around cells (see Review: [11]). Under pathological conditions, sizable changes in the plasma osmolarity are produced in association with a variety of diseases (see Table 1 in [17]). Upon such osmotic perturbation, the volume of animal cells is forced to be altered (Fig. 1) for the following reasons: First, the cell membrane exhibits high water permeability (up to 100 μm/s) and thus behaves as a semipermeable membrane. Second, animal cells, unlike plant cells, do not have a rigid cell wall. Thus, as illustrated in Fig. 1, the cells are rendered to rapidly exhibit osmotic shrinkage or an osmotic volume decrease (OVD) and swelling or an osmotic volume increase (OVI) in response to a hypertonic challenge induced by extracellular hypertonicity or intracellular hypotonicity and a hypotonic challenge by extracellular hypotonicity or intracellular hypertonicity, respectively [17]. In addition, in response to stimulation with secretagogues, secretory cells exhibit shrinkage, called the secretory volume decrease (SVD) [20]. Upon cooling and freezing, cells also show shrinkage, called the cooling and freezing volume decrease (CVD and FVD) [21]. A variety of hormones are known to induce cell volume changes by activating electroneutral ion cotransporters (antiporters and symporters) or by stimulating intracellular metabolism (see Table 2 in [18]). Thus, we term volume changes produced by such hormonal stimulation as the hormonal volume decrease (HVD) and increase (HVI). The most well-known HVD- and HVI-inducing hormones are insulin and glucagon, respectively, in hepatocytes [19]. Although HVD/HVI/SVD and CVD/FVD are both to be, in the broad sense, classified into osmotic volume changes, the former is caused by primary changes in the osmolyte concentrations whereas the latter is by primary changes in the activity of water.

Cell volume changes (shrinkage and swelling) caused by osmotic perturbation and cell volume regulation thereafter. Osmotic volume decrease (OVD), secretory volume decrease (SVD), and cooling/freezing volume decrease (CVD/FVD) are shortly followed by the regulatory volume increase (RVI), whereas osmotic volume increase (OVI) is by the regulatory volume decrease (RVD). Hormonal volume decrease (HVD) and increase (HVI) are given in parenthesis, because it is not known whether they are followed by RVI and RVD, respectively. The key players for RVI and RVD are cationic HICC channels and anionic VSOR/VRAC channels, respectively. (See text for details.)
Cell volume regulation and swelling-activated anion channels
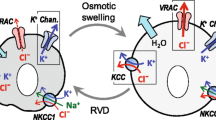
Shortly after cell shrinkage (OVD/SVD/CVD/FVD) and swelling (OVI) forced by non-steady state osmotic perturbation, animal cells can regulate their volume by the mechanisms called the regulatory volume increase and decrease (RVI and RVD) due to water flux mainly driven by uptake of NaCl and release of KCl, respectively [5,6,7] (Fig. 1). However, it is not known whether HVD and HVI are followed by regulatory volume responses. It is noteworthy that arginine vasopressin (AVP) neurons were recently demonstrated to respond to hyperosmotic stimulation not only with osmotic shrinkage (OVD) followed by RVI but also with persistent cell shrinkage (SVD) due to massive AVP release in an additive manner by Sato-Numata et al. [22]. These volume-regulatory NaCl and KCl transports had long been considered to be attained by the operation of electroneutral ion coupled transporters such as the Na+-K+-Cl− symporter or cotransporter (NKCC), Na+-Cl− symporter (NCC), or parallel operation of Na+/H+ antiporter or exchanger (NHE) plus Cl−/HCO3– antiporter (anion exchanger: AE) and K+-Cl− symporter (KCC) or parallel operation of K+/H+ antiporter (KHE) plus AE, respectively (see Reviews: [23, 24]. However, it is now established that these volume-regulatory fluxes of Na+, K+, Cl−, and small negatively-charged organic solutes are predominantly due to the activation of a number of channel-mediated conductive pathways in most cell types. Among them, the main actor for RVI is a shrinkage-activated Na+-permeable cation channel which was, for the first time, observed in 1989 [25] and is nowadays called the hypertonicity-induced cation channel (HICC: [26]; also see Review [27]) (Fig. 1). Meanwhile, the key player for RVD is a swelling-activated anion channel which was originally observed in 1988 by Hazama & Okada in human epithelial Intestine 407 cells [1, 28] and independently by Cahalan & Lewis in human lymphoid cells [2], and is nowadays called the volume-sensitive outwardly rectifying anion channel (VSOR: [3]) or the volume-regulated anion channel (VRAC: [4]) (Fig. 1). These two groups also, for the first time, independently demonstrated that parallel activation of VSOR/VRAC Cl− channels and K+ channels is responsible for the attainment of RVD [1, 2].
In the present Part 1 article, I review the properties and molecular entities of VSOR/VRAC channels from the physiological standpoint. The activation mechanisms and the roles in the transport of organic signal molecules, cell death induction, and anticancer drug resistance of VSOR/VRAC are to be reviewed in the subsequent Part 2 article.
Phenotypical properties of VSOR/VRAC
Since the discovery of VSOR/VRAC, its biophysical and physiological properties have been extensively investigated and well summarized in review articles in the early period [3, 4, 29] and in the last few years [30,31,32,33,34,35] with including information accumulated on the basis of the recent identification of LRRC8 as the core molecule of VSOR/VRAC [8, 9]. The pharmacological properties of VSOR/VRAC have also been extensively studied, as summarized in review articles [3, 4, 32, 34, 36, 37]. Here, these properties are updated by taking the latest studies into consideration.
The physiological and biophysical properties
The phenotypical or physiological and biophysical properties of VSOR/VRAC, which are distinct from other types of anion channels [34], are summarized in Table 1 (A and B, respectively) as the starting or basic point for ongoing and future studies.
Cell volume sensitivity
Cell swelling-induced activation is the most important property of VSOR/VRAC, and this is not directly elicited by membrane stretch, because activation of VSOR/VRAC per se was never elicited by the suction of cell-attached patch membranes [3, 38,39,40]. Under physiological and quasi-physiological conditions, moderate cell swelling (volume expansion) may take place without producing drastic plasma membrane stretch because the cell membrane has, in general, sufficient reserves due to the existence of its infoldings [41, 42]. In fact, the cell surface area estimated from the whole-cell membrane capacitance of human epithelial Intestine 407 cells was more than four-fold larger than that measured by image analysis [43]. Since unitary VSOR/VRAC events could be recorded only when the patch pipette had been attached to the cell after, but never before, the cell was rendered swelling [3, 44], membrane expansion mainly due to unfolding of the invagination is likely to be required for VSOR/VRAC activation. As a matter of fact, human epidermoid KB cells exhibited a nearly linear relationship between the VSOR/VRAC current density and the relative surface area in a certain range above the threshold [45]. Also, it is noteworthy that VSOR/VRAC currents can be activated by cell inflation (swelling) induced by the injection of iso-osmotic solution with identical ionic strength into the cells under whole-cell configuration [46,47,48].
Cytosolic ATP dependence and Mg2+ sensitivity
The presence of intracellular ATP is a prerequisite to VSOR/VRAC activity. This fact was, for the first time, reported by two groups in mouse T lymphocytes [49] and in human epithelial HeLa cells [50]. Independently, such intracellular ATP dependence of VSOR/VRAC was subsequently reported by three groups in rat glioma C6 cells [51], human umbilical endothelial cells [52], and human Intestine 407 cells [53]. Our recent quantitative studies in Intestine 407 cells showed that the half-maximal effective concentration (EC50) for ATP-induced VSOR/VRAC activation was 14 μM in the presence of 1 mM free Mg2+ (Fig. 3 in [34]). Moreover, it was found that VSOR/VRAC activity requires ATP but not its hydrolysis because such an ATP role can be substituted by non-hydrolysable ATP analogs [50,51,52,53,54]. This fact suggests an essential role of some ATP-binding protein(s) in the mechanism of VSOR/VRAC activation. On the other hand, intracellular free Mg2+ was found to suppress VSOR/VRAC activity [53, 55]. The half-maximal inhibitory concentration (IC50) for free Mg2+ was 0.32 and 1.9 mM in the presence of 0.01 and 0.1 mM free ATP, respectively [34]. Such free ATP requirement and free Mg2+ sensitivity suggest that free Mg2+ inhibits VSOR/VRAC channels by forming the Mg-ATP complex.
Intermediate outwardly rectifying unitary conductance
Single-channel recordings of VSOR/VRAC-like intermediate unitary events were, for the first time, observed in human airway epithelial cells [56] and colonic epithelial T84 cells [57]. Subsequently, we provided firm evidence that such intermediate unitary events, in fact, represent VSOR/VRAC activity by performing the double patch-clamp recording, which can simultaneously observe single-channel currents and whole-cell currents, in swollen human Intestine 407 cells [38, 58] and in mouse cortical neurons [59]. Thereafter, intermediate-conductance VSOR/VRAC unitary activities were also observed in a variety of cell types including mouse mammary gland C127 cells [60], mouse thymocytes [61], and mouse cardiomyocytes in primary culture [62].
Three types of voltage dependence
Moderate (but not strong) outward rectification and inactivation kinetics of macroscopic whole-cell VSOR/VRAC currents are distinct from those of other types of anion channels, such as voltage-gated inwardly rectifying ClC-2, large-conductance ohmic (non-rectifying) Maxi-Cl, cAMP-activated ohmic CFTR, acid-sensitive strongly outwardly rectifying anion channel (ASOR) or proton-activated anion channel (PAC), and sharp outward-rectifier Ca2+-activated CaCC [34]. Our double-patch clamp recordings demonstrated that outward rectification observed even under symmetrical Cl− conditions and time-dependent inactivation of outward currents observed only at large positive potentials on the macroscopic whole-cell level are caused by voltage-dependent changes in the single-channel conductance and voltage-dependent stepwise unitary closing events, respectively [38, 59, 63, 64]. Such two types of voltage dependence have been observed by many laboratories in a wide variety of cell types, including human epithelial Intestine 407 [38, 58, 65,66,67,68], T84 [68], HEK293 [69,70,71], and HeLa [6, 72] cells, human epidermal KB and its derivative cells [7, 45, 73], mouse mammary epithelial C127 [60, 74, 75], mouse cortical neurons [59, 63, 76], rat vasopressin (AVP) neurons [77], mouse astrocytes [78,79,80,81], mouse cardiomyocytes [82], mouse adipocytes [83], rat thymocytes [84], and chicken B lymphocyte DT-40 cells [10] so far studied in our own and collaborative laboratories. The third type of voltage dependence of VSOR/VRAC currents is the open-channel blocking by extracellular ATP upon depolarization [85,86,87,88,89,90].
Low-field anion selectivity
The halide anion selectivity sequence (Table 2) is determined by the product of the ratio of equilibrium constant for binding to the positively-charged binding site and the ratio of mobility between two anion species, and it is classified into the high-field strength one dominated by the former ratio and the low-field strength one dominated by the latter ratio, as described in detail by Wright and Diamond [91]. The low-field-strength halide anion selectivity with Eisenman sequence type I (I− > Br− > Cl− > F−) of VSOR/VRAC is distinct from the higher-field anion selectivity of CFTR (Eisenman type III: Br− > Cl− > I− > F−) and ClC-2 (Eisenman type IV: Cl− > Br− > I− > F−) [34]. In our laboratory, this type of anion selectivity was confirmed for VSOR/VRAC currents in human Intestine 407 [66] and KB [73] cells as well as mouse cortical neurons [59] and rat AVP neurons [77].
Severe hypotonicity-activated anion currents
There were several studies suggesting intracellular ATP-independent activation of VSOR/VRAC induced by unphysiologically severe hypotonic stimulation. Jirsch et al. [92] showed that intracellular ATP removal abolished swelling-activated Cl− currents (ICl,swell) caused by exposure to 72% hypotonic solution but not those induced by a 55% hypotonic challenge in human H69AR cancer cells. However, it must be noted that the employed condition was just a nominally ATP-free one (without precluding contamination of residual endogenous ATP), and moreover that these currents were also evoked by suction of the cell-attached patch pipette; that is, mechano-sensitive. Next, Volk et al. [93] and Bond et al. [94] reported that intracellular ATP depletion by using metabolic inhibitors abolished ICl,swell activation induced by a modest (83–86%) hypotonic challenge but failed to abolish ICl,swell activation induced by a severe (50–73%) hypotonic challenge in rat kidney IMCD cells and mouse neuroblastoma N1E115 cells, respectively. However, it is not known whether severe hypotonicity-activated, intracellular ATP-independent ICl,swell exhibited the full set of phenotypes of VSOR/VRAC properties (Table 1) other than intracellular ATP dependence.
Pharmacological properties
None of the VSOR/VRAC blockers are unfortunately selective, as summarized in recent review articles [32, 34, 36, 37]. Most of the VSOR/VRAC blockers can be classified into three groups (Table 3): (1) Conventional type of Cl− channel blockers with two aromatic rings connected with a chain of one to four atoms (see Fig. 9B in [34]); (2) ethacrynic acid derivatives possessing the 2,3-dichloro phenoxyl fragment (see Fig. 9A in [34]); and (3) open-channel blockers.
Conventional VSOR/VRAC blockers
The first group includes a large variety of conventional Cl− channel blockers such as phloretin, SITS, DIDS, NPPB, DPC, niflumic acid, flufenamic acid, NS3728, and glibenclamide. The sensitivity of VSOR/VRAC to the stilbene-derivatives, SITS and DIDS, was systematically demonstrated by Kubo & Okada [65] in human Intestine 407 cells and by Nilius et al. [95] in human umbilical endothelial cells. Blocking efficacy at positive voltages was more prominent than that at negative voltages. Such voltage-dependent VSOR/VRAC sensitivity to SITS/DIDS was confirmed in a wide variety of cell types including mouse cortical neurons [59], human KB and its derivative cells [7, 73], mouse astrocytes [79], and mouse cardiomyocytes [62, 82]. A sulfonylurea, glibenclamide, also exhibited a similar voltage-dependent blocking action to VSOR/VRAC currents in human Intestine 407 cells [96], mouse C127 cells [74], and mouse adipocytes [83]. In contrast, VSOR/VRAC currents were found to be inhibited, in a voltage-independent manner, by carboxylate analogs such as NPPB, 9-AC, and DPC as well as DPC analogs, niflumic acid and flufenamic acid, in a large variety of cell types (see Review [3]) including human Intestine 407 cells [65], HeLa cells [72], KB and its derivative cells [7], mouse cortical neurons [59, 76], rat AVP neurons [77], and mouse adipocytes [83]. A bisphenol, phloretin, was also found to voltage-independently block VSOR/VRAC activity in human colonic epithelial T84 and Intestine 407 cells as well as in mouse C127 cells [68]. Later, similar phloretin sensitivity of VSOR/VRAC was observed in mouse cortical neurons [59, 76], human KB and its derivative cells [7, 73], rat AVP neurons [77], mouse astrocytes [78, 79], and rat thymocytes [84]. Recently, a number of flavonoids, which also have two connected aromatic rings, were reported to inhibit VSOR/VRAC currents [97, 98]. In addition, novel two VSOR/VRAC blockers, pranlukast and zafirlukast, were discovered by a high-throughput screening combined with a YFP quenching assay [99]. These cysteinyl leukotriene 1 (CysLT1) receptor antagonists voltage-independently blocked VSOR/VRAC currents at micromolar concentrations in a manner independent of CysLT1. It is noted that both pranlukast and zafirlukast can also be classified into the two aromatic ring-connected conventional Cl− channel blockers (RZ Sabirov: personal communication). However, the selectivity of pranlukast and zafirlukast to VSOR/VRAC against other ion channels is not known as yet.
Ethacrynic acid-derivative VSOR/VRAC blockers
The second ethacrynic acid-derivative group includes DCPIB, IAA-94, and DIOA. DCPIB was originally found to effectively block VSOR/VRAC currents in a voltage-independent manner in calf endothelial cells [100]. Since then, this indanone compound has been widely employed as a most selective blocker for VSOR/VRAC in many cell types including human epithelial HEK293 [70] and HeLa [72] cells, human epidermoid KB cells [7], rat AVP neurons [77], mouse astrocytes [79,80,81], rat thymocytes [84], and chicken B-lymphocyte DT40 cells [10]. In contrast, DCPIB was shown to be ineffective in blocking CaCC, CFTR, and ClC-1/3/4/5/K1 Cl− channel activities [100] as well as Maxi-Cl [101, 102] and ASOR/PAC [72] anion channel activities. However, it must be noted that even DCPIB exhibits off-target effects on a variety of other ion channels and transporters, as summarized in review articles [34, 37, 99, 103]. In addition, one also needs to bear the possibility in mind that DCPIB may suppress, to a certain extent, some molecularly unidentified Ca2+-activated Cl− currents [104] and acid-activated Cl− currents [105]. IAA-94 was shown to inhibit VSOR/VRAC currents in mouse cortical neurons [76] and rat pyramidal neurons [106]. DIOA was also found to be an effective VSOR/VRAC blocker in microglial BV-2 cells [107] and epithelial HeLa cells [72]. However, DIOA was observed to block not only VSOR/VRAC currents but also ASOR/PAC currents in HeLa cells [72].
Open-channel VSOR/VRAC blockers
The third group is the open-channel blockers that exhibit depolarization-induced suppression of VSOR/VRAC currents only from the extracellular side. As noted above, extracellular free ATP4− was shown to exhibit open-channel blocking actions to VSOR/VRAC currents in a variety of cell types [85,86,87,88,89,90]. Similar open-channel blocking effects were also observed in some of the above conventional Cl− channel blockers. For example, SITS/DIDS and glibenclamide were found to exhibit voltage-dependent blocking action on VSOR/VRAC currents originally by Kubo & Okada [65] and Liu et al. [96], respectively. Suramin6− was also shown to be an open-channel blocker of VSOR/VRAC [64, 72, 90, 108,109,110,111]. The open-channel blocking actions are very interesting from the viewpoint of the pore size estimation. In this regard, the VSOR/VRAC channel should have an outer vestibule larger and a pore smaller than the sizes of ATP, SITS, glibenclamide, and suramin the unhydrated radii of which are approximately 0.58, 0.55, 0.60, and 0.91 nm (see Fig. 15.8 in [64]), respectively. In fact, the effective pore radius was estimated to be 0.57–0.71 nm through the experiments with permeant blockers [108, 112] and around 0.63 nm by the non-electrolyte partitioning method [67].
Other types of VSOR/VRAC blockers
VSOR/VRAC activity was also sensitive to a large variety of other structurally unrelated chemicals (see Review [34]) including one of essential fatty acid, arachidonic acid [65], and a tyrosine kinase inhibitor, genistein [113]. Very recently, dicumarol, which is a known competitive inhibitor of vitamin K epoxide reductase, was shown to be a novel potent VSOR/VRAC inhibitor selective against ASOR and CaCC [114], However, possible off-target effects on other ion channels remain to be examined.
Molecular entities of VSOR/VRAC
Although the phenotypical properties of VSOR/VRAC were well elucidated soon after the functional discovery of VSOR/VRAC in 1988 [1, 2], it took over a quarter of a century to identify LRRC8A, also called SWELL1, as the core component of VSOR/VRAC molecule(s) [8, 9]. In the meantime, there were long-time struggles against many false-positive candidate molecules, as summarized in recent review articles [11, 31, 32]. Among them, the biggest-hyped ones are P-glycoprotein (PGP) proposed in 1992 [115], pICln in 1992 [116], ClC-3 in 1997 [117], and TMEM16F or anoctamin 6 (ANO6) in 2011 [118]. These candidates were, almost one after another, disproved by successive works performed in many laboratories (see [11] in detail), including our works [45, 62, 66, 70, 71, 82, 87, 119, 120]. Such a long time delay for molecular identification of VSOR/VRAC may be caused by the following four factors [31, 33]. First, ubiquitous expression of VSOR/VRAC activity in vertebrate cells precluded the expression cloning; second, all the available chemical inhibitors and activating ligands are not specific enough to perform the affinity purification; third, there exists no sequence homology among known chloride channel families; and fourth, the heterologous expression assay for a single candidate molecule must have been misleading, because the VSOR/VRAC channel turned out to be formed by multiple types, but not single type, of molecules, as described below.
LRRC8 members as the core molecules
Using an unbiased loss-of-function genome-wide RNAi screening method, LRRC8A was demonstrated to be a core molecule for VSOR/VRAC activity in human cells [8, 9] in 2014. Subsequently, a prerequisite role of LRRC8A in VSOR/VRAC activity was confirmed in mouse cells [40, 75, 121, 122], in rat cells [123, 124], in chicken cells [10], and in zebrafish cells [125]. Recent cryo-EM studies demonstrated that not only LRRC8A [126,127,128,129,130] but also another LRRC8 member, LRRC8D [131], form homo-hexameric structures consisting of the transmembrane pore domain, as depicted in Fig. 2 for LRRC8A. However, when overexpressed in cells deficient in all LRRC8 members, LRRC8A homomeric channels exhibited properties different from native VSOR/VRAC channels [132]; namely, insensitivity to osmotic cell swelling and only weak and voltage-dependent sensitivity to DCPIB, which is a known voltage-independent VSOR/VRAC channel blocker [100] and was shown to be most selective to other identified types of anion channel [34].

LRRC8A plus LRRC8C/D/E as the necessary components for VSOR/VRAC channels
The LRRC8 family is composed of five members of A to E with a molecular mass of around 95 kD consisting of around 800 amino acids. Since, collective gene disruption of LRRC8B, 8C, 8D, and 8E was found to abolish VSOR/VRAC activity [9, 133, 134], it is suggested that co-expression of LRRC8A with, at least, one of the other members is required for VSOR/VRAC activity. In fact, co-transfection of LRRC8A and LRRC8C, 8D, or 8E was found to rescue VSOR/VRAC activity in LRRC8−/− HCT116 cells in which all five LRRC8 genes are disrupted [9]. Furthermore, co-immunoprecipitation studies provided evidence for the direct interaction between LRRC8A and LRRC8B/C/D/E in HEK293 cells [9] as well as between LRRC8D and LRRC8A/B/C in HEK293T cells, among LRRC8A, 8C, and 8D with each other in RAW264.7 macrophages, and among LRRC8A, 8B, and 8D with each other in KBM7 cells [135]. In addition, the trafficking of LRRC8B/C/D/E to the plasma membrane was detected when transfected with LRRC8A but not when transfected alone in HEK293 cells [9]. However, it appears that LRRC8B is not involved in the VSOR/VRAC formation in light of the following observations. First, when LRRC8B was co-transfected with LRRC8A, hypotonicity-induced VSOR/VRAC activity was never restored in LRRC8−/− HCT116 cells [9]. Second, using LRRC8 members C-terminally tagged with fluorescent proteins, co-transfection of LRRC8A and LRRC8B failed to induce hypotonicity-induced VSOR/VRAC activity, whereas co-transfection of LRRC8A and LRRC8C/D/E produced the currents in enzymatically defolliculated Xenopus oocytes [136] in which endogenous VSOR/VRAC activity is lacking [137, 138]. Third, endogenous hypotonicity-induced VSOR/VRAC activity was abolished by triple knockdown of LRRC8C, 8D, and 8E in HeLa cells by Sato-Numata et al. [134] and by triple disruption of LRRC8C-8E genes in HEK293 cells by Lutter et al. [139] in 2017.
Most recent cryo-EM studies showed that LRRC8A and LRRC8C, in fact, assemble as a hetero-hexameric structure forming a pore domain [140, 141]. The number of hetero-hexameric complexes of LRRC8A and LRRC8C/D/E was estimated to be on the order of 10,000 by quantitative immunoblot studies [142], which is in good agreement with the functional VSOR/VRAC number estimated by the electrophysiological approach [49]. Mammalian cells may express different VSOR/VRAC channels simultaneously with different combinations of LRRC8A and LRRC8C/D/E. Previous co-immunoprecipitation studies suggested that the incorporation of only one LRRC8A subunit into a hetero-hexamer is sufficient for the formation of a functional VSOR/VRAC channel [142]. In contrast, most recent studies showed that VSOR/VRAC formed by LRRC8A plus LRRC8C exhibits 4:2 and 5:1 stoichiometry based on the data by mass spectrometry analysis [141] and those by single-particle cryo-EM analysis [140], respectively.
Questions about LRRC8A plus LRRC8C/D/E as the sufficient components for VSOR/VRAC channels
It is now obvious that LRRC8A and LRRC8C/D/E provide the necessary components for VSOR/VRAC channels. Then, it must be inquired whether LRRC8A and LRRC8C/D/E are sufficient components for the VSOR/VRAC entity in light of the following criteria for the molecular identification of the VSOR/VRAC channel [143]. Here, I update the criteria as follows: (a) The cells functionally exhibiting VSOR/VRAC activity should endogenously express the candidate mRNA(s) and protein(s); (b) the abolition of expression of the candidate protein(s) abolishes the endogenous VSOR/VRAC currents; (c) transfection with the gene(s) for the candidate protein(s) induces anionic currents with characteristics identical to those of phenotypic properties of VSOR/VRAC (Table 1: A, B); (d) the mutation(s) of the candidate gene(s) gives rise to significant changes in the biophysical pore properties of VSOR/VRAC (Table 1: B); and (e) reconstitution with the candidate protein(s) reproduces anionic currents with exhibiting the phenotypic properties of VSOR/VRAC (Table 1: A, B).
The fact that LRRC8 members are ubiquitously expressed in vertebrate cells [144] is in accord with Criterion-(a). In agreement with Criteria-(b), not only the single abolition of LRRC8A but also the quadruple abolition of LRRC8B/C/D/E were found to abolish the endogenous swelling-activated VSOR/VRAC currents [9, 132, 133]. Concerning Criterion-(c), combined overexpression of LRRC8A and LRRC8C/D/E was shown to induce swelling-induced outwardly rectifying anion currents in LRRC8−/− HCT116 cells [9] and defolliculated Xenopus oocytes [136]. However, it has not been examined as yet whether these anion currents exhibit the full set of phenotypic properties of VSOR/VRAC (Table 1), especially including non-hydrolytic dependence on cytosolic ATP, sensitivity to cytosolic free Mg2+, and low-field Eisenman type-I anion selectivity. Regarding Criterion-(d), a number of point mutation studies have been performed, as given below, on the whole-cell current level, but not on the single-channel level as yet. Outward rectification of whole-cell VSOR/VRAC currents was found to become stronger when the T5R mutants of LRRC8A and LRRC8C were expressed in LRRC8−/− HCT116 cells [145], suggesting that the short hydrophilic N-terminal coil (NTC) region (Fig. 2) is important for the outward-rectifying properties of VSOR/VRAC. The time course of inactivation kinetics of whole-cell currents observed at large positive potentials was found to be affected by charge-modifying mutation of K98 and D100 of LRRC8A (Fig. 2) as well as the corresponding residues (K91 and N93) of LRRC8E in the C-terminal part of the first extracellular loop (EL1), when these mutants of LRRC8A and LRRC8E were both expressed in LRRC8−/− HCT116 cells [145], as follows. The inactivation kinetics becomes much faster by combinations of charge-reversing K98E-LRRC8A and K91E-LRRC8E mutations as well as by those of D100R- or D100K-LRRC8A and N93R- or N93K-LRRC8E mutations. In contrast, the kinetics becomes slower by a combination of charge-neutralizing K98A- or K98N-LRRC8A and K91A- or K91N-LRRC8E mutations as well as by a combination of charge-preserving D100E-LRRC8A and charge-conferring N93E-LRRC8E mutations. In addition, the voltage dependence of the inactivation kinetics was found to be observed at much lesser positive potentials by a combination of charge-neutralizing E6C-LRRC8A and -LRRC8E mutations in the NTC domain. Since the positively charged R103 residue in the extracellular subdomain of LRRC8A forms the narrowest constriction (Fig. 2), the mutation of this residue may be predicted to affect the pore properties of VSOR/VRAC. In fact, the introduction of a charge-neutralizing R103F mutation into LRRC8A was found to abolish the open-channel blocking action of extracellular ATP, when co-expressed with wild-type LRRC8C [128]. Also, the anion selectivity of LRRC8A + LRRC8C channels was shown to be reduced by this mutant of LRRC8A [128]. The introduction of another charge-neutralizing R103A mutation into LRRC8A rendered the LRRC8A + LRRC8C channel slightly permeable to Na+ by changing the PNa/PCl value from 0.02 to 0.28 [126]. In contrast, the positive charge-conferring L105R mutation of the corresponding residue of LRRC8C failed to change the anion selectivity of LRRC8A + LRRC8C channel [126]. Another positively charged K98 residue, existing near R103, in the EL1 domain of LRRC8A (see Fig. 2) appears to be also involved in the anion selectivity filter because the introduction of double charge-reversing K98E and K91E mutations to LRRC8A and LRRC8E, respectively, decreased the PI/PCl value from 1.25 to 1.12 [146]. In addition, it is likely that T5 and E6 in the NTC domain (see Fig. 2) are involved in the anion selectivity because the channels formed by T5R-LRRC8A together with E6C-LRRC8C and by E6C-LRRC8A together with E6C-LRRC8C or E6C-LRRC8E exhibited increased the PI/PCl value from 1.39 to 1.83 [128] and from 1.29 to 2.29 [145], respectively. However, these charge-modified point mutants unfortunately exhibited the same sequence of PI > PCl selectivity though in varying degrees. To assert that the above-studied residues form the anion selectivity filter, much stronger changes in the selectivity sequence, such as reversal of the PI: PCl sequence, that is, transition of types of Eisenman’s halide anion selectivity sequence from type I to types III – VII (Table 2) or just transition of types of the halide anion selectivity sequence from type I to type II (Table 2), are to be observed by the mutational studies by comparing permeabilities to I− and Cl− to Br− at least. However, it must be pointed out that such mutational studies must be performed not only on the whole-cell current level but also on the single-channel level to see the effect on the intermediate outwardly rectifying unitary conductance, in the future. Taken together, these mutational studies indicate that LRRC8A plus LRRC8C/D/E may form the pore of VSOR/VRAC, although the crucial evidence is still missing. Moreover, no mutational studies have been performed as yet for intracellular ATP dependence and intracellular Mg2+ sensitivity.
Requirements of some unidentified additional components for VSOR/VRAC channels
To obtain more direct evidence for the involvement of LRRC8 members, reconstitution studies were recently conducted [127, 129, 133]. The anion channels reconstituted with LRRC8A alone were found to exhibit completely different properties from those of native VSOR/VRAC channels. LRRC8A channels reconstituted in liposomes exhibited homo-hexameric structure and are activated only by reduction of ionic strength in a manner totally independent of ATP [127, 129] and insensitive to Mg2+ [129]. In addition, the unitary conductance of LRRC8A channels looks smaller and more sharply outwardly rectifying than that of native VSOR/VRAC channels; that is, around 50 pS at + 100 mV in 500 mM Cl− conditions [127] and 24 pS at + 100 mV and 5.4 pS at − 100 mV in 70 mM Cl− conditions [129]. When LRRC8A and LRRC8C, 8D, or 8E were reconstituted to form a hetero-hexameric channel in droplet lipid bilayers, a strong hypotonic stimulation (around 47 ~ 77% osmolarity) was found to steadily activate anionic single-channel events, even in the absence of ATP, exhibiting intermediate, but relatively small, unitary conductance of around 8 pS and 50–70 pS in the presence of 70 mM and 500 mM KCl, respectively [133]. However, no single-channel-level studies on the hetero-hexameric channels reconstituted with LRRC8A and LRRC8C/D/E have so far been conducted concerning outward rectification on the single-channel level as well as anion selectivity sequence, Mg2+ sensitivity, and voltage-dependent blocking by ATP on the whole-cell level. Moreover, the unitary events were steadily observed at + 100 mV without exhibiting inactivation kinetics and could be activated preferably by reduction of ionic strength in the absence of ATP in both cis and trans solutions [133]. Above all, the channels were never activated by swelling (droplet inflation) per se [133], in contrast to native VSOR/VRAC channels [46,47,48]. Thus, LRRC8A plus LRRC8C/D/E form anionic channels in the cell-free reconstituted system in a manner completely different from native VSOR/VRAC channels observed in the cell system even though it appears that they participate in the pore formation for the molecular entity of VSOR/VRAC.
Recently proposed molecules required for VSOR/VRAC channel activities
Collectively, it must be concluded that the recent mutational and reconstitution studies with LRRC8 members, unfortunately, failed to alter and reproduce, respectively, all the full set of phenotypical properties of native VSOR/VRAC channels. Also, we here need to remind the following important facts. First, overexpression of LRRC8A alone suppressed endogenous VSOR/VRAC activity [8, 9]. Second, overexpression of LRRC8A plus LRRC8C failed to increase VSOR/VRAC currents by adding new currents to the endogenous currents [9]. Third, cisplatin-resistant KCP-4 cells deficient in endogenous VSOR/VRAC activity exhibit similar protein expression levels of LRRC8 members to those in the parental KB cells as well as in other three (HEK293T, HeLa, and Intestine 407) human epithelial cell lines rich in VSOR/VRAC activity [11, 75]. Fourth, overexpression of LRRC8A plus LRRC8D or LRRC8E in VSOR/VRAC-deficient KCP-4 cells failed to restore VSOR/VRAC activity up to that observed in its parental KB cells [75]. Thus, it is evident that we still miss some as-yet-unidentified additional essential component(s) other than LRRC8 members for VSOR/VRAC activity exhibiting a full set of phenotypical properties (Table 1).
TTYH hypothesis
Following the recent LRRC8 upsurge, two groups [147, 148] proposed TTYH members, the genes of which are homologs of the Drosophila melanogaster tweety, as the pore-forming proteins of VSOR/VRAC in 2019. TTYH1 was initially described as a large-conductance Maxi-Cl anion channel activated by hypotonic stimulation [149, 150] but later this hypothesis was disproven [10, 63]. Han et al. [147] originally reported that TTYH members serve as the core components of VSOR/VRAC channel based on the following observations. First, shRNA-mediated knockdown of LRRC8A and/or LRRC8C failed to abolish ICl,swell in primary mouse astrocytes equilibrated with intracellular (pipette) and extracellular Tris-Cl-rich solutions, although the same knockdown procedure abolished VSOR/VRAC currents in HEK293 cells equilibrated with NaCl-rich solutions. Second, shRNA-mediated triple knockdown of TTYH1, 2, and 3 mostly abolished ICl,swell in mouse astrocytes in primary culture and those in hippocampal slices under Tris-Cl-rich conditions. Third, overexpression of TTYH1, 2, and 3 together with water channel AQP4 enhanced ICl,swell in HEK293 cells under Tris-Cl-rich conditions and rescued the currents in HEK293 and CHO-K1 cells both in which LRRC8A expression was knocked down by treatment with shRNA-LRRC8A. Furthermore, they concluded that TTYH1/2/3 forms the pore of VSOR/VRAC channels because overexpression of the charge-neutralized R165A-TTYH1 and R164A-TTYH2 mutants suppressed ICl,swell in HEK293 and CHO-K1 cells, respectively, under Tris-Cl-rich conditions, and, in addition, because extracellular application of MTSES did not affect ICl,swell recorded in HEK293 cells transfected with WT-TTYH1 plus AQP4 but became suppressive against the currents recorded in those transfected with R165C-TTYH1 plus AQP4 under Tris-Cl-rich conditions. About 1 month later, Bae et al. [148] also deduced that TTYH1 and TTYH2 confer VSOR/VRAC in a manner independent of LRRC8A in gastric cancer cells under the intracellular and extracellular Tris-Cl-rich conditions based on the following observations. First, ICl,swell was not affected by shRNA-mediated knockdown of LRRC8A but, in contrast, was largely abolished by double knockout of TTYH1 and TTYH2 in gastric cancer SNU-601 cells. Second, expression of TTYH1/2, but not LRRC8A, mRNA is lacking in cisplatin-resistant SNU-601-derived R10 cells in which VSOR/VRAC activity is deficient, but ICl,swell and TTYH1/2 mRNA expression were both restored after treatment with a histone deacetylase inhibitor TSA. Third, the knockdown of TTYH1 alone and that of TTYH2 alone inhibited ICl,swell in HepG2 cells which endogenously express TTYH1 but not TTYH2, and in LoVo cells which endogenously express TTYH2 but not TTYH1, respectively. However, it must be noted that a small but significant level of ICl,swell was still observed even after triple knockdown of TTYH1, 2, and 3 in astrocytes [147] and double knockout of TTYH1 and TTYH2 in SNU-601 cells [148]. Moreover, ICl,swell was actually suppressed by knockdown of LRRC8A and/or LRRC8C in HEK293 cells under NaCl-rich conditions [147]. Thus, it is possible that a membrane-impermeable cation, Tris+, somehow impairs the knockdown of LRRC8 expression, and then TTYH blocks this Tris+ effect.
Recent cryo-EM studies provided firm evidence against “TTYH = VSOR/VRAC” hypothesis. First, no pore-like structure was observed in the three-dimensional structure of TTYHs [151, 152], which were shown to be expressed in the plasma membrane [153]. The positively charged residues, such as R165 of TTYH1, R164 of TTYH2 as well as R367 and H370 of TTYH3, that were previously proposed to line a pore [147, 149], exist remote from the membrane [152]. Also, the electrostatic environment at the extracellular side within the transmembrane domain is formed by strongly acidic (negatively charged) residues [151, 152] that should repel permeating anions. Second, hypotonicity-induced VSOR/VRAC-like currents were never observed even under Tris-Cl-rich conditions in HEK293 cells in which TTYH2 alone or together with AQP4 was overexpressed [151], and also never observed under NMDG-Cl-rich conditions in LRRC8−/− HEK293 cells, in which TTYH1, 2, or 3 together with AQP4 were overexpressed [152]. Above all, although expression of TTYH proteins was found to be exclusively in astrocytes in the mouse hippocampus and cortex [147], vigorous VSOR/VRAC activity was actually observed in neurons in the mouse [154] and rat [106, 155] hippocampus as well as in neurons in the mouse cortex [59, 76]. Taken together, it is conceivable that TTYHs represent neither VSOR/VRAC molecules nor its essential core components. However, there remains a possibility that TTYHs play augmenting roles in VSOR/VRAC channel activity only under special (such as Tris-Cl-rich) conditions in some (but not all) cell types including astrocytes and gastric cancer cells.
TRPM7 as the swelling-sensing subcomponent
The physiologically most important phenotype of the VSOR/VRAC is volume expansion sensitivity. During the RVD process, this anion channel participates in the volume-regulatory Cl− efflux [3, 4, 14, 29]. The channels reconstituted with LRRC8A plus LRRC8C/D/E were, however, not activated by cell swelling, though activated by a reduction in the ionic strength [133]. Thus, the volume sensitivity of VSOR/VRAC must be granted by some essential subcomponent other than LRRC8 members. The channel-mediated volume-regulatory KCl efflux was shown to be preceded by the activation of a Ca2+-permeable mechano-sensitive cation channel [156,157,158], which was recently identified as TRPM7 in human epithelial cells [159]. Membrane stretch-induced activation of TRPM7 was demonstrated to cause Ca2+ influx, thereby stimulating Ca2+-activated K+ channels (KCa) [159], that are responsible for volume-regulatory K+ efflux [1, 160]. In light of such a close coupling of VSOR/VRAC, KCa, and TRPM7 functions in the RVD process, there arises a possibility that TRPM7 expressed ubiquitously is involved in the activation not only of KCa but also of VSOR/VRAC. Recently, Numata et al. [10], in fact, provided firm evidence that TRPM7 functionally regulates VSOR/VRAC activity by molecularly interacting with LRRC8A and by sensing volume expansion, as follows. First, shRNA-mediated knockdown of TRPM7 prominently reduced both swelling-activated VSOR/VRAC currents and human LRRC8A mRNA expression in HeLa cells. In addition, the knockout of gallus TRPM7 (gTRPM7) abolished both VSOR/VRAC currents and gLRRC8A mRNA expression in chicken DT40 cells. Since 2 day treatment with a TRPM7 blocker NS8593 or a Ca2+ chelator EGTA was found to suppress not only TRPM7 currents and the cytosolic Ca2+ level but also VSOR/VRAC currents, it appears that the regulatory effect of TRPM7 on VSOR/VRAC activity is mediated by steady-state Ca2+ influx through TRPM7 channels. Second, plasmalemmal colocalization and physical interaction between LRRC8A and TRPM7 were observed by immunostaining and coimmunoprecipitation studies, respectively, in osmotically swollen HeLa cells. Such colocalization and physical interaction were also observed between LRRC8A and the K1648R construct of TRPM7, in which enzyme activity of the C-terminal α-kinase domain was inactivated by the point mutation of the ATP-binding site, but were never observed between LRRC8A and the Δ-kinase construct of TRPM7, in which the entire α-kinase domain was deleted. Third, it appears that VSOR/VRAC activity is exhibiting a functional coupling, in real-time, with TRPM7 activity, because linear relationships were observed between TRPM7-mediated cationic current densities and VSOR/VRAC-mediated anionic current densities simultaneously measured after hypotonic stimulation in the same DT40 cells as well as in the same gTRPM7-deficient DT40 cells complemented with wild-type hTRPM7. Taken together, it is concluded that TRPM7 serves as an essential volume-sensitive subcomponent of VSOR/VRAC first by enhancing LRRC8A mRNA expression via steady-state Ca2+ influx, second by stabilizing the plasmalemmal expression of LRRC8A protein via physical interaction, and third by exhibiting a real-time functional coupling with VSOR/VRAC activity.
Conclusions and perspectives
The volume-sensitive/regulatory outwardly rectifying anion channel, VSOR/VRAC, functionally discovered in 1988 exhibits phenotypical properties such as volume expansion sensitivity, cytosolic ATP dependence, intracellular Mg2+ sensitivity, and low-field anion selectivity, those of which are distinct from other anion channels. Unfortunately, specific VSOR/VRAC blockers are not available as yet and are to be surveyed or developed by taking two basic structures of known effective blockers into consideration. Therefore, one could not perform a search for the ubiquitous VSOR/VRAC molecule by affinity purification using any specific inhibitors. By an unbiased genome-wide siRNA screening approach, then, the hetero-multimeric complex of LRRC8A and LRRC8C/D/E was identified as the core, highly possibly pore-forming, component of VSOR/VRAC in 2014. However, the LRRC8 channel per se was shown to be insensitive to osmotic swelling but activated by the reduction of ionic strength even in the absence of cytosolic ATP. Recently, a Ca2+-permeable mechano-sensitive non-selective cation channel TRPM7 was identified as the essential subcomponent that endows the anionic VSOR/VRAC channel with volume expansion sensitivity. It must be emphasized that some other subcomponent(s) giving cytosolic ATP dependence and Mg2+ sensitivity are still missing. Thus, studies still need to keep in progress to identify the molecular entity of VSOR/VRAC channels exhibiting the full set of phenotypical properties.
From not only physiological but also pathological and clinical perspectives, elucidation of the activation mechanisms and the roles in release of organic signals and in cell death induction of VSOR/VRAC is very important. In this regard, the molecular entity of VSOR/VRAC is to be reassessed. These topics will be described in the subsequent Part 2 article.
Availability of data and materials
The data underlying this article will be obtained via PubMed and Google Scholar or available from the author upon reasonable request.
Abbreviations
- RVD:
-
Regulatory volume decrease
- CaCC:
-
Ca2+-activated Cl− channel
- VSOR:
-
Volume-sensitive outwardly rectifying anion channel
- VRAC:
-
Volume-regulated anion channel
- OVD:
-
Osmotic volume decrease
- OVI:
-
Osmotic volume increase
- HVD:
-
Hormonal volume decrease
- HVI:
-
Hormonal volume increase
- SVD:
-
Secretory volume decrease
- CVD:
-
Cooling volume decrease
- FVD:
-
Freezing volume decrease
- RVI:
-
Regulatory volume increase
- NKCC:
-
Na+-K+-Cl− symporter or cotransporter
- NCC:
-
Na+-Cl+ symporter
- NHE:
-
Na+/H+ antiporter or exchanger
- AE:
-
Cl−/HCO3– antiporter (anion exchanger)
- KCC:
-
K+-Cl− symporter
- KHE:
-
K+/H+ antiporter
- HICC:
-
Hypertonicity-induced cation channel
- EC50 :
-
Half-maximal effective concentration
- IC50 :
-
Half-maximal inhibitory concentration
- ASOR:
-
Acid-sensitive outwardly rectifying anion channel
- PAC:
-
Proton-activated anion channel
- ICl,swell :
-
Swelling-activated Cl− currents
- CysLT1:
-
Cysteinyl leukotriene 1
- PGP:
-
P-glycoprotein
- ANO6:
-
Anoctamin 6
- EL1:
-
First extracellular loop
- NTC:
-
N-Terminal coil
- KCa :
-
Ca2+-activated K+ channel
References
Hazama A, Okada Y (1988) Ca2+ sensitivity of volume-regulatory K+ and Cl– channels in cultured human epithelial cells. J Physiol 402:687–702. https://doi.org/10.1113/jphysiol.1988.sp017229
Cahalan MD, Lewis RS (1988) Role of potassium and chloride channels in volume regulation by T lymphocytes. Soc Gen Physiol Ser 43:281–301
Okada Y (1997) Volume expansion-sensing outward-rectifier Cl– channel: fresh start to the molecular identity and volume sensor. Am J Physiol 273(3 Pt 1):C755–C789. https://doi.org/10.1152/ajpcell.1997.273.3.C755
Nilius B, Eggermont J, Voets T, Buyse G, Manolopoulos V, Droogmans G (1997) Properties of volume-regulated anion channels in mammalian cells. Prog Biophys Mol Biol 68(1):69–119. https://doi.org/10.1016/s0079-6107(97)00021-7
Maeno E, Ishizaki Y, Kanaseki T, Hazama A, Okada Y (2000) Normotonic cell shrinkage due to disordered volume regulation is an early prerequisite to apoptosis. Proc Natl Acad Sci USA 97:9487–9492. https://doi.org/10.1073/pnas.140216197
Shimizu T, Numata T, Okada Y (2004) A role of reactive oxygen species in apoptotic activation of volume-sensitive Cl– channel. Proc Natl Acad Sci USA 101(17):6770–6773. https://doi.org/10.1073/pnas.0401604101
Lee EL, Shimizu T, Ise T, Numata T, Kohno K, Okada Y (2007) Impaired activity of volume-sensitive Cl– channel is involved in cisplatin resistance of cancer cells. J Cell Physiol 211:513–521. https://doi.org/10.1002/jcp.20961
Qiu Z, Dubin AE, Mathur J, Tu B, Reddy K, Miraglia LJ, Reinhardt J, Orth AP, Patapoutian A (2014) SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell 157(2):447–458. https://doi.org/10.1016/j.cell.2014.03.024
Voss FK, Ullrich F, Münch J, Lazarow K, Lutter D, Mah N, Andrade-Navarro MA, von Kries JP, Stauber T, Jentsch TJ (2014) Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 344(6184):634–638. https://doi.org/10.1126/science.1252826
Numata T, Sato-Numata K, Hermosura MC, Mori Y, Okada Y (2021) TRPM7 is an essential regulator for volume-sensitive outwardly rectifying anion channel. Commun Biol 4(1):599. https://doi.org/10.1038/s42003-021-02127-9
Okada Y, Okada T, Islam MR, Sabirov RZ (2018) Molecular identities and ATP release activities of two types of volume-regulatory anion channels, VSOR and Maxi-Cl. Curr Top Membr 81:125–176. https://doi.org/10.1016/bs.ctm.2018.07.004
Wehner F, Olsen H, Tinel H, Kinne-Saffran E, Kinne RKH (2003) Cell volume regulation: osmolytes, osmolyte transport, and signal transduction. Rev Physiol Biochem Pharmacol 148:1–80. https://doi.org/10.1007/s10254-003-0009-x
Okada Y (2004) Ion channels and transporters involved in cell volume regulation and sensor mechanisms. Cell Biochem Biophys 41(2):233–258. https://doi.org/10.1385/CBB:41:2:233
Hoffmann EK, Lambert IH, Pedersen SF (2009) Physiology of cell volume regulation in vertebrates. Physiol Rev 89(1):193–277. https://doi.org/10.1152/physrev.00037.2007
Okada Y (1998) Cell volume regulation: the molecular mechanism and volume sensing machinery. Elsevier, Amsterdam
Lang F (1998) Cell volume regulation. Karger, Basel
Okada Y, Numata T, Sato-Numata K, Sabirov RZ, Liu H, Mori S, Morishima S (2019) Roles of volume-regulatory anion channels, VSOR and Maxi-Cl, in apoptosis, cisplatin resistance, necrosis, ischemic cell death, stroke and myocardial infarction. Curr Top Membr 83:205–283. https://doi.org/10.1016/bs.ctm.2019.03.001
Lang F, Busch GL, Ritter M, Völkl H, Waldegger S, Gulbins E, Häussinger D (1998) Functional significance of cell volume regulatory mechanisms. Physol Rev 78(1):247–306. https://doi.org/10.1152/physrev.1998.78.1.247
Häussinger D, Lang F, Gerok W (1994) Regulation of cell function by the cellular hydration state. Am J Physiol 267(3 Pt 1):E343–E355. https://doi.org/10.1152/ajpendo.1994.267.3.E343
Manabe K, Shimizu T, Morishima S, Okada Y (2004) Regulatory volume increase after secretory volume decrease in colonic epithelial cells under muscarinic stimulation. Pflügers Arch Eur J Physiol 448(6):596–604. https://doi.org/10.1007/s00424-004-1301-6
Okada Y (2016) Channelling frozen cells to survival after thawing: opening the door to cryo-physiology. J Physiol 594(6):1523–1524. https://doi.org/10.1113/JP271842
Sato-Numata K, Numata T, Ueta Y, Okada Y (2021) Vasopressin neurons respond to hyperosmotic stimulation with regulatory volume increase and secretory volume decrease by activating ion transporters and Ca2+ channels. Cell Physiol Biochem 55(S1):119–134. https://doi.org/10.33594/000000342
Hoffmann EK, Simonsen LO (1989) Membrane mechanisms in volume and pH regulation in vertebrate cells. Physiol Rev 69(2):315–382. https://doi.org/10.1152/physrev.1989.69.2.315
Grinstein S, Foskett JK (1990) Ionic mechanisms of cell volume regulation in leukocytes. Annu Rev Physiol 52:399–414. https://doi.org/10.1146/annurev.ph.52.030190.002151
Okada Y, Hazama A (1989) Volume-regulatory ion channels in epithelial cells. News Physiol Sci 4:238–242
Shimizu T, Wehner F, Okada Y (2006) Inhibition of hypertonicity-induced cation channels sensitizes HeLa cells to shrinkage-induced apoptosis. Cell Physiol Biochem 18:295–302. https://doi.org/10.1159/000097607
Wehner F, Bondarava M, Veld FT, Endl E, Nürnberger HR, Li T (2016) Hypertonicity-induced cation channels. Acta Physiol 187(1–2):21–25. https://doi.org/10.1111/j.1748-1716.2006.01561.x
Okada Y, Hazama A (1988) Cytosolic Ca2+ activates volume-regulatory K+ channels but not Cl– channels in cultured epithelial cells exposed to a hypotonic solution. Biomed Res 9(Suppl. 2):161–165
Strange K, Emma F, Jackson PS (1996) Cellular and molecular physiology of volume-sensitive anion channels. Am J Physiol 270(3 Pt 1):C711–C730. https://doi.org/10.1152/ajpcell.1996.270.3.C711
Akita T, Okada Y (2014) Characteristics and roles of the volume-sensitive outwardly rectifying (VSOR) anion channel in the central nervous system. Neuroscience 275:211–231. https://doi.org/10.1016/j.neuroscience.2014.06.015
Jentsch TJ, Lutter D, Lanells-Cases R, Ullrich F, Voss FK (2016) VRAC: molecular identification as LRRC8 heteromers with differential functions. Pflügers Arch Eur J Physiol 468(3):385–393. https://doi.org/10.1007/s00424-015-1766-5
Pedersen SF, Okada Y, Nilius B (2016) Biophysics and physiology of the volume-regulated anion channel (VRAC)/volume-sensitive outwardly rectifying anion channel (VSOR). Pflügers Arch Eur J Physiol 468(3):371–383. https://doi.org/10.1007/s00424-015-1781-6
Osei-Owusu J, Yang J, Vitery MDC, Qiu Z (2018) Molecular biology and physiology of volume-regulated anion channel (VRAC). Curr Top Membr 81:177–203. https://doi.org/10.1016/bs.ctm.2018.07.005
Okada Y, Okada T, Sato-Numata K, Islam MR, Ando-Akatsuka Y, Numata T, Kubo M, Shimizu T, Kurbannazarova RS, Marunaka M, Sabirov RZ (2019) Cell volume-activated and -correlated anion channels in mammalian cells: their biophysical, molecular and pharmacological properties. Pharmacol Rev 71:49–88. https://doi.org/10.1124/pr.118.015917
Strange K, Yamada T, Denton JS (2019) A 30-year journey from volume-regulated anion currents to molecular structure of the LRRC8 channel. J Gen Physiol 151(2):100–117. https://doi.org/10.1085/jgp.201812138
Figueroa EE, Denton JS (2022) A SWELL time to develop the molecular pharmacology of the volume-regulated anion channel (VRAC). Channels 16(1):27–36. https://doi.org/10.1080/19336950.2022.2033511
Liu T, Li Y, Wang D, Stauber T, Zhao J (2023) Trends in volume-regulated anion channel (VRAC) research: visualization and bibliometric analysis from 2014 to 2022. Front Pharmacol 14:1234885. https://doi.org/10.3389/fphar.2023.1234885
Okada Y, Petersen CC, Kubo M, Morishima S, Tominaga M (1994) Osmotic swelling activates intermediate-conductance Cl– channels in human intestinal epithelial cells. Jpn J Physiol 44(4):403–409. https://doi.org/10.2170/jjphysiol.44.403
Mao J, Xu B, Li H, Chen L, Jin X, Zhu J, Wang W, Zhu L, Zuo W, Chen W, Wang L (2011) Lack of association between stretch-activated and volume-activated Cl– currents in hepatocellular carcinoma cells. J Cell Physiol 226(5):1176–1185. https://doi.org/10.1002/jcp.22443
Behe P, Foote JR, Levine AP, Platt CD, Chou J, Benavides F, Geha RS, Segal AW (2017) The LRRC8A mediated “swell activated” chloride conductance is dispensable for vacuolar homeostasis in neutrophils. Front Pharmacol 8:262. https://doi.org/10.3389/fphar.2017.00262
Groulx N, Boudreault F, Orlov SN, Grygorczyk R (2006) Membrane reserves and hypotonic cell swelling. J Membr Biol 214(1):43–56. https://doi.org/10.1007/s00232-006-0080-8
Pangrsic T, Potokar M, Haydon PG, Zorec R, Kreft M (2006) Astrocyte swelling leads to membrane unfolding, not membrane insertion. J Neurochem 99(2):514–523. https://doi.org/10.1111/j.1471-4159.2006.04042.x
Morishima S, Shimizu T, Kida H, Okada Y (2000) Volume expansion sensitivity of swelling-activated Cl– channel in human epithelial cells. Jpn J Physiol 50:277–280. https://doi.org/10.2170/jjphysiol.50.277
Okada Y (1998) Cell volume-sensitive chloride channels. Contrib Nephrol 123:21–33. https://doi.org/10.1159/000059920
Miwa A, Ueda K, Okada Y (1997) Protein kinase C-independent correlation between P-glycoprotein expression and volume sensitivity of Cl– channel. J Membr Biol 157:63–69. https://doi.org/10.1007/s002329900216
Hagiwara N, Masuda H, Shoda M, Irisawa H (1992) Stretch-activated anion currents of rabbit cardiac myocytes. J Physiol 456:285–302. https://doi.org/10.1113/jphysiol.1992.sp019337
Doroshenko P (1998) Pervanadate inhibits volume-sensitive chloride current in bovine chromaffin cells. Pflügers Arch Eur J Physiol 435(2):303–309. https://doi.org/10.1007/s004240050516
Best L, Brown P (2009) Studies of the mechanism of activation of the volume-regulated anion channel in rat pancreatic beta-cells. J Membr Biol 230(2):83–91. https://doi.org/10.1007/s00232-009-9189-x
Lewis RS, Ross PE, Cahalan MD (1993) Chloride channels activated by osmotic stress in T lymphocytes. J Gen Physiol 101(6):801–826. https://doi.org/10.1085/jgp.101.6.801
Díaz M, Valverde MA, Higgins CF, Rucăreanu C, Sepúlveda FV (1993) Volume-activated chloride channels in HeLa cells are blocked by verapamil and dideoxyforskolin. Pflügers Arch Eur J Physiol 422(4):347–353. https://doi.org/10.1007/BF00374290
Jackson PS, Morrison R, Strange K (1994) The volume-sensitive organic osmolyte-anion channel VSOAC is regulated by nonhydrolytic ATP binding. Am J Physiol 267(5 Pt 1):C1203–C1209. https://doi.org/10.1152/ajpcell.1994.267.5.C1203
Oike M, Droogmans G, Nilius B (1994) The volume-activated chloride current in human endothelial cells depends on intracellular ATP. Pflugers Arch 427(1–2):184–186. https://doi.org/10.1007/BF00585960
Oiki S, Kubo M, Okada Y (1994) Mg2+ and ATP-dependence of volume-sensitive Cl– channels in human epithelial cells. Jpn J Physiol 44(Suppl 2):S77–S79
Bryan-Sisneros A, Sabanov V, Thoroed SM, Doroshenko P (2000) Dual role of ATP in supporting volume-regulated chloride channels in mouse fibroblasts. Biochim Biophys Acta 1468(1–2):63–72. https://doi.org/10.1016/s0005-2736(00)00243-1
Oiki S, Kubo M, Okada Y (1998) Electrophysiological properties of volume-regulated Cl– channels in intestinal epithelial cells. In: Okada Y (ed) Cell volume regulation: the molecular mechanism and volume sensing machinery. Elsevier, Amsterdam, pp 125–129
McCann JD, Li M, Welsh MJ (1989) Identification and regulation of whole-cell chloride currents in airway epithelium. J Gen Physiol 94(6):1015–1036. https://doi.org/10.1085/jgp.94.6.1015
Worrell RT, Butt AG, Cliff WH, Frizzell RA (1989) A volume-sensitive chloride conductance in human colonic cell line T84. Am J Physiol 256(6 Pt 1):C1111–C1119. https://doi.org/10.1152/ajpcell.1989.256.6.C1111
Petersen CC, Kubo M, Morishima S, Tominaga M, Okada Y (1994) Single-channel recordings of volume-sensitive Cl- channels in human intestinal epithelial cells. Jpn J Physiol 44(Suppl 2):S73–S75
Inoue H, Mori S, Morishima S, Okada Y (2005) Volume-sensitive chloride channels in mouse cortical neurons: characterization and role in volume regulation. Eur J Neurosci 21(6):1648–1658. https://doi.org/10.1111/j.1460-9568.2005.04006.x
Abdullaev IF, Sabirov RZ, Okada Y (2003) Upregulation of swelling-activated Cl channel sensitivity to cell volume by activation of EGF receptors in murine mammary cells. J Physiol 549(Pt 3):749–758. https://doi.org/10.1113/jphysiol.2003.039784
Kurbannazarova RS, Bessonova SV, Okada Y, Sabirov RZ (2011) Swelling-activated anion channels are essential for volume regulation of mouse thymocytes. Int J Mol Sci 12(12):9125–9137. https://doi.org/10.3390/ijms12129125
Wang J, Xu H, Morishima S, Tanabe S, Jishage K, Uchida S, Sasaki S, Okada Y, Shimizu T (2005) Single-channel properties of volume-sensitive Cl channel in ClC-3-deficient cardiomyocytes. Jpn J Physiol 55:379–383. https://doi.org/10.2170/jjphysiol.S655
Okada Y (2006) Cell volume-sensitive chloride channel: phenotypic properties and molecular identity. Contrib Nephrol 152:9–24. https://doi.org/10.1159/000096285
Okada Y, Sato K, Toychiev AH, Suzuki M, Dutta AK, Inoue H, Sabirov RZ (2009) The puzzles of volume-activated anion channels. In: Alvarez-Leefmans FJ, Delpire E (eds) Physiology and pathology of chloride transporters and channels in the nervous system. From molecules to diseases. Elsevier, San Diego, pp 283–306
Kubo M, Okada Y (1992) Volume-regulatory Cl– channel currents in cultured human epithelial cells. J Physiol 456:351–371. https://doi.org/10.1113/jphysiol.1992.sp019340
Tominaga M, Horie M, Sasayama S, Okada Y (1995) Glibenclamide, an ATP-sensitive K+ channel blocker, inhibits the cardiac cyclic AMP-activated Cl– conductance. Circ Res 77:417–423. https://doi.org/10.1161/01.res.77.2.417
Ternovsky VI, Okada Y, Sabirov RZ (2004) Sizing the pore of the volume-sensitive anion channel by differential polymer partitioning. FEBS Lett 576(3):433–436. https://doi.org/10.1016/j.febslet.2004.09.051
Fan HT, Morishima S, Kida H, Okada Y (2001) Phloretin differentially inhibits volume-sensitive and cAMP-activated, but not Ca-activated, Cl– channels. Br J Pharmacol 133:1096–1106. https://doi.org/10.1038/sj.bjp.0704159
Ando-Akatsuka Y, Abdullaev IF, Lee EL, Okada Y, Sabirov RZ (2002) Down-regulation of volume-sensitive Cl– channels by CFTR is mediated by the second nucleotide-binding domain. Pflügers Arch Eur J Physiol 445:177–186. https://doi.org/10.1007/s00424-002-0920-z
Shimizu T, Iehara T, Sato K, Fujii T, Sakai H, Okada Y (2013) TMEM16F is a component of a Ca2+-activated Cl– channel but not a volume-sensitive outwardly rectifying Cl– channel. Am J Physiol Cell Physiol 304:C748–C759. https://doi.org/10.1152/ajpcell.00228.2012
Okada T, Akita T, Sato-Numata K, Islam MR, Okada Y (2014) A newly cloned ClC-3 isoform, ClC-3d, as well as ClC-3a mediates Cd2+-sensitive outwardly rectifying anion currents. Cell Physiol Biochem 33:539–556. https://doi.org/10.1159/000358633
Sato-Numata K, Numata T, Inoue R, Okada Y (2016) Distinct pharmacological and molecular properties of the acid-sensitive outwardly rectifying (ASOR) anion channel from those of the volume-sensitive outwardly rectifying (VSOR) anion channel. Pflügers Arch Eur J Physiol 468:795–803. https://doi.org/10.1007/s00424-015-1786-1
Ise T, Shimizu T, Lee EL, Inoue H, Kohno K, Okada Y (2005) Roles of volume-sensitive Cl– channel in cisplatin-induced apoptosis in human epidermoid cancer cells. J Membr Biol 205(3):139–145. https://doi.org/10.1007/s00232-005-0779-y
Hazama A, Fan HT, Abdullaev I, Maeno E, Tanaka S, Ando-Akatsuka Y, Okada Y (2000) Swelling-activated, cystic fibrosis transmembrane conductance regulator-augmented ATP release and Cl– conductances in murine C127 cells. J Physiol 523(Pt 1):1–11. https://doi.org/10.1111/j.1469-7793.2000.t01-6-00001.x
Okada T, Islam MR, Tsiferova NA, Okada Y, Sabirov RZ (2017) Specific and essential but not sufficient roles of LRRC8A in the activity of volume-sensitive outwardly rectifying anion channel (VSOR). Channels 11:109–120. https://doi.org/10.1080/19336950.2016.1247133
Inoue H, Okada Y (2007) Roles of volume-sensitive chloride channel in excitotoxic neuronal injury. J Neurosci 27(6):1445–1455. https://doi.org/10.1523/jneurosci.4694-06.2007
Sato K, Numata T, Saito T, Ueta Y, Okada Y (2011) V2 receptor-mediated autocrine role of somatodendritic release of AVP in rat vasopressin neurons under hypo-osmotic conditions. Sci Signal 4:ra5. https://doi.org/10.1126/scisignal.2001279
Liu H-T, Tashmukhamedov BA, Inoue H, Okada Y, Sabirov RZ (2006) Roles of two types of anion channels in glutamate release from mouse astrocytes under ischemic or osmotic stress. Glia 54:343–357. https://doi.org/10.1002/glia.20400
Liu H-T, Akita T, Shimizu T, Sabirov RZ, Okada Y (2009) Bradykinin-induced astrocyte-neuron signaling: glutamate release is mediated by ROS-activated volume-sensitive outwardly rectifying anion channels. J Physiol 587:2197–2209. https://doi.org/10.1113/jphysiol.2008.165084
Akita T, Okada Y (2011) Regulation of bradykinin-induced activation of volume-sensitive outwardly rectifying anion channels by Ca2+ nanodomains in mouse astrocytes. J Physiol 589:3909–3927. https://doi.org/10.1113/jphysiol.2011.208173
Akita T, Fedorovich SV, Okada Y (2011) Ca2+ nanodomain-mediated component of swelling-induced volume-sensitive outwardly rectifying anion current triggered by autocrine action of ATP in mouse astrocytes. Cell Physiol Biochem 28(6):1181–1190. https://doi.org/10.1159/000335867
Gong W, Xu H, Shimizu T, Morishima S, Tanabe S, Tachibe T, Uchida S, Sasaki S, Okada Y (2004) ClC-3-independent, PKC-dependent activity of volume-sensitive Cl– channel in mouse ventricular cardiomyocytes. Cell Physiol Biochem 14:213–224. https://doi.org/10.1159/000080330
Inoue H, Takahashi N, Okada Y, Konishi M (2010) Volume-sensitive outwardly rectifying chloride channel in white adipocytes from normal and diabetic mice. Am J Physiol Cell Physiol 298:C900–C909. https://doi.org/10.1152/ajpcell.00450.2009
Sabirov RZ, Kurbannazarova RS, Melanova NR, Okada Y (2013) Volume-sensitive anion channels mediate osmosensitive glutathione release from rat thymocytes. PLoS ONE 8(1):e55646. https://doi.org/10.1371/journal.pone.0055646
Nilius B, Sehrer J, Droogmans G (1994) Permeation properties and modulation of volume-activated Cl– currents in human endothelial cells. Br J Pharmacol 112(4):1049–1056. https://doi.org/10.1111/j.1476-5381.1994.tb13189.x
Ackerman MJ, Wickman KD, Clapham DE (1994) Hypotonicity activates a native chloride current in Xenopus oocytes. J Gen Physiol 103(2):153–179. https://doi.org/10.1085/jgp.103.2.153
Tsumura T, Oiki S, Ueda S, Okuma M, Okada Y (1996) Sensitivity of volume-sensitive Cl– conductance in human epithelial cells to extracellular nucleotides. Am J Physiol 271:C1872–C1878. https://doi.org/10.1152/ajpcell.1996.271.6.C1872
Jackson PS, Strange K (1995) Characterization of the voltage-dependent properties of a volume-sensitive anion conductance. J Gen Physiol 105(5):661–676. https://doi.org/10.1085/jgp.105.5.661
Hisadome K, Koyama T, Kimura C, Droogmans G, Ito Y, Oike M (2002) Volume-regulated anion channels serve as an auto/paracrine nucleotide release pathway in aortic endothelial cells. J Gen Physiol 119(6):511–520. https://doi.org/10.1085/jgp.20028540
Poletto Chaves LA, Varanda WA (2008) Volume-activated chloride channels in mice Leydig cells. Pflügers Arch Eur J Physiol 457(2):493–504. https://doi.org/10.1007/s00424-008-0525-2
Wright EM, Diamond JM (1977) Anion selectivity in biological systems. Physiol Rev 57(1):109–156. https://doi.org/10.1152/physrev.1977.57.1.109
Jirsch JD, Loe DW, Cole SP, Deeley RG, Fedida D (1994) ATP is not required for anion current activated by cell swelling in multidrug-resistant lung cancer cells. Am J Physiol 267(3 Pt 1):C688–C699. https://doi.org/10.1152/ajpcell.1994.267.3.C688
Volk KA, Zhang C, Husted RF, Stokes JB (1996) Cl− current in IMCD cells activated by hypotonicity: time course, ATP dependence, and inhibitors. Am J Physiol 271(3 Pt 2):F552–F559. https://doi.org/10.1152/ajprenal.1996.271.3.F552
Bond T, Basavappa S, Christensen M, Strange K (1999) ATP dependence of the ICl, swell channel varies with rate of cell swelling. Evidence for two modes of channel activation. J Gen Physiol 113(3):441–456. https://doi.org/10.1085/jgp.113.3.441
Nilius B, Oike M, Zahradnik I, Droogmans G (1994) Activation of a Cl– current by hypotonic volume increase in human endothelial cells. J Gen Physiol 103(5):787–805. https://doi.org/10.1085/jgp.103.5.787
Liu Y, Oiki S, Tsumura T, Shimizu T, Okada Y (1998) Glibenclamide blocks volume-sensitive Cl– channels by dual mechanisms. Am J Physiol 275:C343–C351. https://doi.org/10.1152/ajpcell.1998.275.2.C343
Xue Y, Li H, Zhang Y, Han X, Zhang G, Li W, Zhang H, Lin Y, Chen P, Sun X, Liu Y, Chu L, Zhang J, Zhang M, Zhang X (2018) Natural and synthetic flavonoids, novel blockers of the volume-regulated anion channels, inhibit endothelial cell proliferation. Pflügers Arch Eur J Physiol 470(10):1473–1483. https://doi.org/10.1007/s00424-018-2170-8
Rustamova SI, Tsiferova NA, Khamidova OJ, Kurbannazarova RS, Merzlyak PG, Khushbaktova ZA, Syrov VN, Botirov EK, Eshbakova KA, Sabirov RZ (2019) Effect of plant flavonoids on the volume regulation of rat thymocytes under hypoosmotic stress. Pharmacol Rep 71(6):1079–1087. https://doi.org/10.1016/j.pharep.2019.05.023
Figueroa EE, Kramer M, Strange K, Denton JS (2019) CysLT1 receptor antagonists pranlukast and zafirlukast inhibit LRRC8-mediated volume regulated anion channels independently of the receptor. Am J Physiol Cell Physiol 317(4):C857–C866. https://doi.org/10.1152/ajpcell.00281.2019
Decher N, Lang HJ, Nilius B, Brüggemann A, Busch AE, Steinmeyer K (2001) DCPIB is a novel selective blocker of I(Cl, swell) and prevents swelling-induced shortening of guinea-pig atrial action potential duration. Br J Pharmacol 134(7):1467–1479. https://doi.org/10.1038/sj.bjp.0704413
Sabirov RZ, Okada Y (2005) ATP release via anion channels. Purinergic Signal 1:311–328. https://doi.org/10.1007/s11302-005-1557-0
Sabirov RZ, Merzlyak PG, Islam MR, Okada T, Okada Y (2016) The properties, functions and pathophysiology of maxi-anion channels. Pflügers Arch Eur J Physiol 468:405–420. https://doi.org/10.1007/s00424-015-1774-5
Kasuya G, Nureki O (2022) Recent advances in the structural biology of the volume-regulated anion channel LRRC8. Front Pharmacol 13:896532. https://doi.org/10.3389/fphar.2022.896532
Bowens NH, Dohare P, Kuo YH, Mongin AA (2013) DCPIB, the proposed selective blocker of volume-regulated anion channels, inhibits several glutamate transport pathways in glial cells. Mol Pharmacol 83(1):22–32. https://doi.org/10.1124/mol.112.080457
Kittl M, Winklmayr M, Helm K, Lettner J, Gaisberger M, Ritter M, Jakab M (2020) Acid- and volume-sensitive chloride currents in human chondrocytes. Front Cell Dev Biol 8:583131. https://doi.org/10.3389/fcell.2020.583131
Zhang H, Cao HJ, Kimelberg HK, Zhou M (2011) Volume regulated anion channel currents of rat hippocampal neurons and their contribution to oxygen-and-glucose deprivation induced neuronal death. PLoS ONE 6(2):e16803. https://doi.org/10.1371/journal.pone.0016803
Harl B, Schmölzer J, Jakab M, Ritter M, Kerschbaum HH (2013) Chloride channel blockers suppress formation of engulfment pseudopodia in microglial cells. Cell Physiol Biochem 31(2–3):319–337. https://doi.org/10.1159/000343370
Droogmans G, Maertens C, Prenen J, Nilius B (1999) Sulphonic acid derivatives as probes of pore properties of volume-regulated anion channels in endothelial cells. Br J Pharmacol 28(1):35–40. https://doi.org/10.1038/sj.bjp.0702770
Galietta LJ, Falzoni S, Di Virgilio F, Romeo G, Zegarra-Moran O (1997) Characterization of volume-sensitive taurine- and Cl–-permeable channels. Am J Physiol 273(1 Pt 1):C57–C66. https://doi.org/10.1152/ajpcell.1997.273.1.C57
Darby M, Kuzmiski JB, Panenka W, Feighan D, MacVicar BA (2003) ATP released from astrocytes during swelling activates chloride channels. J Neurophysiol 89(4):1870–1877. https://doi.org/10.1152/jn.00510.2002
Yang X, Zhu L, Lin J, Liu S, Luo H, Mao J, Nie S, Chen L, Wang L (2015) Cisplatin activates volume-sensitive like chloride channels via purinergic receptor pathways in nasopharyngeal carcinoma cells. J Membr Biol 248(1):19–29. https://doi.org/10.1007/s00232-014-9724-2
Droogmans G, Prenen J, Eggermont J, Voets T, Nilius B (1998) Voltage-dependent block of endothelial volume-regulated anion channels by calix[4]arenes. Am J Physiol 275(3):C646–C652. https://doi.org/10.1152/ajpcell.1998.275.3.C646
Inoue H, Ohtaki H, Nakamachi T, Shioda S, Okada Y (2007) Anion channel blockers attenuate delayed neuronal cell death induced by transient forebrain ischemia. J Neurosci Res 85:1427–1435. https://doi.org/10.1002/jnr.21279
Chu J, Yang J, Zhou Y, Chen J, Chen KH, Zhang C, Cheng HY, Koylass N, Liu JO, Guan Y, Qiu Z (2023) ATP-releasing SWELL1 channel in spinal microglia contributes to neuropathic pain. Sci Adv 9(13):9931. https://doi.org/10.1126/sciadv.ade9931
Valverde MA, Díaz M, Sepúlveda FV, Gill DR, Hyde SC, Higgins CF (1992) Volume-regulated chloride channels associated with the human multidrug-resistance P-glycoprotein. Nature 355(6363):830–833. https://doi.org/10.1038/355830a0
Paulmichl M, Li Y, Wickman K, Ackerman M, Peralta E, Clapham D (1992) New mammalian chloride channel identified by expression cloning. Nature 356(6366):238–241. https://doi.org/10.1038/356238a0
Duan D, Winter C, Cowley S, Hume JR, Horowitz B (1997) Molecular identification of a volume-regulated chloride channel. Nature 390(6658):417–421. https://doi.org/10.1038/37151
Martins JR, Faria D, Kongsuphol P, Reisch B, Schreiber R, Kunzelmann K (2011) Anoctamin 6 is an essential component of the outwardly rectifying chloride channel. Proc Natl Acad Sci USA 108(44):18168–18172. https://doi.org/10.1073/pnas.1108094108
Okada Y, Oiki S, Tominaga M, Kubo M, Miwa A, Tominaga T, Tsumura T, Ueda K (1997) Volume-sensitive Cl– channel in human epithelial cells: regulation by ATP and relation to P-glycoprotein. Jpn J Physiol 47(Suppl. 1):S19–S20
Takahashi N, Wang X-M, Tanabe S, Uramoto H, Jishage K, Uchida S, Sasaki S, Okada Y (2005) ClC-3-independent sensitivity of apoptosis to Cl channel blockers in mouse cardiomyocytes. Cell Physiol Biochem 15:263–270. https://doi.org/10.1159/000087236
Platt CD, Chou J, Houlihan P, Badran YR, Kumar L, Bainter W, Poliani PL, Perez CJ, Dent SYR, Clapham DE, Benavides F, Geha RS (2017) Leucine-rich repeat containing 8A (LRRC8A)-dependent volume-regulated anion channel activity is dispensable for T-cell development and function. J Allergy Clin Immunol 140(6):1651-1659.e1. https://doi.org/10.1016/j.jaci.2016.12.974
Wang R, Lu Y, Gunasekar S, Zhang Y, Benson CJ, Chapleau MW, Sah R, Abboud FM (2017) The volume-regulated anion channel (LRRC8) in nodose neurons is sensitive to acidic pH. JCI Insight 2(5):e90632. https://doi.org/10.1172/jci.insight.90632
Schober AL, Wilson CS, Mongin AA (2017) Molecular composition and heterogeneity of the LRRC8-containing swelling-activated osmolyte channels in primary rat astrocytes. J Physiol 595(22):6939–6951. https://doi.org/10.1113/JP275053
Formaggio F, Saracino E, Mola MG, Rao SB, Amiry-Moghaddam M, Muccini M, Zamboni R, Nicchia GP, Caprini M, Benfenati V (2019) LRRC8A is essential for swelling-activated chloride current and for regulatory volume decrease in astrocytes. FASEB J 33(1):101–113. https://doi.org/10.1096/fj.201701397RR
Yamada T, Wondergem R, Morrison R, Yin VP, Strange K (2016) Leucine-rich repeat containing protein LRRC8A is essential for swelling-activated Cl currents and embryonic development in zebrafish. Physiol Rep 4(19):e12940. https://doi.org/10.14814/phy2.12940
Deneka D, Sawicka M, Lam AKM, Paulino C, Dutzler R (2018) Structure of a volume-regulated anion channel of the LRRC8 family. Nature 558(7709):254–259. https://doi.org/10.1038/s41586-018-0134-y
Kasuya G, Nakane T, Yokoyama T, Jia Y, Inoue M, Watanabe K, Nakamura R, Nishizawa T, Kusakizako T, Tsutsumi A, Yanagisawa H, Dohmae N, Hattori M, Ichijo H, Yan Z, Kikkawa M, Shirouzu M, Ishitani R, Nureki O (2018) Cryo-EM structures of the human volume-regulated anion channel LRRC8. Nat Struct Mol Biol 25(9):797–804. https://doi.org/10.1038/s41594-018-0109-6
Kefauver JM, Saotome K, Dubin AE, Pallesen J, Cottrell CA, Cahalan SM, Qiu Z, Hong G, Crowley CS, Whitwam T, Lee W-H, Ward AB, Patapoutian A (2018) Structure of the human volume regulated anion channel. Elife 7:e38461. https://doi.org/10.7554/eLife.38461
Kern DM, Oh S, Hite RK, Brohawn SG (2019) Cryo-EM structures of the DCPIB-inhibited volume-regulated anion channel LRRC8A in lipid nanodiscs. Elife 8:e42636. https://doi.org/10.7554/eLife.42636
Okada Y, Sabirov RZ, Merzlyak PG, Numata T, Sato-Numata K (2021) Properties, structures and physiological roles of three types of anion channels molecularly identified in the 2010’s. Front Physiol 12:805148. https://doi.org/10.3389/fphys.2021.805148
Nakamura R, Numata T, Kasuya G, Yokoyama T, Nishizawa T, Kusakizako T, Kato T, Hagino T, Dohmae N, Inoue M, Watanabe K, Ichijo H, Kikkawa M, Shirouzu M, Jentsch TJ, Ishitani R, Okada Y, Nureki O (2020) Cryo-EM structure of the volume-regulated anion channel LRRC8D isoform. Commun Biol 3:240. https://doi.org/10.1038/s42003-020-0951-z
Yamada T, Figueroa EE, Denton JS, Strange K (2012) LRRC8A homohexameric channels poorly recapitulate VRAC regulation and pharmacology. Am J Physiol Cell Physiol 320(3):C293–C303. https://doi.org/10.1152/ajpcell.00454.2020
Syeda R, Qiu Z, Dubin AE, Murthy SE, Florendo MN, Mason DE, Mathur J, Cahalan SM, Peters EC, Montal M, Patapoutian A (2016) LRRC8 proteins form volume-regulated anion channels that sense ionic strength. Cell 164(3):499–511. https://doi.org/10.1016/j.cell.2015.12.031
Sato-Numata K, Numata T, Inoue R, Sabirov RZ, Okada Y (2017) Distinct contributions of LRRC8A and its paralogs to the VSOR anion channel from those of the ASOR anion channel. Channels 11:167–172. https://doi.org/10.1080/19336950.2016.1230574
Lee CC, Freinkman E, Sabatini DM, Ploegh HL (2014) The protein synthesis inhibitor blasticidin S enters mammalian cells via leucine-rich repeat-containing protein 8D. J Biol Chem 289(24):17124–17131. https://doi.org/10.1074/jbc.M114.571257
Gaitán-Peñas H, Gradogna A, Laparra-Cuervo L, Solsona C, Fernández-Dueñas V, Barrallo-Gimeno A, Ciruela F, Lakadamyali M, Pusch M, Estévez R (2016) Investigation of LRRC8-mediated volume-regulated anion currents in Xenopus oocytes. Biophys J 111(7):1429–1443. https://doi.org/10.1016/j.bpj.2016.08.030
Arellano RO, Miledi R (1995) Functional role of follicular cells in the generation of osmolarity-dependent Cl currents in Xenopus follicles. J Physiol 488(Pt 2):351–357. https://doi.org/10.1113/jphysiol.1995.sp020971
Voets T, Buyse G, Tytgat J, Droogmans G, Eggermont J, Nilius B (1996) The chloride current induced by expression of the protein pICln in Xenopus oocytes differs from the endogenous volume-sensitive chloride current. J Physiol 495(Pt 2):441–447. https://doi.org/10.1113/jphysiol.1996.sp021605
Lutter D, Ullrich F, Lueck JC, Kempa S, Jentsch TJ (2017) Selective transport of neurotransmitters and modulators by distinct volume-regulated LRRC8 anion channels. J Cell Sci 130(6):1122–1133. https://doi.org/10.1242/jcs.196253
Kern DM, Bleier J, Mukherjee S, Hill JM, Kossiakoff AA, Isacoff EY, Brohawn SG (2023) Structural basis for assembly and lipid-mediated gating of LRRC8A: C volume-regulated anion channels. Nat Struct Mol Biol 30(6):841–852. https://doi.org/10.1038/s41594-023-00944-6
Rutz S, Deneka D, Dittmann A, Sawicka M, Dutzler R (2023) Structure of a volume-regulated heteromeric LRRC8A/C channel. Nat Struct Mol Biol 30(1):52–61. https://doi.org/10.1038/s41594-022-00899-0
Pervaiz S, Kopp A, von Kleist L, Stauber T (2019) Absolute protein amounts and relative abundance of volume-regulated anion channel (VRAC) LRRC8 subunits in cells and tissues revealed by quantitative immunoblotting. Int J Mol Sci 20(23):5879. https://doi.org/10.3390/ijms20235879
Okada Y, Oiki S, Hazama A, Morishima S (1998) Criteria for the molecular identification of the volume-sensitive outwardly rectifying Cl– channel. J Gen Physiol 112:365–367. https://doi.org/10.1085/jgp.112.3.365
Abascal F, Zardoya R (2012) LRRC8 proteins share a common ancestor with pannexins, and may form hexameric channels involved in cell-cell communication. BioEssays 34(7):551–560. https://doi.org/10.1002/bies.201100173
Zhou P, Polovitskaya MM, Jentsch TJ (2018) LRRC8 N termini influence pore properties and gating of volume-regulated anion channels (VRACs). J Biol Chem 293(35):13440–13451. https://doi.org/10.1074/jbc.RA118.002853
Ullrich F, Reincke SM, Voss FK, Stauber T, Jentsch TJ (2016) Inactivation and anion selectivity of volume-regulated anion channels (VRACs) depend on C-terminal residues of the first extracellular loop. J Biol Chem 291(33):17040–17048. https://doi.org/10.1074/jbc.M116.739342
Han YE, Kwon J, Won J, An H, Jang MW, Woo J, Lee JS, Park MG, Yoon BE, Lee SE, Hwang EM, Jung JY, Park H, Oh SJ, Lee CJ (2019) Tweety-homolog (Ttyh) family encodes the pore-forming subunits of the swelling-dependent volume-regulated anion channel (VRACswell) in the brain. Exp Neurobiol 28(2):183–215. https://doi.org/10.5607/en.2019.28.2.183
Bae Y, Kim A, Cho C-H, Kim D, Jung H-G, Kim S-S, Yoo J, Park J-Y, Hwang EM (2019) TTYH1 and TTYH2 serve as LRRC8A-independent volume-regulated anion channels in cancer cells. Cells 8(6):562. https://doi.org/10.3390/cells8060562
Suzuki M, Mizuno A (2004) A novel human Cl− channel family related to Drosophila flightless locus. J Biol Chem 279(21):22461–22468. https://doi.org/10.1074/jbc.M313813200
Suzuki M (2006) The Drosophila tweety family: molecular candidates for large-conductance Ca2+-activated Cl– channels. Exp Physiol 91(1):141–147. https://doi.org/10.1113/expphysiol.2005.031773
Li B, Hoel CM, Brohawn SG (2021) Structures of tweety homolog proteins TTYH2 and TTYH3 reveal a Ca2+-dependent switch from intra- to intermembrane dimerization. Nat Commun 12(1):6913. https://doi.org/10.1038/s41467-021-27283-8
Sukalskaia A, Straub MS, Deneka D, Sawicka M, Dutzler R (2021) Cryo-EM structures of the TTYH family reveal a novel architecture for lipid interactions. Nat Commun 12(1):4893. https://doi.org/10.1038/s41467-021-25106-4
Melvin E, Kalaninová Z, Shlush E, Man P, Giladi M, Haitin Y (2022) TTYH family members form tetrameric complexes at the cell membrane. Commun Biol 5(1):886. https://doi.org/10.1038/s42003-022-03862-3
Zhou JJ, Luo Y, Chen SR, Shao JY, Sah R, Pan HL (2020) LRRC8A-dependent volume-regulated anion channels contribute to ischemia-induced brain injury and glutamatergic input to hippocampal neurons. Exp Neurol 332:113391. https://doi.org/10.1016/j.expneurol.2020.113391
Li G, Olson JE (2004) Extracellular ATP activates chloride and taurine conductances in cultured hippocampal neurons. Neurochem Res 29:239–246. https://doi.org/10.1023/b:nere.0000010452.26022.a7
Christensen O (1987) Mediation of cell volume regulation by Ca2+ influx through stretch-activated channels. Nature 330(6143):66–68. https://doi.org/10.1038/330066a0
Okada Y, Hazama A, Yuan W-L (1990) Stretch-induced activation of Ca2+-permeable ion channels is involved in the volume regulation of hypotonically swollen epithelial cells. Neurosci Res 12:S5–S13. https://doi.org/10.1016/0921-8696(90)90004-m
Christensen O, Hoffmann EK (1992) Cell swelling activates K+ and Cl– channels as well as nonselective, stretch-activated cation channels in Ehrlich ascites tumor cells. J Membr Biol 129(1):13–36. https://doi.org/10.1007/BF00232052
Numata T, Shimizu T, Okada Y (2007) TRPM7 is a stretch- and swelling-activated cation channel involved in volume regulation in human epithelial cells. Am J Physiol Cell Physiol 292:C460–C467. https://doi.org/10.1152/ajpcell.00367.2006
Wang J, Morishima S, Okada Y (2003) IK channels are involved in the regulatory volume decrease in human epithelial cells. Am J Physiol Cell Physiol 284:C77–C84. https://doi.org/10.1152/ajpcell.00132.2002
Acknowledgements
The author thanks R.Z. Sabirov, T. Numata, and K. Sato-Numata for their discussion and suggestions for this manuscript. Thanks are also due to all the previous and present coworkers for their nice collaboration, technical assistance, and secretarial help.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
YO conceptualized the study; prepared an original draft; reviewed and edited the manuscript; and read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This article does not contain any studies with human participant or animals performed by any of authors.
Consent for publication
Consent for publication is not required for this article.
Competing interests
The author declares no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Okada, Y. Physiology of the volume-sensitive/regulatory anion channel VSOR/VRAC. Part 1: from its discovery and phenotype characterization to the molecular entity identification. J Physiol Sci 74, 3 (2024). https://doi.org/10.1186/s12576-023-00897-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12576-023-00897-x