
Abstract
Background
Asian rice (Oryza sativa L.) has been a model plant but its cultivation history is inadequately understood, and its origin still under debate. Several enigmas remain, including how this annual crop shifted its growth habit from its perennial ancestor, O. rufipogon, why genetic divergence between indica and japonica appears older than the history of human domestication, and why some domestication genes do not show signals of introgression between subgroups. Addressing these issues may benefit both basic research and rice breeding.
Results
Gene genealogy-based mutation (GGM) analysis shows that history of Asian rice is divided into two phases (Phase I and II) of about equal lengths. Mutations occurred earlier than the partition of indica and japonica to Os genome mark Phase-I period. We diagnosed 91 such mutations among 101 genes sampled across 12 chromosomes of Asian rice and its wild relatives. Positive selection, detected more at 5′ regions than at coding regions of some of the genes, involved 22 loci (e.g., An-1, SH4, Rc, Hd3a, GL3.2, OsMYB3, OsDFR, and OsMYB15), which affected traits from easy harvesting, grain color, flowering time, productivity, to likely taste and tolerance. Phase-I mutations of OsMYB3, OsHd3a and OsDFR were experimentally tested and all caused enhanced functions of the genes in vivo. Phase-II period features separate cultivations, lineage-specific selection, and expanded domestication to more genes. Further genomic analysis, along with phenotypic comparisons, indicates that O. sativa is hybrid progeny of O. rufipogon and O. nivara, inherited slightly more genes of O. rufipogon. Congruently, modern alleles of the sampled genes are approximately 6% ancient, 38% uni-specific, 40% bi-specific (mixed), and 15% new after accumulating significant mutations. Results of sequencing surveys across modern cultivars/landraces indicate locus-specific usages of various alleles while confirming the associated mutations.
Conclusions
Asian rice was initially domesticated as one crop and later separate selection mediated by human resulted in its major subgroups. This history and the hybrid origin well explain previous puzzles. Positive selection, particularly in 5′ regions, was the major force underlying trait domestication. Locus-specific domestication can be characterized and the result may facilitate breeders in developing better rice varieties in future.
Similar content being viewed by others
Background
Early agricultural varieties represent a major milestone of human civilization (Diamond 2002), but phenotypes and genotypes of which are generally little known in the absence of historical records or archeological materials. Inferring historical changes from the standing genetic variation has been challenging, as cumulative results of past mutations, segregation/recombination, drift, and selection can be difficult to tease apart according to the causes. Much effort has been put to describe relationships among homologous haplotypes in phylogenetic trees (Felsenstein 1988) or cladograms using the cladistic principal and statistical tests (Posada et al. 2006; Templeton et al. 1992), but these methods do not show directly the temporal distribution of individual mutations on a gene genealogy, as their major issue is on estimation of degrees of relatedness of biological groups. We argue that knowing mutational distribution on a gene genealogy is more relevant to addressing issues on domestication than knowing degrees of closeness of the groups.
Issues of domestication of a crop typically include its origin, selection, selected traits, and cultivation history, and much of the information has been embedded in the temporal distributions of past mutations across genomic regions. Early mutations, in particular, can indicate the nature of historical selection for crops of long histories. Knowledge of the mutations can also help assess how a subset of genetic variation of the wild predecessor was retained in the successor crops (Doebley et al. 2006). During domestication, phenotypic differentiations are accelerated by human selection and the underlined mutation patterns inevitably left prints in genomes of crops in forms of selective sweep (Olsen et al. 2006; Sweeney et al. 2007), or/and positive selection (Koenig et al. 2013; Rubin et al. 2012), which can be readily detected. Nonetheless, duration of detected selection has rarely been articulated.
Since early mutations must have been frequent enough to transcend domestication history, there is a good chance for them to be recognized in a small number of high-quality genomes. This notion is supported by a recent finding that a high percentage (98%) of common alleles has been shared between wild and domesticated populations of strawberry (Hardigan et al. 2021). With increasingly known genomes of wild relatives of crops, gene genealogies can be constructed at genomic level to help grasp a full picture of past variation. Here we show that early mutations can be diagnosed in Asian rice (Oryza sativa L.).
Asian rice has been well sequenced, but its origin is still in debate after intensive studies in the recent decade (Choi et al. 2017; Civan and Brown 2018; Gross and Zhao 2014; Huang and Han 2016; Huang et al. 2012; Ishikawa et al. 2020). Its closest wild relatives are the tall, aquatic perennial, O. rufipogon Griff. (Or), and the short, drought resistant annual, O. nivara Sharma & Shastry (On). The perennial has a mixed-mating system, and the annual performs self-fertilization and has been considered derived from the perennial (Chang 1976). The wild species can interbreed with each other (Oka 1974) and with Asian rice (Thomson et al. 2003; Mahmoud et al. 2008). Within Asian rice, two traditional subspecies, indica (O. sativa ssp. indica) and japonica (O. sativa ssp. japonica), were further divided into subgroups. Japonica rice (sensu lato) refers to varieties of temperate japonica, tropical japonica, and aromatic subgroups, whereas indica rice (sensu lato) includes those of indica (sensu stricto) and aus subgroups (Garris et al. 2005). The origins of indica and japonica are also in debate. Ecologically, cultivars of japonica can grow in regions broader than those of indica (Khush 1997). The two subspecies differ in physiological traits (Oka and Morishima 1982) and genomic expression (Liu et al. 2007), but culinary differences are the most familiar– rice from indica is less sticky than that of japonica and easily solidifies at a low temperature.
The origin of Asian rice has been hypothesized in various forms. Patterns of phylogeography (Londo et al. 2006; Gutaker et al. 2020) and population structure (Civan et al. 2015) support multiple domestications, but trait analysis (Oka and Morishima 1982), genetic variations in microsatellite patterns (Gao and Innan 2008), phylogenetic analysis (Molina et al. 2011), and genomic variation (Huang et al. 2012) generally support a single domestication, with O. rufipogon frequently taken as the immediate ancestor of O. sativa. Meanwhile, distribution of retroposon p-SINE1 across accessions suggests close associations of O. nivara with O. sativa indica and O. rufipogon with O. sativa japonica (Cheng et al. 2003). Some of the hypotheses can be tested with patterns in mutation distributions. For instance, Huang et al. (2012) proposed that the ancient Asian rice (early japonica) could first come from O. rufipogon and then cross with O. nivara to form indica. If it is true, no genetic signatures of O. nivara are to be found in genomes of japonica. The prediction can be addressed in genomic analysis, but no relevant report has been seen in the literature.
Other puzzles about Asian rice (Sang and Ge 2007) also persist. One is that nuclear divergence between indica and japonica is older than the history of human domestication itself (Ma and Bennetzen 2004). This divergence is also seen in genomes of chloroplasts (Tong et al. 2016) and mitochondria (Cheng et al. 2019). The second one is the lack of introgressions for major domestication genes such as SH4 and Rc (Civan and Brown 2018). The third puzzle is the annual habit of rice, which cannot be generated from a cross between indica and O. rufipogon (Xiong et al. 1999) or between O. nivara and O. rufipogon in F2 generation (Grillo et al. 2009), as the perennial growth is dominant in F1 and appears persistent in F2 generation as well.
In this study, we tried to address the above conflicts by reconstructing mutation-based gene genealogies, focusing on identification of early mutations and evaluation of their functions. The result and associated tests help dissipate the previous dilemma, broaden the scope of domestication genes, and shed light on the early domestication history as well as the later selection in Asian rice. More importantly, the origin of Asian rice is clarified with little ambiguity for the first time.
Results
Early Mutations Show Pervasive Positive Selection During Early Domestication
We took members of two major subgroups (indica and temperate japonica) as representatives of indica and japonica of O. sativa, showing, from five high-quality genomes including those of the closest wild species (O. nivara and O. rufipogon), relationships of orthologs of each sampled gene by two gene genealogies — one based on 5′-sequences and the other on coding-sequences — to capture possibly varying signals of selection across genic regions. A total of 101 genes (Additional file 1: Table S1) sampled in parallel from these genomes across all chromosomes gave rise to 202 gene genealogies (Additional file 2: Fig. S1a–l). The topology of each gene genealogy, based on carefully aligned orthologous sequences, is determined by polymorphisms of oldest mutations including both substitutions and insertion/deletions (indels). A total of 756 mutations are present in the sampled genes of O. sativa (Os mutations), of which 478 dispersed in the 1 Kb 5′ regions and 278 scattered in exon regions of various lengths (Fig. 1a). Significantly, 91 early Os mutations accumulated at 30 loci in the lineage prior to the partition of indica and japonica but after the divergence of O. nivara from O. rufipogon (Additional file 2: Fig. S1, Additional file 3: Table S2), which we call the early phase (Phase I) of domestication.

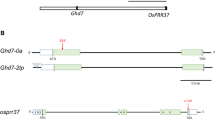
Identification of early mutations in O. sativa. a Manhattan plot of 756 Os mutations on 101 genes distributed across rice chromosomes (chr1–chr12). Details on sampled genes see Additional file 1: Table S1. b–h Gene genealogies of seven genes showing early mutations (in asterisks). Gene genealogies connecting indica (I) and japonica (J) of O. sativa to its wild relatives (O. rufipogon (Or) and O. nivara (On)) are shown in bar graph or tree. A graph of horizontal bars shows orthologs in bars and similarities in similar colors, with substitutions coded by colored sticks (A in green, T in red, C in blue, and G in black, and thick ones for nonsynonymous changes) and indels by arrows. A tree-like gene genealogy also uses colored sticks as above for substitutions but circles for indels. Aligned sticks refer to changes at the same nucleotide site. Number of repeated and adjacent changes is shown before x for simplicity. Allele of indica (I) is shown as the identical sequence among three indica genomes or for genome in the parentheses (Shuhui for Shuhui-498); allele of japonica (J) is from the japonica genome. Only the genic regions having early mutations are shown here
To detect signals of positive selection in 5′ regions during Phase-I domestication, we examined the mutations occurred to Os introns within the same period (Additional file 4: Table S3). The intronic mutational rate (0.56 per kilo-nucleotides per period), taken as a proxy of background mutation rate of the Os genomes, was used to establish the threshold of two mutations over a 1 K-nucleotide 5′ region during the period as the signal of positive selection. Under the criteria, 5′ regions of 18 Os loci were under positive selection (Table 1). For coding regions, 8 genes (4 overlapping with ones involved also in the 5′ selection) displayed positive selection under the stringent test of dN/dS > 1 (Nielsen and Yang 1998) or amino acid changes of more than three-fold the background mutation for cases of dS being zero and/or indels. The analysis generated a conserved estimate of 22 Os loci under early positive selection, which involved at least six categories of traits (Table 1).
Agriculture Traits, Tolerance, and Flowering Time were Targeted During the Early Domestication
The inclusion of known domestication genes (e.g., SH4, Rc), which served as internal references for the detective method above, indicates that the positively selected loci are candidates of domestication genes. Most loci on the list are previously unknown of their roles in domestication. For instance, three genes of the shikimate pathway were under positive selection, including 5′ regions of OsEPSPS, which encodes 5-enolpyruvylshikimate 3-phosphate synthase (Xu et al. 2002; Zhou et al. 2006), and OsDAHPS2 (Fig. 1b), which presumably encodes 3-deoxy-7-phosphoheptulonate synthase, and both 5′ and coding regions of OsMYB15 (Fig. 1c), a homolog of which in Arabidopsis thaliana, AtMYB15, may regulate the shikimate synthesis (Chen et al. 2006) while affecting cold tolerance (Agarwal et al. 2006) and immunity (Chezem et al. 2017). Positively selected mutations at the shikimate loci could have altered the tolerance of early Asian rice. Genes associated with the anthocyanin pathway were also positively selected—one encoding dihydroflavonol 4-reductase named OsDFR (Shih et al. 2008) and the other a regulatory gene OsC1 (Zheng et al. 2019). The two loci influence synthesis of anthocyanins and possibly other classes of flavonoids. Since content change of phenylpropanoids may influence flavors of cooked rice (Bett-Garber et al. 2013), mutations at OsDFR could affect the taste of early Asian rice. Given that shikimate pathway supplies precursors for biosynthesis of aromatic amino acids and flavonoids (reviewed by Maeda and Dudareva (2012)), mutated enzymes of the two pathways above or their altered expressions could bring correlated changes on traits of tolerance, taste, and nutrition of early Asian rice.
Three flowering-time genes, Hd1 in the coding regions (Fig. 1d) and Hd6 (Additional file 2:Fig. S1c) and Hd3a (Additional file 2: Fig. S1f) in their 5′ regions, were under early positive selection. So were genes for awn development, such as An-1 (Fig. 1e) and Rae2 (Additional file 2: Fig. S1h), and for awn-length, such as An-2 (Fig. 1f). Early positive selection detected on the 5′ region of PROG1 (Fig. 1g), which affects plant architecture (Jin et al. 2008), clarified its role in domestication, since previously suspected A → T substitution in coding is shared between O. nivara and O. sativa thus unlikely the target of human selection. Early positive selection also strongly acted on productivity genes such as GL3.2 (Fig. 1h). Only continuous selection on the same materials would result in the fixation and accumulation of the mutations and documented positive selection involved agriculture-related traits, we thus conclude that the positive selection was largely the result of human selection during this early period of domestication.
Early mutations identified for known domestication genes may explain their lack of introgressions between subgroups. SH4 is a key gene for grain shattering (Li et al. 2006b) with the non-shattering mutant allele (sh4) nearly fixed in the modern rice (Zhang et al. 2009). Its lysine-to-asparagine substitution was demonstrated to affect the shattering phenotype (Li et al. 2006b). We show that this substitution occurred during the early domestication (Additional file 2: Fig. S1d), along with six 5′ mutations fixed in the same period. Rc, a domestication gene related to grain-color (Sweeney et al. 2007, 2006), was also intensely selected during the early period in both coding and 5′ regions (Additional file 2: Fig. S1g). Along with other 20 Os loci, a conserved estimate of 54 mutations in 5′ regions and 17 in the coding regions have been accumulated under positive selection during the early period (Table 1). These results are consistent with the existence of one crop and its continuous human selection. Validity of identified mutations was subsequently tested for their functional impacts at some newly recognized loci of domestication.
Positive Selection on Anthocyanin- and Flowering-Related Genes were Experimentally Supported
Mutations at three loci were evaluated for their possible impacts on gene function. The first case was early mutations in the coding sequence of OsMYB3, a gene that may affect color of seeds (Zheng et al. 2021). Two mutations introduced an early stop codon in the last exon (Fig. 2a), making OsMYB3 shorter than otherwise identical OrMYB3 (Fig. 2b). We investigated the capacity of OsMYB3 as a single regulator, using one Kb-long 5′ regions of chalcone-synthase encoding OsCHS and OsDFR (Shih et al. 2008) as baits. The results indicate that OsMYB3 not only trans-activated OsCHS (Fig. 2c) and OsDFR (Fig. 2d) but also gained a regulatory capacity stronger than that of its ancestor, OrMYB3 from O. rufipogon.

Functional analysis of early mutations. a Partial alignment of Oryza MYB3 exon3s. Early mutations are by the asterisks. b Gene genealogy based on the entire coding regions of MYB3. The format follows Fig. 1. c Activation capacities by three MYB3s on the 1 Kb-long Nipponbare promoter of OsCHS (OsCHSpro1K). The promoter activity was measured by the ratio of two fluorescent luciferases (LUC/RUC) in three biological replicates (n = 3, but 5 for the negative control). A significant t-test (P < 0.001) on mean difference was shown by different letters. Data are represented as mean ± SE here and below. d Activations of MYB3s on the 1041 bp-long Nipponbare promoter of OsDFR (OsDFRpro1K). Each test had six biological replicates (four for negative control). Comparisons of averages by different letters were significant (t-test, P < 0.008 in both cases). e Impact of two early mutations (in square; comma refers to an indel) within 5′ region of OsDFR. Each test of transient expression had six biological replicates (four for negative control). Treatment comparisons in different letters were significant (t-tests, all P < 0.003). f Impact of one substitution (by the folded arrow and red letter) in the 5′ UTR of Hd3a. The lower panel shows the synthetic 5′UTR, with partial 5′UTR of OsDFR promoter (upper case and underlined) replaced by the 5′ UTR fragment (lower case) of Hd3a. The upper and italic sequences indicate restriction enzymes between the reporter and the tested 5′UTR region. The right panel shows reporter activities driven by OsC1/OsB2/OsTTG1 (TF complex) and the negative controls (n = 3). The mean difference is supported by a significant t-test (P < 0.001)
The second test was on early mutations in the 5′ region of OsDFR. Two mutations—one indel and one substitution—about 370 bp upstream the translation-starting site set OsDFRpro370 apart from OrDFRpro369 (Fig. 2e). When driven by OsMYB3 in vivo, OsDFRpro370 caused a significantly more transcription of the reporter gene than OrDFRpro369 did (Fig. 2e), indicating enhanced gene expression due to the mutations.
The third test was on an early substitution in 5′ UTR of Hd3a (Fig. 2f), a homolog of Flowering Locus T gene of Arabidopsis (Kojima et al. 2002). The substitution led to 65% more transcripts for OsHd3a than those for OrHd3a under the same regulatory complex of OsC1/OsB2/OsTTG1 (Fig. 2f). Since elevated OsHd3a could promote early flowering in rice (Kojima et al. 2002; Takahashi et al. 2009), the mutant carrier could set mature seeds earlier and became an easy target of human in grain harvesting. All three cases suggest that the detected positive selection on these gene regions were due to their functional impacts, which were noticed and selected by early breeders.
Two-Phased History of Rice Domestication
Phase-II domestication of rice began when different mutations started to accumulate in indica or japonica lineage (branch) on a gene genealogy. Of all positively selected 22 Os loci of Phase I (Table 1), 14 loci were continually under positive selection, sometimes variably, along lineages of indica and japonica in Phase II (Additional file 3: Table S2). For instance, An-1 underwent a stronger selection in japonica than in indica or Phase I (Fig. 1e). Meanwhile, positive selection specific to Phase II was observed at 5′ or coding regions of additional 18 loci of indica or japonica genomes (Table 2). Six of the loci appeared to be associated with yield, including grain-filling- and grain-size-related GS5, grain-length-related GL6, grain-width-related SPL16, grain-number-related NOG1, and yield-related SD1 and Dst. Two additional flowering-time loci, Ehd1 and Hd5, were positively selected. Six loci experienced positive selection only in indica while other seven loci only in japonica (Table 2). The lineage-specific selection is seen also at unreported or less studied loci, from chorismate-mutase-encoding OsCM3 (Additional file 2: Fig. S1a), to alcohol dehydrogenase 2-encoding OsADH2 and unknown (Additional file 2: Fig. S1k). These non-overlapping loci between early and later lineages and between indica and japonica signify that not only selection in phase II differed from that of Phase I and within Phase II, selection also varied between indica and japonica. A major shift of rice cultivation and domestication occurred in Phase II.
The shift of selection is particularly illuminating in the case of flowering-time gene Hd3a. The Phase-I selection occurred only at the 5′ region, which presumably affected only the gene expression (Fig. 2f), but Phase-II selection switched to the coding regions in japonica, leaving indica keeping the protein sequence identical to that of the early rice (Additional file 2: Fig. S1f). An opposite example is Chalk5, which controls chalkiness of the endosperm (Li et al. 2014) and has not changed from Phase I to japonica lineage but accumulated four mutations in the 5′ region and three in the coding regions of the gene in indica (Additional file 2:Fig. S1e).
Besides positive selection, human influence sometimes led to negative selection. One candidate example is the 5′ region of OsC1, which had two early mutations during Phase I but none for Phase II. We examined this peculiar pattern by sequencing 5′ regions of additional cultivars, including Heidao of temperate japonica subgroup and Jixuenuo of indica subgroup, and obtained the same results. It appears that after positive selection of Phase I, further changes in expression of OsC1 were discouraged during Phase-II domestication, which suggests negative selection. Similar deficiency of mutations in the phase II period was seen also at the coding regions of OsMYB3 (Additional file 2: Fig. S1c), OsMYB15 (Additional file 2: Fig. S1d), and OsPROG1 (Additional file 2: Fig. S1g).
The Emergence Time of Japonica and Indica
The annual growth habit and presumably uninterrupted planting history of Asian rice could bring about somewhat steady selection under traditional breeding, as a more or less fixed proportion of the grains were selected each year for the next generation. To estimate roughly the relative durations of Phase I and Phase II, we sampled the Os loci that showed mutations throughout rice history (Additional file 3: Table S2), with an exception of AGO2, a gene affecting grain length and salt tolerance (Yin et al. 2020) but considered an outlier here due to its disproportional influence on the total number of mutations in Phase II. The remaining 29 loci accumulated 87 mutations in the early period and 88 in the lineage of indica and 83 in the lineage of japonica (Additional file 3: Table S2) at 5′ regions. Using our model-based estimation method (see Methods) and taking the sum of mutations observed on the two continuing lineages (after the separation of O. sativa from its closest ancestor on a gene genealogy) as a measure of the entire history of rice domestication, the estimates show that Phase I constitutes approximately half of the domestication history (49.7% by the lineage of indica or 51.2% by the lineage of japonica). Estimates based on only mutations in the coding regions gave similar results, ranging from 47.0 to 49.2%. Consistently, the number of positively selected loci (22) in Phase I was close to those (22 in japonica and 21 in indica) in Phase II, based on our sampled genes. Collectively, the estimates indicate that the history of rice domestication can be divided into Phase I and II almost evenly in time.
O. sativa was Originated from Hybridization Between O. nivara and O. rufipogon
Genome-wide sampling of genealogies indicates that some Os loci are closer to the orthologs of O. nivara and others to those of O. rufipogon (Additional file 2: Fig. S1). Further pursuit of the affinities led to the finding of eight types of Os genes across 12 chromosomes (Fig. 3a; Additional file 5: Table S4). Type 1 designates ancient genes (Fig. 3b) and their slight variants in O. sativa. Nine genes have identical protein sequences and four have nearly unchanged amino-acid sequences among all four Oryza groups. These are the old and conserved genes in the Oryza groups. Type 2 is an On-like gene, as exemplified by PGMp (Fig. 3c), which encodes a plastidic form of phosphoglucomutase. 16 Os regions at 12 Os loci have sequences of 5′ or/and coding regions identical or similar to O. nivara. Type 3 is a discernibly Or-like gene and found in 30 Os regions involving 18 Os loci, as shown by OsGS3 (Fig. 3d), a gene involved in grain size (Fan et al. 2006). Type 4 is for genes of well mixed sequences (Fig. 3e), found at 79 rice regions (involving 56 Os loci) showing both features of O. rufipogon and O. nivara. Genes of Type 5 show associations between Or and japonica orthologs and between On and indica orthologs (Fig. 3f), as displayed at 29 Os regions (involving 20 Os loci) where japonica is closer to O. rufipogon and indica to O. nivara. Type 6 exhibits the reversal of Type 5, showing Or-indica association and On-japonica association instead, as seen at the 5′ regions of two loci, one is a regulatory bHLH gene B2 and the other unknown (Fig. 3g). Type 7 is unique in having separate origins between the 5′ and coding regions, as shown by OsC1, which has an Or-like 5′ region but On-like coding regions (Fig. 3h). Type 8 refers to new variants, when allelic mutations outnumbered five in 5′ region or two in coding region in O. sativa, as seen at OsHd3a and OsDFR of japonica (Fig. 3i). New alleles emerged in 31 cases among sampled genes, involving 24 Os loci (CKX2 (Ashikari et al. 2005), NOG1, CM3, SD1, SK2, DAHPS1, GL3.2, Dst, An-1, B2, AGO2, F3H, GS5, EPSPS, Hd3a, Hd1, GL6, bZIP58, SSH1, RAE2, Ehd1, ADH2) and three unknowns (Additional file 1: Table S1). All of the loci were under positive selection. Together, these Os gene types suggest contributions of both O. rufipogon and O. nivara to the rice genomes, arguing strongly for a hybrid origin of rice. Consistent with the origin, the most frequently observed gene type, the mixed-type, disperses across all rice chromosomes (Additional file 1: Table S1 and Additional file 5: Table S4).

Types of genes in O. sativa. a A flow chart showing the movement of parental genes into genomes of O. sativa and eight gene types in standing rice. The left panel classifies the parental orthologs into Or (O. rufipogon, in yellow) and On (O. nivara, in dark green) and their common ones (in dark red). The middle and right panels show the current types of rice genes observed in genomes of indica (i) and japonica (j), with the integrated types further broken down into four classes, along with new alleles, in the right panel. b A genealogy of Type 1 gene. The example by EPSPS of the shikimate pathway shows no changes. c A genealogy of Type 2 gene. A resemblance to On sequence is seen at PGMp of glycolysis. d A genealogy of Type 3 gene. A resemblance to Or sequence is shown by GS3, a regulator of grain size. e A genealogy of Type 4 gene. The well-mixed type is found in the coding regions of SSY3 in starch synthesis. f Type-5 gene genealogies with Or-j and On-i associations. It appears in both 5′ and coding regions of ACC7, a gene likely involved in ethylene synthesis. g Type-6 gene genealogies with Or-i and On-j associations. 5′ regions of two genes, B2 (a regulator on the anthocyanin pathway) and an unknown gene, show the associations. h Genealogies of a Type 7 gene. Different origins of 5′ and coding regions is at C1. i Genealogies of Type 8 genes. Mutations causing multiple amino-acid changes in Hd3a led to the Nipponbare allele of Hd3a. And an early stop codon led to a new allele of OsDFR in japonica lineage. Gene genealogies follow the format of Fig. 1
Since a hybrid origin is expected to show certain patterns, we examined some expected patterns in Asian rice. The first one is recombinant. Though many historical recombinants have developed mutations to be less recognizable, some accumulated none. We found three Os loci in the sample showing recombinants (Fig. 4a). They are APS1, which encodes a protein resembling the small unit of glucose-1-phosphate adenylyltransferase in starch synthesis of Arabidopsis (Crevillen et al. 2005), iPGAM1, a gene presumably encoding phosphoglycerate mutase, and PK1, a pyruvate-kinase gene (Additional file 2: Fig. S1k). The second pattern is segregation between the parental genomes. Though the immediate results of segregation are no longer available, the associated patterns remain, as shown at loci of Type 5 (japonica-Or and indica-On associations) and Type 6 (japonica-On and indica-Or associations). The two reversal types imply that chromosomal segregation did occur in both directions during the initial synapsis after pairing of the parental genomes, but one type (Type 5) became more common. A reason could be segregation distortion, which was previously reported on hybrid chromosomes of O. nivara and O. rufipogon created in the laboratory (Grillo et al. 2009). The third pattern is functional complementation, by which we refer to the presence of genes from both ancestors in forming a genetic or biochemical unit. For instance, a metabolic pathway in O. sativa may contain enzymes encoded by genes from either O. nivara or O. rufipogon, as shown in the shikimate pathway and the anthocyanin pathway (Fig. 4b).

The hybrid origin of O. sativa. a Gene genealogies showing two recombinant alleles identified in O. sativa. One is japonica allele at OsAPS1, and the other indica allele at iPGAM1. Both can be generated by one crossing-over event during synapsis between chromatids of parental genomes O. rufipogon (Or) and O. nivara (On), as shown to the right. b Origins of structural genes of two metabolic pathways in rice. c The distribution of plant height in O. sativa. Plant height was measured at harvest on F2 progeny (N = 305) of a cross between Heidao as pollen receiver and Jixuenuo as pollen donor. See Additional file 6: Data 1 for detail. d Variation of the anther length just before dispersal of pollen. A total of 65 plants from the F2 population, one japonica, and three indica cultivars were sampled in two days at noon when flowers (N = 466) were open. See Additional file 7: Data 2 for detail. e Degree of panicle exsertion in O. sativa. A random sample of 232 plants from 46 accessions of indica, japonica, and their crossed progeny were measured at 696 panicles for stem length beyond the sheath of flag leaf. Details see Additional file 7: Data 2. f A brief summary of rice domestication. The green rectangle indicates the period when rice was selected as one crop. The colored arrows show contributions of parental genomes, and the bar of the same color indicates slight more contribution of the species to the subgroup. The dots represent other subgroups
The hybrid hypothesis also predicts intermediate phenotypes of rice between their parental ancestors. Certain morphological traits vary largely between O. rufipogon and O. nivara (Grillo et al. 2009; Xiong et al. 1999), allowing a test on these traits. Given that japonica genome resembles more Or genome while indica genome is closer to On genome, we created crossed progeny between Heidao (japonica) and Jixuenuo (indica). Plant height in the F2 generation was more variable than those of their parental plants, with the extremes comparable to heights of their annual and perennial ancestors (Fig. 4c). Similarly, anther length (Fig. 4d) and degree of panicle exsertion (Fig. 4e) measured in the filial generations of the cross and cultivars of rice indicated that they were intermediate between those of O. rufipogon and O. nivara and close to the hybrids of O. rufipogon and O. nivara created artificially (Grillo et al. 2009). All evidence thus far concurs with the hybrid origin of cultivated rice and two-stage domestication (Fig. 4f).
Co-existence of Ancestral and Derived Alleles in Current Rice Genomes
The hybrid origin and long history of domestication of Asian rice may create locus-specific assortments of alleles. Can Os loci that harbored alleles of different origins use them effectively in modern cultivars? To seek the relevant evidence while evaluating precisions of mapped mutations, we sequenced a subset of the sampled loci across diverse cultivars and landraces of rice using Sanger′s method (Additional file 8: Table S5). At OsAPS1 locus, the recombinant Nipponbare allele (44%) coexist with the indica allele (32%), which is identical to OnAPS1 in 5′ and coding regions (Table 3). At OsSSY3 locus (Additional file 9: Table S6), however, the most abundant allele (OsSSY3_b, 59%) is a derived one from O. nivara, with ancestral alleles of O. rufipogon (6%) and O. nivara (1%) rarely observed. For OsPGMp (Additional file 10: Table S7), a glycolysis gene, the ancestral OnPGMp is the most frequent allele (44%), with O. rufipogon contributing little to this locus. This pattern is consistent with its uni-specific (On) origin based on gene genealogy (Additional file 2: Fig. S1j). At OsCKX2 locus, which influences grain production (Ashikari et al. 2005), newly evolved OsCKX2_a (Nipponbare allele, 41%) and OsCKX2_b (indica allele, 40%) have taken the central stage (Additional file 11: Table S8). These data not only confirmed the accuracy of mapped mutations (42 checked and all confirmed) but also indicated that the gene pool of modern rice primarily consists of recombinant, uni-specific, and new alleles.
In addition to data collected here, we consulted previously reported data at NCBI. From the mutations listed in Table 1, we randomly picked 12 early and 3 later mutations at six loci and compared them against multiple rice varieties reported (Additional file 12: Table S9). The independent data sets largely agree with our identification of the early and later mutations. We then test whether or not the gene types identified here are specific to one subgroup, taking the 5′ region of Hd3a (identified as mixed-type above) as an example. Independent data sets from NCBI indicate that the mixed-type is not specific to a subgroup but a feature of the locus (Additional file 13: Table S10).
Discussion
The GGM analysis above is simple but highly effective for sorting out fixed early polymorphisms, providing a temporal framework for lineage-specific mutations and allowing detection of selection and functional tests of specific mutations. Though early-stage genetic modifications can also be gathered from archeological remains, as shown for maize (Jaenicke-Despres et al. 2003) and grapes (Ramos-Madrigal et al. 2019), these materials are generally scarce. For O. sativa, our approach revealed its origin, along with a better understanding of how genetic diversity has accumulated in rice, both of which were thorny issues previously.
The Hybrid Origin and Subsequent Selection of Asian Rice Explain the Previous Puzzles
Since O. nivara can easily contribute pollen and O. rufipogon can accept outside pollen (Vaughan et al. 2008), a natural hybridization event between O. rufipogon as maternal plant and O. nivara as paternal plant was the most likely scenario for the origin of Asian rice. This scenario is compatible with at least three known patterns—its typically watery environment of growth, chloroplast genomes of O. rufipogon being basal to those of cultivated rice (Moner et al. 2018), and lack of trait segregation of annual growth when O. sativa was crossed with O. nivara (Li et al. 2006a). Once O. nivara passed down its selfing trait to early rice, further crossing of the progeny as maternal plant with O. nivara became less likely. Meanwhile, grain-harvesting and subsequent cultivation by human also prevented crossing of early rice with wild O. rufipogon. The hybrid origin gave the early rice rich genetic diversity through recombination and segregation. New mutations occurred to inherited alleles and their recombinants were subject to selection by human and environment over the long history of cultivation, which transformed Asian rice, creating its numerous landraces and cultivars along the way.
From the surveyed 101 genes, 12 On-like loci and 18 Or-like loci were identified (Additional file 1: Table S1). The compositional bias of Os genomes toward O. rufipogon could be caused by selection favoring Or genes in the filial generations, as they supposedly grew in the maternal habitat of O. rufipogon after the initial hybridization. This pattern may explain the higher average genetic similarity between O. rufipogon and O. sativa than one between O. nivara and O. sativa. More frequent Type-5 loci (20 sampled) than Type-6 loci (2 sampled) in genomes of Asian rice suggest easily detected japonica-Or and indica-On associations, as noticed (Cheng et al. 2003). Consequently, part of genetic differentiation between O. rufipogon and O. nivara at Type-5 loci was carried over to subgroups of O. sativa, causing the divergence between indica and japonica older than the domestication history.
The hybrid origin of Asian rice can explain both trait segregation and lack of trait segregation in the literature. In the former category, traits such as cold resistance and awn length are bi-specifically determined in rice (as shown by OsMYB15 and OsAn-2, respectively), which permits segregation of a japonica morph in progeny of crosses between an indica rice and O. nivara (Oka and Morishima 1982). In the latter category, traits such as selfing and annual growth of rice were supposedly inherited from O. nivara, thus no segregation of the traits would follow in F2 generation of a cross between an indica cultivar and O. nivara (Li et al. 2006a). Though perennial habit of O. rufipogon is dominant to the annual habit of O. nivara in the first two generations (Grillo et al. 2009), annual growth could appear in further generations and be selected by early breeders. Selected early mutations and their hitchhiking sites necessarily transcended the early rice stock, leaving no signals of introgression at loci of SH4, Rc, and other 20 Os loci (Table 1) during the early domestication.
If the hybrid origin of rice is considered hypothesis A here, it has gained all support so far. An alternative and broadly referenced hypothesis (B) by Huang et al. (2012) finds no support in our GGM analysis (Additional file 14: Fig. S2). Under the hypothesis B, none of On alleles or recombinant between On and Or alleles are expected in japonica genomes, and significantly higher genetic diversity is expected to be seen in genomes of indica than in those of japonica due to the proposed hybrid origin of indica. These predictions, however, are rejected by genomic data (Additional file 14: Fig. S2). Furthermore, the gene genealogies of numerous genomic regions, as shown by those of the coding regions of C1, Hd3a, and DFR (Fig. 3h, i), would have been impossible under the hypothesis B, as the hypothesis denies the presence of O. nivara mutations in genomes of japonica (Additional file 2: Fig. S1a–l). Since neither indica nor japonica displays strong evidence for having undergone a longer domestication history than the other, the mutation distribution of O. sativa supports little of hypothesis-B-like scenarios, i.e. one subspecies was domesticated first and then crossed with a wild species to give rise to the other subspecies.
Unknown History of Rice Domestication
The single domestication of early rice matches the archeological evidence showing that rice collected from 6600 to 6900 years ago at a south site (Tianluoshan) of the Yangtze River had a gradual increase of non-shattering grains from 27 to 39% (Fuller et al. 2009). If the domestication started about eight to nine thousand years ago (Gross and Zhao 2014), this documented archeological period fell in Phase I domestication when OsSH4 was still under active selection. Rice in this period had undergone much domestication but still differed from later rice. As mutations were gradually accumulated over time, the current sh4 allele is evidently not one that contributed to non-shattering grains thousands of years ago.
Detected positive selection and functional validations of some of the early mutations suggest that the early human selection on Asian rice had concentrated on traits of not only grain shattering, color, or awn length, but also flowering, yield, and likely rice taste and tolerance. This extensive range of trait modifications by human selection had likely made the early rice an attractive crop by the end of Phase I. Then, the split domestication came about 4–5 millennia ago, which could be either based on phenotypic differences (including taste), if distorted segregation could lead to some progeny having more genes from O. nivara (proto-indica) and others having more genes from O. rufipogon (proto-japonica), or phenotype-irrelevant, since different environments and/or selection criteria could also lead to the differentiation of subgroups. Under either scenario, separate cultivations of Asian rice were inevitably an event of historical importance, as it involved divided labor and possibly people(s), though the cause is unclear.
Following the historical event, selections over the second stage of domestication were largely isolated from each other, judging from fixation of lineage-specific mutations in genomes of nucleus and organelles, causing phylogenetic divergence between indica and japonica within Asian rice (Tong et al. 2016; Choi et al. 2017; Cheng et al. 2019). This period lasted until the modern age, and recent hybridizations between indica and japonica by human, though increasing quickly, are still relatively rare among rice varieties and can hardly alter the historical patterns presented here.
Genomic Basis of Selected Phenotypes
Since a significant portion of positive selection was operated on 5′ mutations, phenotypic alterations during rice domestication were more likely through adjusting gene expression than by modifying protein. For instance, transcript enhancements of Hd3a and OsDFR were positively selected during the early domestication (Table1; Fig. 2). This domestication history of Hd3a, along with that of Hd1, expands a recent report of their positive selections in japonica cultivars (Liu et al. 2021a). In another example, positively selected four 5′ mutations of Chalk5 happened only in Phase II in indica, two (thymines) of which were previously demonstrated to abate expression levels of Chalk5 (Li et al. 2014). We show that, as a Type-5 gene, Chalk5 inherited different alleles from O. nivara and O. rufipogon and passed them down to indica and japonica lineages, respectively (Additional file 2: Fig. S1e). Whether or not the ancestral differences also contributed to texture dissimilarity between indica and japonica requires additional investigations.
Certain patterns have been made clear for known Os genes—loci influencing productivity and under positive selection increased from four in Phase I to ten in Phase II. Meanwhile, positive selection was expanded from three to five loci for flowering time. These patterns reflect increased breeding effort for high yield and broader regions of rice planting in Phase II, a trend continued until this date. Consistently, domestication has promoted emergence of new alleles at Os loci affected.
Promises and Caveats
The major advantage of obtaining a mutation-based gene genealogy is its robustness to species boundaries, selection, recombination, and not much influenced by random events such as bottleneck or incomplete lineage sorting, which make the approach particularly fit for understanding domestication of crops. Depending on availability of genomes from various lineages, mutations can be allocated on inner branches of a gene-genealogy, providing information on largely fixed mutations at earlier stages for an evolutionarily recent process of differentiation (e.g. domestication).
Cautions need to be exercised, however, in applying the method. One is the lack of resolution for detecting the temporal order of single mutations within a branch when multiple mutations occurred within the period. The second is that detection of positive selection based on mutation rate of introns can be highly conservative when introns themselves are influenced by selective processes, leaving human selection at a moderate or low intensity less likely to be detected using the criteria given here. One can either use a less stringent threshold or switch to other appropriate base-line for comparison. Due to different criteria used for detecting positive selection across regions, an inference about inter-region comparison of positive selection should be taken as a hypothesis and subject to more evidence. The third caution is on the evaluation of new mutations in the focal group. Only substitutions or/and indels present in crop (offpring) genomes and absent in relatives′ (ancestral) genomes are classified as new mutations, the genomes of wild relatives thus serve as references for each monomorphic site in crops, leaving polymorphic sites between wild relatives only used for inferring the topology of a gene genealogy. For evaluating fixed polymorphisms (McDonald and Kreitman 1991) early in domestication, the minimum sample size for our method to be informative consists of two high-quality genomes representing diverse groups of a crop and two high-quality genomes from its wild relatives.
If the goal of an investigation is to know historical events/patterns, a small to moderate number of high-quality and independent genomes are often sufficient. If a sample of all mutations follows the binomial distribution and everything else is equal, two independent genomes, when being the same at a given nucleotide site, may give 75% assurance for identifying a monomorphic site and four such genomes raise the probability to 94%. The probability can be much higher if the substitution models are taken into account. Increasing genome number may, of course, lead to more mutations identified, but transient mutations (ones of low frequencies) tend to rise disproportionally, too, which may inflate end-branch length when the base line of comparison is the historical and fixed mutation. To illustrate this point, we compared the genome of Kitaake (PRJNA448171), another well-sequenced japonica, against our earlier classification of mutations of the 101 genes. The Kitaake genome differs from the Nipponbare genome at 43 of the 202 regions but agrees with all of the early fixed mutations except one indel (1/91) and most (652/665) of the later mutations (Additional file 15: Table S11). The disagreements (< 2%) have a negligible effect on our analysis. Among the 43 regions, 3 in 2 genes are identical to those of 9311, suggesting some level of genetic introgression; one region (the coding of RAE2) is a recombinant between indica and japonica, and the rest appears largely transient.
The biggest caveat, nonetheless, comes from sequencing errors or missing sites, which can be corrected by re-sequencing critical sites if necessary. Clearly, genomes of uncertain alignments are not fit for the method. This makes the GGM method particularly fit for studies on a process of recent differentiation.
Conclusions
We show that domestication history of Asian rice can be divided into two phases of similar lengths. The rice in the first or early phase was a single crop and the rice in the later or divergent phase was represented by two subspecies, referred to as indica and japonica. Early mutations of domestication can now be delineated by stratifying mutations on an inner branch of gene genealogy, enabling researchers to evaluate impacts of specific mutations on phenotypic traits over a historical period. This has led to better understanding of how selection has transformed Asian rice and broadening of the genetic basis for future breeding. One message brought by the history of domestication is that positive selection at 5′ regions may occur more frequently than one at coding regions. This selective strategy is mild to plant and likely healthy to the Os genomes as well. Knowing the origin of allelic diversity and history of human selection helps reconstruct past achievements of agriculture, while assisting better varieties to be created in future.
Methods
Gene Sampling and Alignment of Orthologs
Sampled genes (Additional file 1: Table S1) should cover all chromosomes in order to capture genome-wide patterns, though the proportions of unknown and known genes can be arbitrary. Known genes are essential, as they allow inferences on mutations and traits of human selection. Including unknown genes is to make sure that no great bias is introduced by sampling known genes. Information of selection can be gathered regardless of the status of gene.
For the purpose of identifying fixed mutations within a domesticated species, diverse genomes should be included, along with genomes of wild relative(s). The specific period of domestication is defined by selected genomes, thus sampling of genomes are not totally random. For instance, if the early period before subgroups is targeted, samples from subgroups are needed. A minimum of two diverse genetic groups per domesticate should be included in order to provide a topology needed for inferring early mutations. For rice, high-quality nuclear genomes were chosen at the NCBI web site (https://www.ncbi.nlm.nih.gov/, accessed 2019.4.21–2021.2.18), including O. rufipogon (PRJEB4137, strain W1943), O. nivara (PRJNA48107, strain IRGC 100897), one cultivar (PRJDB1747, Nipponbare) of japonica and two cultivars, Shuhui (PRJNA302543, Shuhui-498) and 9311(PRJNA427873), of indica. A third indica cultivar Bio226 (PRJNA285384, RP Bio-226) was consulted only in the case of insufficient information in the previous two genomes, as its coverage (~ 20 ×) was relatively low. For Asian rice, a total of 101 loci were sampled to represent 12 chromosomes, genetic pathways of interest, and ones related to agricultural traits. Following BLAST searches of the model genomes using the reference genes as baits, we obtained homologous sequences and identified the best matches as candidate orthologs. The homology of the candidate sequences was further examined for chromosomal location, gene structure, and similarity to the reported reference to ensure orthologs of one another. Two genealogies, one based on the 5′ regions (5′ genealogy) and the other on the entire coding regions (coding genealogy), were then built. This dual representation of a gene is based on their different functions, the former influencing protein quantity and the latter affecting protein function (quality).
Fragments of genomic sequences covering the whole gene from 1 Kb 5′ upstream to about 200 bp of 3′ downstream were sliced in Sequencher (version 4.10.1, Gene Codes) or directly from the genome using online tools at the NCBI website. The 1 Kb 5′ regions are generally well aligned, but can be shorter due to difficulty in alignments in some cases. We aligned sequences in MegAlign (version 8+, DNASTAR) using ClustalW and adjusted the alignment manually when necessary. For many genes of small variation, manual alignments are sufficient.
Construction of Gene Genealogy and Identification of Early Mutations
A gene genealogy here can be viewed as a graphic translation of an unambiguous alignment of DNA sequences for the purposes of inferring mutation distribution. The oldest mutations on the gene genealogy are those polymorphic between O. rufipogon and O. nivara, which are mainly for setting the topology of a gene genealogy. The targeted mutations are those within lineages of O. sativa. We mapped these mutations in two interchangeable forms of genealogy. The traditional tree-like gene genealogy shows better the temporal distribution of mutations and suits for small numbers of mutations such as those of coding sequences. A gene genealogy in bar graph displays better the spatial distributions of mutations and fits for a large number of mutations as typically seen at 5′ region. All genealogies had at least four terminal units connecting orthologs of O. rufipogon, O. nivara, O. sativa japonica, and O. sativa indica. As O. nivara was considered derived from O. rufipogon, we labeled nonsynonymous changes in the lineage of O. nivara when the situation was permitted. This labeling is tentative and has no direct impact on the subsequent analysis here. The second category of mutations includes ones shared between japonica and indica but absent in both O. rufipogon and O. nivara, which were the early mutations. Mutations emerged after the split of japonica and indica within O. sativa belong to the third category, which may include a small quantity of transient ones by chance. Recurrent mutations are unlikely due to the shallow history of domestication, thus not considered.
Detection of Positive Selection
For the 5′ gene genealogy, an intron-based test of positive selection was developed. Relative to 5′ regions, introns seldom influence gene function and are less likely subject to selection. To estimate the background mutation rates (u, in the unit of mutations per 1 Kb nucleotides over the period (lineage) considered) of the Oryza genomes, we collected mutations observed in a random sample of introns located within the sampled genes and longer than 800 bp. The purpose of sampling long introns was to make them more comparable to the lengths of 5′ region and average coding regions. Naturally occurring mutations in Os introns were averaged as a proxy for the background mutations (the higher of the two estimates between indica and japonica lineages was taken for Phase II). Since positive selection can elevate mutation rate, the cut-off value for the signal of positive selection in 5′ regions was set at a stringent level of more than three times the background-level mutation rate (3u), which includes both substitutions and indels.
For the coding gene genealogy, codon-based detection of positive selection (Nielsen and Yang 1998; Yang et al. 2000) can be readily applied to cases when substitutions are sufficient to permit the test. The average rate of synonymous substitutions per site (dS) and that of nonsynonymous substitutions per site (dN) can be computed in DNAsp (Rozas et al. 2017) or PAML (Yang 2007). We applied the stringent condition of dN/dS > 1 for positive selection (Nielsen and Yang 1998) on the japonica and indica lineages. Because mutation numbers on the early lineage are often low and dS can be zero, we applied a simple test using a cut-off value of two amino-acid changes per 1 Kb sequence as the signal of positive selection in these cases. The test is more stringent than that of dN/dS, as it is equivalent to regarding all sites as nonsynonymous.
Estimation of the Relative Duration of a Recent Lineage
Because one plant of the Oryza groups can generate a reasonable gamete pool, genetic drift is expected to be small. Only mutations and selection under the infinite site model are considered for Asian rice. Let the background mutation rate be v per generation, effective size of rice population be Ne, neutral mutations are expected to occur in a quantity of 2vNe per generation. If mutations were under selection at intensity s, their fixation probability (P) would be (1−e−4sqNe)/(1−e−4sNe) according to Kimura (1962), where q is the initial frequency of the mutation. Let q = 1/(2 N), here N denotes the census population size, P = (1−e−(2sNe)/N)/(1−e−4sNe), which can be approximated by 2 s when s is small and Ne equals N (Li 1997). However, since Ne in a selfing population is reduced to 1/2 of N (Nordborg and Donnelly 1997), P ≈ 1−e−s in Asian rice. It suggests that P is largely s-dependent. The expected number (m) of fixed mutations per lineage can be approximated by 2vNePT, where T denotes the total generations spent in a lineage. For the early lineage where mutations were expected to accumulate in the quantity of me in Te generations, Te can be computed by me/(2vNeP). Similarly, for the lineage of indica, where mi mutations are expected to be fixed, Ti (time in indica lineage) can be computed by mi/(2vNeP). The whole history of rice (Tw) can thus be estimated up by (Te + Ti) or (Te + Tj), where Tj refers to the generation time in japonica lineage. The relative duration of Te becomes Te/Tw, which can be measured simply by me/(mi + me) or me/(mj + me), where mj is the number of expected mutations fixed in japonica lineage. By letting the expected mutation numbers equal to the observed ones (assuming transient mutations are rare in the samples and negligible), the relative duration of the early lineage (Te/Tw) can be estimated for rice. We used only loci that accumulated mutations throughout domestication for the inference to reduce bias of estimation.
Nucleotide Extractions and cDNA Amplifications
Total RNAs were obtained from fresh grains collected in a Paddy field using TRNzol total RNA Reagent (Tiangen, Beijing, China) following the method of Wang et al. (2012). About 2 µg RNAs was then used for the cDNA synthesis via TIANScript II RT Kit (Tiangen, Beijing, China). For each PCR reaction, about 100–500 ng cDNA was taken as the template to amplify targeted genes with specific primers. Total genomic DNA was gathered from young leaves freshly collected from the field by the CTAB method.
Constructs Used in Functional Experiments
To examine whether OsMYB3 was able to activate OsDFR in vivo, we constructed CaMV 35S-driven-transcription factor (TF) vector pOsMYB3 by replacing the coding sequence of pJIT163 (Guerineau et al. 1992) with that of OsMYB3 from cDNAs, which was prepared from RNAs extracted from developing grains of Heidao. On the same backbone, we built reporter vector pOsDFRpro1K from 1041-bp long 5′ region of OsDFR, which was amplified from leaf gDNA of Heidao using primers OsDFRpro0921-F and OsDFRpro0921-R (Additional file 16: Table S12). The second reporter vector pOsCHSpro1K was based on 1 Kb-long 5′ sequence of OsCHS1 from Heidao′s gDNA using amplification primers (OsCHSpro-EcoRIF and OsCHSpro-MluIR).
Further TF-vectors were built from the same backbone above using the coding sequences of OrMYB3 or OnMYB3. OrMYB3 were adapted from OsMYB3 by introducing needed changes with linkers (Orlink1r1, Orlink1r2, and Orlink2f) and primers (OsTT2-163-SalIF and OsTT2-163-EcoRI_R). OnMYB3 was similarly obtained with linkers (OnTT2cag-f, OnTT2cag-r, OnTT2tlnk-r, OnTT2tlnk-f, Onlink1r, and Onlink2) and primers (OnTT2-163TF-F and OnTT2-163TF-R).
Mutation-specific reporters were constructed for pOsDFRpro-370 from 370 bp 3′ end of pOsDFRpro1K using primers (OsDFRp370f and OsDFRpro0921-R) and for pOrDFRpro-369, which hosted that same 5′ sequences except at the two mutated sites, using primers (OrDFRp369f and OsDFRpro0921-R).
For 5′ UTR mutation in Hd3a, a single substitution (T in Or → A in Os) at the TATA site of OsHd3a was designed into two reporter vectors based on pOsDFRpro1K by replacing the local 5′UTR of OsDFR with that of OsHd3a or OrHd3a. To build the nested vector pOsHd3a, we took pOsDFRpro1K as the template and two linkers (OsHd3alinker1 and Hd3alinker2) and primers OsDFRpro0921-F and OsDFRpro0921-R as interacting components in PCR reactions. The desired 5′ sequence was inserted back to pJIT163 at enzyme cut sites. Similarly, pOrHd3a was created from two linkers (orHd3alinker1and Hd3alinker2) and the same primers. With pJIT163, we further synthesized three vectors from the coding sequences of OsC1, OsB2, and OsTTG1. OsC1 and OsTTG1 were cloned from the cDNAs of Heidao and OsB2 from that of Jixuenuo. All primers were synthesized by Sangon (Shanghai, China). Constructs were all sequenced to ensure correct alignments and sequences.
Transient Expressions
We used a yellow-husked indica material (B16-44) to prepare the protoplasts for the following tests of transient gene expression using fluorescent firefly luciferase (LUC) as the reporter gene. Protoplast isolation and transformation followed the previous protocol of Yoo et al. (Yoo et al. 2007) but the incubation temperature used 28 °C. The two constructs, pOsMYB3 and pOsDFRpro1K (2 μg of each), were co-introduced into rice protoplasts, along with 0.2 μg renilla-luciferase (RUC)-expressing vector pRUC as internal control. After incubating at 28 °C for 16 h, the culture was analyzed via the dual luciferase reporter assay system (Promega, Madison, WI. USA), following the manufactory′s protocol. The experiment was repeated with pOsMYB3 and pOsCHSpro1K using the same routine.
After knowing the activation capacity of OsMYB3, we compared it with OnMYB3 and OrMYB3 in their activation capacities on the same promoter of pOsDFRpro1K. For two promoter mutations within 370 bp 5′ region of OsDFR, we compared their activation levels measured after co-infections of pOsDFRpro-370 (or pOrDFRpro-369) and TF-vector pOsMYB3 (2 μg of each), along with the internal control as above.
Effect of the 5′UTR mutation in Hd3a on gene expression was compared between two reporters, pOsHd3a and pOrHd3a, by subjecting them to the regulatory complex consisted of products from three regulatory vectors. The experimental procedure followed the same protocol mentioned above.
Morphological Traits
Besides nuclear data, morphological traits were collected in two field experiments and compared to reported values on O. rufipogon (Xiong et al. 1999), O. nivara (Grillo et al. 2009; Li et al. 2006a), and their hybrids (Grillo et al. 2009). In the first experiment, we measured plant height in F2 generation of a cross between Heidao of japonica and Jixuenuo of indica. Mature plants were harvested just above ground and hang upside down to allow measuring of height using a meter ruler (1 mm). In the second experiment, length of an anther was measured on unopened pollen sacs of freshly opened flowers by a caliper (0.01 mm) on 26 accessions of the filial generation of the cross. Toward grain maturation, 48 accessions were sampled for degree of panicle exsertion, measured as the distance from the basal node of a panicle to the top of leaf sheath surrounding the panicle by a ruler (1 mm).
Sequencing Surveys
Current allelic diversity was surveyed in polymerase chain reactions using leaf gDNAs and gene-specific primers (Additional file 16: Table S12). The gDNAs were extracted from seedlings of rice cultivars with the conventional CTAB method. The 5′ region of C1 gene was similarly amplified from leaf gDNA by specific primers (Additional file 16: Table S12). All sequences were edited against valid chromatograms when necessary.
Availability of Data and Materials
All data generated or analyzed during this study are included in this publication [and its Additional files]. The experimental materials are available from the corresponding author on reasonable request.
Abbreviations
- B16-44:
-
An accession from interbreeding of two white-rice cultivars
- d S :
-
The average rate of synonymous substitutions per site
- d N :
-
The average rate of nonsynonymous substitutions per site
- GGM:
-
Gene genealogy-based mutation analysis
- Kb:
-
Kilobase
- m:
-
The expected number of fixed mutations in a lineage
- LUC:
-
Luciferase of firefly
- N:
-
Population size by census
- N e :
-
Effective population size
- NCBI:
-
National Center for Biotechnology Information, National Institute of Health, USA
- Or :
-
Oryza rufipogon
- On :
-
Oryza nivara
- Os :
-
Oryza sativa
- P:
-
Fixation probability
- RUC:
-
Renilla luciferase
- s :
-
Selection intensity
- T:
-
Total generations spent in a lineage
- TF:
-
Transcription factor
- 5′ UTR:
-
Untranslated segment at the 5′ region of a gene
- u :
-
Background mutation rate over a lineage (or period)
- v :
-
Background mutation rate per generation
References
Agarwal M, Hao YJ, Kapoor A, Dong CH, Fujii H, Zheng XW, Zhu JK (2006) A R2R3 type MYB transcription factor is involved in the cold regulation of CBF genes and in acquired freezing tolerance. J Biol Chem 281:37636–37645
Ashikari M, Sasaki A, Ueguchi-Tanaka M, Itoh H, Nishimura A, Datta S, Ishiyama K, Saito T, Kobayashi M, Khush GS, Kitano H, Matsuoka M (2002) Loss-of-function of a rice gibberellin biosynthetic gene, GA20 oxidase (GA20ox-2), led to the rice “green revolution.” Breed Sci 52:143–150
Ashikari M, Sakakibara H, Lin SY, Yamamoto T, Takashi T, Nishimura A, Angeles ER, Qian Q, Kitano H, Matsuoka M (2005) Cytokinin oxidase regulates rice grain production. Science 309:741–745
Bett-Garber KL, Lea JM, McClung AM, Chen M-H (2013) Correlation of sensory, cooking, physical, and chemical properties of whole grain rice with diverse bran color. Cereal Chem 90:521–528
Chang TT (1976) Origin, evolution, cultivation, dissemination, and diversification of Asian and African rices. Euphytica 25:425–441
Chen YH, Zhang XB, Wu W, Chen ZL, Gu HY, Qu LJ (2006) Overexpression of the wounding-responsive gene AtMYB15 activates the shikimate pathway in Arabidopsis. J Integr Plant Biol 48:1084–1095
Cheng CY, Motohashi R, Tsuchimoto S, Fukuta Y, Ohtsubo H, Ohtsubo E (2003) Polyphyletic origin of cultivated rice: Based on the interspersion pattern of SINEs. Mol Biol Evol 20:67–75
Cheng L, Kim KW, Park YJ (2019) Evidence for selection events during domestication by extensive mitochondrial genome analysis between japonica and indica in cultivated rice. Sci Rep 9:9
Chezem WR, Memon A, Li FS, Weng JK, Clay NK (2017) SG2-type R2R3-MYB transcription factor MYB15 controls defense-induced lignification and basal immunity in Arabidopsis. Plant Cell 29:1907–1926
Choi JY, Platts AE, Fuller DQ, Hsing Y-I, Wing RA, Purugganan MD (2017) The rice paradox: multiple origins but single domestication in Asian rice. Mol Biol Evol 34:969–979
Civan P, Brown TA (2018) Role of genetic introgression during the evolution of cultivated rice (Oryza sativa L.). BMC Evol Biol 18:57
Civan P, Craig H, Cox CJ, Brown TA (2015) Three geographically separate domestications of Asian rice. Nat Plants 1:15164
Crevillen P, Ventriglia T, Pinto F, Orea A, Merida A, Romero JM (2005) Differential pattern of expression and sugar regulation of Arabidopsis thaliana ADP-glucose pyrophosphorylase-encoding genes. J Biol Chem 280:8143–8149
Diamond J (2002) Evolution, consequences and future of plant and animal domestication. Nature 418:700–707
Doebley JF, Gaut BS, Smith BD (2006) The molecular genetics of crop domestication. Cell 127:1309–1321
Doi K, Izawa T, Fuse T, Yamanouchi U, Kubo T, Shimatani Z, Yano M, Yoshimura A (2004) Ehd1, a B-type response regulator in rice, confers short-day promotion of flowering and controls FT-Iike gene expression independently of Hd1. Genes Dev 18:926–936
Fan CH, Xing YZ, Mao HL, Lu TT, Han B, Xu CG, Li XH, Zhang QF (2006) GS3, a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theor Appl Genet 112:1164–1171
Felsenstein J (1988) Phylogenies from molecular sequences - inference and reliability. Annu Rev Genet 22:521–565
Fujino K, Yamanouchi U, Yano M (2013) Roles of the Hd5 gene controlling heading date for adaptation to the northern limits of rice cultivation. Theor Appl Genet 126:611–618
Fuller DQ, Qin L, Zheng YF, Zhao ZJ, Chen X, Hosoya LA, Sun GP (2009) The domestication process and domestication rate in rice: spikelet bases from the lower Yangtze. Science 323:1607–1610
Furukawa T, Maekawa M, Oki T, Suda I, Iida S, Shimada H, Takamure I, Kadowaki KI (2007) The Rc and Rd genes are involved in proanthocyanidin synthesis in rice pericarp. Plant J 49:91–102
Gao LZ, Innan H (2008) Nonindependent domestication of the two rice subspecies, Oryza sativa ssp indica and ssp japonica, demonstrated by multilocus microsatellites. Genetics 179:965–976
Garris AJ, Tai TH, Coburn J, Kresovich S, McCouch S (2005) Genetic structure and diversity in Oryza sativa L. Genetics 169:1631–1638
Grillo MA, Li CB, Fowlkes AM, Briggeman TM, Zhou AL, Schemske DW, Sang T (2009) Genetic architecture for the adaptive origin of annual wild rice, Oryza nivara. Evolution 63:870–883
Gross BL, Zhao ZJ (2014) Archaeological and genetic insights into the origins of domesticated rice. Proc Natl Acad Sci USA 111:6190–6197
Gu BG, Zhou TY, Luo JH, Liu H, Wang YC, Shangguan YY, Zhu JJ, Li Y, Sang T, Wang ZX, Han B (2015) An-2 encodes a cytokinin synthesis enzyme that regulates awn length and grain production in rice. Mol Plant 8:1635–1650
Guerineau F, Lucy A, Mullineaux P (1992) Effect of 2 consensus sequences preceding the translation initiator codon on gene-expression in plant-protoplasts. Plant Mol Biol 18:815–818
Gutaker RM, Groen SC, Bellis ES, Choi JY, Pires IS, Bocinsky RK, Slayton ER, Wilkins O, Castillo CC, Negrao S, Oliveira MM, Fuller DQ, Guedes JAD, Lasky JR, Purugganan MD (2020) Genomic history and ecology of the geographic spread of rice. Nat Plants 6:492–502
Hardigan MA, Lorant A, Pincot DDA, Feldmann MJ, Famula RA, Acharya CB, Lee S, Verma S, Whitaker VM, Bassil N, Zurn J, Cole GS, Bird K, Edger PP, Knapp SJ (2021) Unraveling the complex hybrid ancestry and domestication history of cultivated strawberry. Mol Biol Evol 38:2285–2305
Huang XH, Han B (2016) Rice domestication occurred through single origin and multiple introgressions. Nat Plants 2:16207
Huang XH, Kurata N, Wei XH, Wang ZX, Wang A, Zhao Q, Zhao Y, Liu KY, Lu HY, Li WJ, Guo YL, Lu YQ, Zhou CC, Fan DL, Weng QJ, Zhu CR, Huang T, Zhang L, Wang YC, Feng L, Furuumi H, Kubo T, Miyabayashi T, Yuan XP, Xu Q, Dong GJ, Zhan QL, Li CY, Fujiyama A, Toyoda A, Lu TT, Feng Q, Qian Q, Li JY, Han B (2012) A map of rice genome variation reveals the origin of cultivated rice. Nature 490:497–503
Huo X, Wu S, Zhu ZF, Liu FX, Fu YC, Cai HW, Sun XY, Gu P, Xie DX, Tan LB, Sun CQ (2017) NOG1 increases grain production in rice. Nat Commun 8:11
Ishikawa R, Castillo CC, Fuller DQ (2020) Genetic evaluation of domestication-related traits in rice: implications for the archaeobotany of rice origins. Archaeol Anthropol Sci 12:14
Jaenicke-Despres V, Buckler ES, Smith BD, Gilbert MTP, Cooper A, Doebley J, Paabo S (2003) Early allelic selection in maize as revealed by ancient DNA. Science 302:1206–1208
Jiang L, Ma X, Zhao S, Tang Y, Liu F, Gu P, Fu Y, Zhu Z, Cai H, Sun C, Tan L (2019) The APETALA2-like transcription factor SUPERNUMERARY BRACT controls rice seed shattering and seed size. Plant Cell 31:17–36
Jin J, Huang W, Gao JP, Yang J, Shi M, Zhu MZ, Luo D, Lin HX (2008) Genetic control of rice plant architecture under domestication. Nat Genet 40:1365–1369
Khush GS (1997) Origin, dispersal, cultivation and variation of rice. Plant Mol Biol 35:25–34
Kimura M (1962) On probability of fixation of mutant genes in a population. Genetics 47:713–719
Koenig D, Jimenez-Gomez JM, Kimura S, Fulop D, Chitwood DH, Headland LR, Kumar R, Covington MF, Devisetty UK, Tat AV, Tohge T, Bolger A, Schneeberger K, Ossowski S, Lanz C, Xiong GY, Taylor-Teeples M, Brady SM, Pauly M, Weigel D, Usadel B, Fernie AR, Peng J, Sinha NR, Maloof JN (2013) Comparative transcriptomics reveals patterns of selection in domesticated and wild tomato. Proc Natl Acad Sci USA 110:E2655–E2662
Kojima S, Takahashi Y, Kobayashi Y, Monna L, Sasaki T, Araki T, Yano M (2002) Hd3a, a rice ortholog of the Arabidopsis FT gene, promotes transition to flowering downstream of Hd1 under short-day conditions. Plant Cell Physiol 43:1096–1105
Li W-H (1997) Molecular evolution. Sinauer Associates, Sunderland
Li CB, Zhou AL, Sang T (2006a) Genetic analysis of rice domestication syndrome with the wild annual species, Oryza nivara. New Phytol 170:185–193
Li CB, Zhou AL, Sang T (2006b) Rice domestication by reducing shattering. Science 311:1936–1939
Li YB, Fan CC, Xing YZ, Jiang YH, Luo LJ, Sun L, Shao D, Xu CJ, Li XH, Xiao JH, He YQ, Zhang QF (2011) Natural variation in GS5 plays an important role in regulating grain size and yield in rice. Nat Genet 43:1266–1269
Li SY, Zhao BR, Yuan DY, Duan MJ, Qian Q, Tang L, Wang B, Liu XQ, Zhang J, Wang J, Sun JQ, Liu Z, Feng YQ, Yuan LP, Li CY (2013) Rice zinc finger protein DST enhances grain production through controlling Gn1a/OsCKX2 expression. Proc Natl Acad Sci USA 110:3167–3172
Li YB, Fan CC, Xing YZ, Yun P, Luo LJ, Yan B, Peng B, Xie WB, Wang GW, Li XH, Xiao JH, Xu CG, He YQ (2014) Chalk5 encodes a vacuolar H+-translocating pyrophosphatase influencing grain chalkiness in rice. Nat Genet 46:398–404
Liu XH, Lu TT, Yu SL, Li Y, Huang YC, Huang T, Zhang L, Zhu JJ, Zhao Q, Fan DL, Mu J, Shangguan YY, Feng Q, Guan JP, Ying K, Zhang Y, Lin ZX, Sun ZX, Qian Q, Lu YP, Han B (2007) A collection of 10,096 indica rice full-length cDNAs reveals highly expressed sequence divergence between Oryza sativa indica and japonica subspecies. Plant Mol Biol 65:403–415
Liu C, Peng P, Li W, Ye C, Zhang S, Wang R, Li D, Guan S, Zhang L, Huang X, Guo Z, Guo J, Long Y, Li L, Pan G, Tian B, Xiao J (2021a) Deciphering variation of 239 elite japonica rice genomes for whole genome sequences-enabled breeding. Genomics 113:3083–3091
Liu Y, Wang H, Jiang Z, Wang W, Xu R, Wang Q, Zhang Z, Li A, Liang Y, Ou S, Liu X, Cao S, Tong H, Wang Y, Zhou F, Liao H, Hu B, Chu C (2021b) Genomic basis of geographical adaptation to soil nitrogen in rice. Nature 590:600–605
Londo JP, Chiang YC, Hung KH, Chiang TY, Schaal BA (2006) Phylogeography of Asian wild rice, Oryza rufipogon, reveals multiple independent domestications of cultivated rice, Oryza sativa. Proc Natl Acad Sci USA 103:9578–9583
Luo JH, Liu H, Zhou TY, Gu BG, Huang XH, Shangguan YY, Zhu JJ, Li Y, Zhao Y, Wang YC, Zhao Q, Wang AH, Wang ZQ, Sang T, Wang ZX, Han B (2013) An-1 encodes a basic helix-loop-helix protein that regulates awn development, grain size, and grain number in rice. Plant Cell 25:3360–3376
Ma JX, Bennetzen JL (2004) Rapid recent growth and divergence of rice nuclear genomes. Proc Natl Acad Sci USA 101:12404–12410
Maeda H, Dudareva N (2012) The shikimate pathway and aromatic amino acid biosynthesis in plants. Annu Rev Plant Biol 63:73–105
Mahmoud AA, Sukumar S, Krishnan HB (2008) Interspecific rice hybrid of Oryza sativa x Oryza nivara reveals a significant increase in seed protein content. J Agric Food Chem 56:476–482
McDonald JH, Kreitman M (1991) Adaptive protein evolution at the adh locus in Drosophila. Nature 351:652–654
Molina J, Sikora M, Garud N, Flowers JM, Rubinstein S, Reynolds A, Huang P, Jackson S, Schaal BA, Bustamante CD, Boyko AR, Purugganan MD (2011) Molecular evidence for a single evolutionary origin of domesticated rice. Proc Natl Acad Sci USA 108:8351–8356
Moner AM, Furtado A, Henry RJ (2018) Chloroplast phylogeography of AA genome rice species. Mol Phylogenet Evol 127:475–487
Nielsen R, Yang ZH (1998) Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148:929–936
Nordborg M, Donnelly P (1997) The coalescent process with selfing. Genetics 146:1185–1195
Oka HI (1974) Experimental studies on origin of cultivated rice. Genetics 78:475–486
Oka HI, Morishima H (1982) Phylogenetic differentiation of cultivated rice.23. potentiality of wild progenitors to evolve the indica and japonica types of rice cultivars. Euphytica 31:41–50
Olsen KM, Caicedo AL, Polato N, McClung A, McCouch S, Purugganan MD (2006) Selection under domestication: evidence for a sweep in the rice Waxy genomic region. Genetics 173:975–983
Onodera Y, Suzuki A, Wu CY, Washida H, Takaiwa F (2001) A rice functional transcriptional activator, RISBZ1, responsible for endosperm-specific expression of storage protein genes through GCN4 motif. J Biol Chem 276:14139–14152
Posada D, Crandall KA, Templeton AR (2006) Nested clade analysis statistics. Mol Ecol Notes 6:590–593
Ramos-Madrigal J, Runge AKW, Bouby L, Lacombe T, Castruita JAS, Adam-Blondon AF, Figueiral I, Hallavant C, Martinez-Zapater JM, Schaal C, Topfer R, Petersen B, Sicheritz-Ponten T, This P, Bacilieri R, Gilbert MTP, Wales N (2019) Palaeogenomic insights into the origins of French grapevine diversity. Nat Plants 5:595–603
Reddy VS, Scheffler BE, Wienand U, Wessler SR, Reddy AR (1998) EMBL accession #: Y15219. Plant Mol Biol 36:497–498
Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sanchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34:3299–3302
Rubin C-J, Megens H-J, Barrio AM, Maqbool K, Sayyab S, Schwochow D, Wang C, Carlborg O, Jern P, Jorgensen CB, Archibald AL, Fredholm M, Groenen MAM, Andersson L (2012) Strong signatures of selection in the domestic pig genome. Proc Natl Acad Sci USA 109:19529–19536
Sakamoto W, Ohmori T, Kageyama K, Miyazaki C, Saito A, Murata M, Noda K, Maekawa M (2001) The Purple leaf (Pl) locus of rice: the Pl(w) allele has a complex organization and includes two genes encoding basic helix-loop-helix proteins involved in anthocyanin biosynthesis. Plant Cell Physiol 42:982–991
Sang T, Ge S (2007) The puzzle of rice domestication. J Integr Plant Biol 49:760–768
Shih CH, Chu H, Tang LK, Sakamoto W, Maekawa M, Chu IK, Wang M, Lo C (2008) Functional characterization of key structural genes in rice flavonoid biosynthesis. Planta 228:1043–1054
Sweeney MT, Thomson MJ, Pfeil BE, McCouch S (2006) Caught red-handed: Rc encodes a basic helix-loop-helix protein conditioning red pericarp in rice. Plant Cell 18:283–294
Sweeney MT, Thomson MJ, Cho YG, Park YJ, Williamson SH, Bustamante CD, McCouch SR (2007) Global dissemination of a single mutation conferring white pericarp in rice. PLoS Genet 3:1418–1424
Takahashi Y, Shomura A, Sasaki T, Yano M (2001) Hd6, a rice quantitative trait locus involved in photoperiod sensitivity, encodes the alpha subunit of protein kinase CK2. Proc Natl Acad Sci USA 98:7922–7927
Takahashi Y, Teshima KM, Yokoi S, Innan H, Shimamoto K (2009) Variations in Hd1 proteins, Hd3a promoters, and Ehd1 expression levels contribute to diversity of flowering time in cultivated rice. Proc Natl Acad Sci USA 106:4555–4560
Templeton AR, Crandall KA, Sing CF (1992) A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics 132:619–633
Thomson MJ, Tai TH, McClung AM, Lai XH, Hinga ME, Lobos KB, Xu Y, Martinez CP, McCouch SR (2003) Mapping quantitative trait loci for yield, yield components and morphological traits in an advanced backcross population between Oryza rufipogon and the Oryza sativa cultivar Jefferson. Theor Appl Genet 107:479–493
Tong W, Kim TS, Park YJ (2016) Rice chloroplast genome variation architecture and phylogenetic dissection in diverse Oryza species assessed by whole-genome resequencing. Rice 9:13
Vaughan DA, Lu BR, Tomooka N (2008) The evolving story of rice evolution. Plant Sci 174:394–408
Wang ZY, Zheng FQ, Shen GZ, Gao JP, Snustad DP, Li MG, Zhang JL, Hong MM (1995) The amylose content in rice endosperm is related to the posttranscriptional regulation of the waxy gene. Plant J 7:613–622
Wang E, Wang J, Zhu XD, Hao W, Wang LY, Li Q, Zhang LX, He W, Lu BR, Lin HX, Ma H, Zhang GQ, He ZH (2008) Control of rice grain-filling and yield by a gene with a potential signature of domestication. Nat Genet 40:1370–1374
Wang GF, Wang G, Zhang XW, Wang F, Song RT (2012) Isolation of high quality RNA from cereal seeds containing high levels of starch. Phytochem Anal 2:159–163
Wang SK, Li S, Liu Q, Wu K, Zhang JQ, Wang SS, Wang Y, Chen XB, Zhang Y, Gao CX, Wang F, Huang HX, Fu XD (2015) The OsSPL16-GW7 regulatory module determines grain shape and simultaneously improves rice yield and grain quality. Nat Genet 47:949–955
Wang AH, Hou QQ, Si LZ, Huang XH, Luo JH, Lu DF, Zhu JJ, Shangguan YY, Miao JS, Xie YF, Wang YC, Zhao Q, Feng Q, Zhou CC, Li Y, Fan DL, Lu YQ, Tian QL, Wang ZX, Han B (2019) The PLATZ transcription factor GL6 affects grain length and number in rice. Plant Physiol 180:2077–2090
Xiong LX, Liu KD, Dai XK, Xu CG, Zhang QF (1999) Identification of genetic factors controlling domestication-related traits of rice using an F-2 population of a cross between Oryza sativa and O. rufipogon. Theor Appl Genet 98:243–251
Xu JW, Feng DJ, Li XG, Chang TJ, Zhu Z (2002) Cloning of genomic DNA of rice 5-enolpyruvylshikimate 3-phosphate synthase gene and chromosomal localization of the gene. Sci China Ser C Life Sci 45:251–259
Xu F, Fang J, Ou SJ, Gao SP, Zhang FX, Du L, Xiao YH, Wang HR, Sun XH, Chu JF, Wang GD, Chu CC (2015) Variations in CYP78A13 coding region influence grain size and yield in rice. Plant Cell Environ 38:800–811
Yang ZH (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591
Yang ZH, Nielsen R, Goldman N, Pedersen AMK (2000) Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155:431–449
Yano M, Katayose Y, Ashikari M, Yamanouchi U, Monna L, Fuse T, Baba T, Yamamoto K, Umehara Y, Nagamura Y, Sasaki T (2000) Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene CONSTANS. Plant Cell 12:2473–2483
Yin WC, Xiao YH, Niu M, Meng WJ, Li LL, Zhang XX, Liu DP, Zhang GX, Qian YW, Sun ZT, Huang RY, Wang SP, Liu CM, Chu CC, Tong HN (2020) ARGONAUTE2 enhances grain length and salt tolerance by activating BIG GRAIN3 to modulate cytokinin distribution in rice. Plant Cell 32:2292–2306
Yoo SD, Cho YH, Sheen J (2007) Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat Protoc 2:1565–1572
Zhang LB, Zhu QH, Wu ZQ, Ross-Ibarra J, Gaut BS, Ge S, Sang T (2009) Selection on grain shattering genes and rates of rice domestication. New Phytol 184:708–720
Zheng J, Wu H, Zhu HB, Huang CY, Liu C, Chang YS, Kong ZC, Zhou ZH, Wang GW, Lin YJ, Chen H (2019) Determining factors, regulation system, and domestication of anthocyanin biosynthesis in rice leaves. New Phytol 223:705–721
Zheng J, Wu H, Zhao MC, Yang ZN, Zhou ZH, Guo GM, Lin YJ, Chen H (2021) OsMYB3 is a R2R3-MYB gene responsible for anthocyanin biosynthesis in black rice. Mol Breed 41:51
Zhou M, Xu HL, Wei XL, Ye ZQ, Wei LP, Gong WM, Wang YQ, Zhu Z (2006) Identification of a glyphosate-resistant mutant of rice 5-enolpyruvylshikimate 3-phosphate synthase using a directed evolution strategy. Plant Physiol 140:184–195
Acknowledgements
We thank Enlai Zhang, Guangzhu Zhang, Bin Lv, Weiren Meng, and Baosheng Xie for sharing seeds and Ruijuan Zhang, Yiting Wang, Zhixin Zhu, Lulu Xie, and Hongling Zhou for help with sequencing surveys.
Funding
This work was supported by National Natural Science Foundation of China (91331116), Chinese Academy of Sciences (XDA08020204), and State Key Laboratory of Systematic Botany and Evolution.
Author information
Authors and Affiliations
Contributions
YL developed the conceptual framework and tests, sampled genomes, and analyzed data, YX conducted sequencing and transient experiments, NL participated in genotyping and construct-building, all authors collected phenotypic data. YL wrote the manuscript with inputs of YX and NL. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Information on 101 genes analyzed in this study.
Additional file 2: Fig. S1.
Mapping of mutational events. a Chromosome 1. b Chromosome 2. c Chromosome 3. d Chromosome 4. e Chromosome 5. f Chromosome 6. g Chromosome 7. h Chromosome 8. i Chromosome 9. j Chromosome 10. k Chromosome 11. l Chromosome 12.
Additional file 3: Table S2.
A list of 30 loci showing early and later mutations in rice.
Additional file 4: Table S3.
Estimation of background-mutation rates from intron mutations of four genomes (Or, On, Indica (I) and Nipponbare (J)).
Additional file 5: Table S4.
Distribution of eight gene types (shown in Fig. 3) among 101 sampled loci of O. sativa.
Additional file 6: Data 1.
Plant height of F2 population in the field experiment 1.
Additional file 7: Data 2.
Morphological data in the field experiment 2.
Additional file 8: Table S5.
Genotypes at four Os loci across accessions.
Additional file 9: Table S6.
Comparisons of the model genomes (upper panel) with alleles of OsSSY3 (lower panel) in rice cultivars.
Additional file 10: Table S7.
Comparisons of model genomes (upper panel) with alleles of OsPGMp (lower panel) surveyed in rice materials.
Additional file 11: Table S8.
Comparisons of model genomes (upper panel) with alleles of OsCKX2 (lower panel) surveyed in rice.
Additional file 12: Table S9.
Random samples on identified polymorphisms from NCBI.
Additional file 13: Table S10.
Population-level examinations of mixed contributions of Or and On to OsHd3a based on NCBI data.
Additional file 14: Fig. S2.
Comparisons of predictions under different hypotheses.
Additional file 15: Table S11.
Genomic comparisons of Kitaake with reference genomes across 101 genes.
Additional file 16: Table S12.
List of primers used in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lu, Y., Xu, Y. & Li, N. Early Domestication History of Asian Rice Revealed by Mutations and Genome-Wide Analysis of Gene Genealogies. Rice 15, 11 (2022). https://doi.org/10.1186/s12284-022-00556-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12284-022-00556-6