
Abstract
With the first RNA interference (RNAi) drug (ONPATTRO (patisiran)) on the market, we witness the RNAi therapy field reaching a critical turning point, when further improvements in drug candidate design and delivery pipelines should enable fast delivery of novel life changing treatments to patients. Nevertheless, ignoring parallel development of RNAi dedicated in vitro pharmacological profiling aiming to identify undesirable off-target activity may slow down or halt progress in the RNAi field. Since academic research is currently fueling the RNAi development pipeline with new therapeutic options, the objective of this article is to briefly summarize the basics of RNAi therapy, as well as to discuss how to translate basic research into better understanding of related drug candidate safety profiles early in the process.
Similar content being viewed by others

Introduction
Francis Crick’s 1957 central dogma lecture changed the course of modern biology and the pharmaceutical industry by placing proteins at the end of the biological information transfer [1,2,3]. Consequently, perturbations in protein levels and function contribute to pathomechanisms of human diseases, despite their molecular, genetic and physiological origins. Hence, restoring human protein homeostasis has become one of the main goals of research into post-genomic therapeutic strategies. However, it quickly became clear that only some disease-related proteins have the ability to bind small chemical molecules, being potential drugs. Indeed, as estimated in the early 2000 s, among the approximately 3000 disease-related proteins encoded in the human genome, only 600-1500 are potential small-molecule drug targets (proteins with enzymatic function or a conformation that is accessible to traditional drug molecules) [4,5,6]. Similarly, the highly specific, protein-based drugs including monoclonal antibodies are mainly limited to cell-surface receptors or circulating proteins [7, 8]. Notably, about 80% of the proteins involved in human diseases execute two or more biochemical functions [9], and thus their precise chemical targeting can be very difficult or impossible due to potential adverse effects. Furthermore, pharmacologically relevant small molecule-mediated therapeutic effects often rely on maximizing drug-receptor effects (at above 90% target engagement), requiring high dosing levels and thus reduced safety [10]. Thus, the discovery and development of alternate therapeutic strategies addressing and exploiting chemically “undrugabble” proteins have remained a challenge for the industry.
The 2006 Nobel prize crowned the discovery of RNA interference (RNAi) [11] as a pathway in which small non-coding RNA molecules, by controlling mRNA stability and translation, modulate protein cellular levels. Furthermore, subsequent reports that short (21 and 22 nucleotide) double stranded RNAs (dsRNAs) may enter the RNAi silencing pathway in mammalian cells [12,13,14] opened new prospects for the pharmaceutical industry. Initially, the opportunity for rational drug design to treat diseases that were once thought to be untreatable was well received by drug developers. However, subsequent unsuccessful clinical trials revealed numerous limitations of RNAi application, including: dose-limiting and immune-related toxicities, insufficient therapeutic efficacy, poor metabolic stability, as well as off-targets effects [15,16,17,18,19,20]. Hence, despite confirming efficient RNAi therapy in humans, the mainstream pharmacological industry withdrew from the RNAi field in the 2010s [20,21,22].
However, despite this excessive skepticism toward RNAi therapy, in August 2018 a small interfering RNA (siRNA) against transthyretin (TTR) mRNA, ONPATTRO (patisiran) was proven to be an effective therapy for hereditary transthyretin amyloidosis (hATTR) and approved as the first RNAi drug by both the US Food and Drug Administration (FDA) and the European Medicine Agency (EMA) [23,24,25]. Furthermore, multiple RNAi drug candidates are currently progressing through clinical trials, with many of them excelling and reaching phase III [25]. Hence, we witness the RNAi therapy field reaching a critical turning point, when further improvements in drug candidate design and delivery pipelines should enable fast delivery of novel life changing treatments to patients. Furthermore, microRNA (miRNA) based drug candidates promise not only elimination of erratic proteins (such as siRNA), but also provide tools to restore missing proteins to physiological levels [26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44]. Importantly, since mammalian miRNAs are not perfectly complementary to their target mRNA sequences and have multiple targets, this directly translates into a higher attrition rate in related drug discovery. Hence, ignoring parallel development of RNAi dedicated in vitro pharmacological profiling [45] aiming to identify undesirable off-target activity may slow down or even halt progress in the RNAi field.
Since academic research is currently fueling the RNAi development pipeline with new therapeutic options, the objective of this article is to briefly summarize the basics of RNAi therapy, as well as to discuss how to translate basic research into better understanding of related drug candidate safety profiles early in the process.
RNA interference
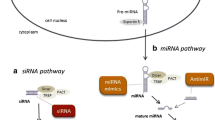
RNA interference is a native gene silencing pathway of most eukaryotic cells that utilizes non-coding RNA (ncRNA) molecules (produced by various mechanisms) to obtain efficient post-transcriptional repression of homologous sequences [46,47,48]. ncRNA molecules act on specific mRNAs through short guide strands that recognize complementary bases in the target RNAs. With an 8 nucleotide (nt) long region called the “seed sequence,” the guide strands must have significant homology to their target strand(s) in order to allow the RNAi mechanism to affect gene expression. The guide strands, depending on their biogenesis and actions on the intended mRNAs, can be broken up into the three categories of RNAi.
-
(i)
miRNAs are short (approx. 22 nt) endogenous non-coding single substrates for the RNAi machinery [49]. microRNAs are encoded in both introns and intergenic clusters and these genes are first transcribed by RNA polymerase II into long primary miRNA (pri-miRNA) transcripts. Next, the pri-miRNA are processed by the the double-strand-specific ribonuclease Drosha-DGCR8 complex transcripts into precursor miRNA (pre-miRNA) stem loop structures [50] that, following their transport to the cytoplasm, are further dissected by the Dicer RNAase III endonuclease to deliver mature 21-23 nucleotide microRNAs [50,51,52,53,54,55,56]. Notably, an alternate, Dicer-independent miRNA biogenesis pathway has also been reported [57]. Mature miRNAs strands are associated with Argonaute 2 (Ago2) containing RNA-induced silencing complexes (RISC) that can diminish a specific target mRNA by Ago2-catalyzed degradation of the mRNA and down regulate specific target gene expression via either reducing the transcript levels or by translational repression [52,53,54,55,56, 58,59,60,61,62,63]. Notably, in humans, only Ago2 carries catalytic cleavage activity [64, 65]. microRNAs perceive their target mRNAs through base-pairing interactions between nucleotide numbers 2 and 8 of the miRNA (the seed sequence) and the complementary nucleotides in the 3′-untranslated region (3′-UTR) of the mRNAs [66,67,68,69]. Importantly, nuclear mammalian miRNAs mediated nuclear chromatin silencing at specific loci by base pairing to nascent transcripts has also been reported [70,71,72].
-
(ii)
Small interfering RNAs (siRNA) being ∼21-22 bp long dsRNA with 3′ two-nucleotide overhangs originate from cytosolic Dicer mediated processing of 30 to 100 bp dsRNA that are either transcribed from cellular genes or introduced into the cells by infecting pathogens, or artificially via transfection or transduction by a viral-derived vector [12, 47, 73, 74]. siRNA interacts with and activates RISC (Ago2 cleaves and releases the “passenger” siRNA strand (sense strand), while the “guide” strand (antisense strand) remains associated with the complex) [73, 74]. The single “guide” strand of siRNA directs the specificity of the mRNA target recognition and cleavage by Ago2 by intermolecular base pairing [74]. mRNA targets that bind the “guide” strand with perfect or near-perfect complementarity are then degraded by Ago2, and thus specific gene expression silencing is obtained [27, 75]. In some cases, however, imperfect complementarity between the “guide” strand and target mRNA may mimic miRNAs’ mediated translational repression [76]. Importantly, RISC can also mediate transcriptional gene silencing using the siRNA specificity to direct silent chromatin modifications over homologous DNA loci [77]. Natural siRNAs likely originated as a defense mechanism against viruses and foreign DNA elements, allowing their elimination [47, 78].
-
(iii)
piwi-interacting RNAs (piRNAs) are small, ∼23-30-nucleotide, endogenous RNAs that are issued from long single stranded precursors-a Dicer-independent mechanism [79]-and serve as repressors of transposable elements (TE) [79]. Hence, piRNA safeguard mammalian germ cells from deleterious effects of transposons and preserve chromatin structure [79]. piRNAs guided silencing is analogous to the other RNAi mechanism in that piRNAs guide PIWI proteins to target mRNAs through RNA base pairing and the mRNAs are then dissected by the endonuclease activity of the PIWI proteins [80,81,82,83]. Although initial studies assigned piRNAs activity with mammalian germ cells, emerging evidence suggests that they may be functional in somatic cells as well [79, 81, 82, 84,85,86,87,88,89,90,91]. However, despite the increasing interest in piRNA mediated mechanisms, knowledge about their functional roles remains fairly limited. Furthermore, related experimental approaches are limited by the plethora of unique piRNAs sequences [92,93,94,95,96,97]. and the lack of easily available verified piRNAs analogs and inhibitors. Hence, although piRNAs may eventually be included in RNAi therapy, this is not going to be quick or easy process.
siRNAs are highly specific with only one mRNA target and generally allow effective gene silencing. This makes siRNAs the leading branch of the developing specific RNAi therapies. However, these therapies are limited to the elimination of target proteins. In contrast, miRNAs have multiple targets, and consequently specific miRNAs can modulate transcriptional networks involving diverse autonomous targets such as transcription factors [98,99,100], and thus avoiding off-target effects can be extremely difficult. Furthermore, despite the fact that some miRNAs have large switch-like effects reported under conditions of stress or disease [101,102,103,104,105,106,107,108,109,110,111,112,113], these RNAs instead modulate protein levels than serve as strong post-transcriptional repressors [114]. Hence, the miRNA-based drug discovery process seems very challenging, as is reflected by the limited number of drug candidates undergoing clinical trials [20, 25,26,27]. Nevertheless, miRNA and their analogs (antagomiRs) and agonists for RNA (target protectors/block-miRs) provide a therapeutic opportunity for not only eliminating proteins but also restoring their physiological levels and thus should be considered as the future of RNAi therapies [115].
Design of RNAi drug candidates
Although the therapeutic potential of RNAi drugs is evident, their formulation must overcome different sets of hurdles impeding their development into clinical use, including: off-target activity, immunogenic reactions to foreign dsRNA, immunogenic as well as non-immunogenic effects of delivery chemicals, specific tissue delivery, as well as obtaining desired drug candidate pharmacokinetics and bioavailability (stability, competition with endogenous RNA, cellular uptake, endosomal escape) [19, 25,26,27, 35, 41]. To date, numerous design and delivery strategies have been developed to address these obstacles and to enhance RNAi drug candidate efficacy and specificity.
Entering the RNAi pathway
RNAi drug candidates triggered by synthetic RNAs channel into the RISC pathway at the cytoplasmic stage. Minimal 15-30 bp, fully complementary dsRNAs or short hairpin RNAs (shRNAs) are most commonly used. dsRNAs longer than 30 bp have increased propensity for off-targeting and inducing nonspecific cytotoxicity via activating interferon pathway [116]. dsRNAs shorter than 15 bp are not recognized by RNAi machinery. Importantly, dsRNAs shorter than 21 bp do not require Dicer processing prior to association with RISC [117, 118]. However, it has been reported that Dicer processing of RNAi drug candidates results in their increased potency and better specificity (it has been reported that dsRNAs with 27 nucleotides are up to 100 times more efficient than typical siRNAs with 21 nucleotides) [119,120,121,122]. In contrast, dsRNAs that bypass Dicer processing provide the opportunity for more extensive chemical modification of such RNAs and thus obtaining better metabolic stability [123]. Numerous chemical and design strategies have been tested in combine Dicer processing-related potency with increased stability of RNAi drug candidates that include small segmented siRNAs (division into 2 fragments precedes their association with RISC) or incorporating motives that eliminate the Dicer cleavage requirement, but sustain Dicer-RISC interaction [25, 124]. Single-stranded RNAs (ssRNAs) may also be used as RNAi triggers, but their potencies are usually much lower than those reported for dsRNAs [125, 126]. Nevertheless, recent reports show that ssRNAs offer enhanced delivery properties (even entering cells via gymnosis), due to their amphiphilic nature and enhanced structural flexibility (ssRNA vs dsRNA) [125,126,127]. A similar rule applies to synthetic miRNAs (mimics), and despite ssRNAs containing the sequences that are identical to the guide strands of the mature miRNAs that can function as miRNA mimics, their potency is 100 to 1000 times lower than that of dsRNAs containing miRNAs’ guide and passenger strands [27, 115, 128]. Other strategies include designing longer synthetic miRNA precursors such as pre-miRNA (that will undergo Dicer processing in the cytoplasm) and pri-miRNA (that will require delivery to the nucleus for processing) [129,130,131].
Another family of RNAi drug candidates, antagomiRs (antimiRs), are synthetic chemically modified ssRNA, about 21-23 nucleotides long, which fully complement miRNAs and effectively sequester mature miRNA in competition with cellular target mRNAs leading to functional inhibition of miRNA [132,133,134,135,136,137]. However, assessing antagomiRs efficiency in preventing miRNAs activity may be very challenging since their mechanism of miRNA inhibition depends on the type of chemical modifications used. Two types of modified antimiRs can be discussed here: high affinity oligo nucleotides which sequester the targeted miRNA in a heteroduplex, and lower affinity oligonucleotides which promote miRNA degradation as also do cholesterol-conjugated antimiRs [138,139,140,141].
Finally, an alternate and more explicit concept relies on the prevention of miRNA interaction with an individual seed sequence of a specific mRNA using target protectors [142]. Target protectors (morpholinos) are chemically modified ssRNAs (~ 25 base) complementary to an mRNA target sequence (at least 14-15 contiguous bases) that prevent the interaction of the miRNA with its specific target, and assembly of the RISC complex [142,143,144,145]. The chemical modification of target protectors prevents them from triggering the RNAi pathway, whereas their uncharged backbone facilitates their delivery by non-toxic endocytosis assisted delivery reagents [146].
Sequence optimization
The potency of the RNAi drug candidate varies greatly depending on its sequence and this ensures specific selection of an antisense strand and minimal off-target effects [147]. Hence, the first concern is the quality of the software package for designing RNAi drug candidates and to predict their efficacy [31, 148,149,150,151]. Particularly important in this design procedure is avoiding sequence related off-target effects that may result from partial homology to other transcripts and thus induce miRNA-like activity [152,153,154]. It has been reported that siRNA duplexes can have differing activities contingent upon the number, position, and base-pair composition of mismatches with respect to the target RNA [155], but so far this problem remains mostly unsolved. Notably, siRNAs seed regions consist of 7 nucleotides, which often results in a large number of partially complementary off-target transcripts. However, modern algorithms often include and develop filtering of siRNA with seed regions that mirror naturally occurring miRNAs and select these with the fewest seed region matches in the 3′ UTRs of off-target transcripts [148, 156].
Since the majority of RNAi drug candidates are dsRNA, both strands can enter RISC. However, on-target silencing requires the guide strand (antisense strand) to remain associated to the active RISC to guide it to the target mRNA, while the passenger strand is degraded and discarded [157]. An improper RISC loading orientation causes the expected guide strand to be neglected and off-target effects are created as the remaining strand is complementary to the unintended transcripts resulting in off-target effects. The same problems apply to synthetic miRNAs, where wrong strand selection at RISCs results in the other miRNA (star form) to be a guide RNAi toward its targets [158, 159]. However, the strand with weaker base pairing at the 5′ terminus of an miRNA or siRNA duplex will be preferred as a guide strand [158, 160]. Furthermore, since a strand with a relatively unstable 5′ end is selected as the guide strand while the strand with a more stable 5′ end is discarded as the passenger strand, the so-called “asymmetry rule” can be applied, by designing a 5′ of the antisense (guide) strand more AU rich than the corresponding end of the sense strand (5′) [157]. Furthermore, AGO proteins display a preference for selecting, as the guide strand, the strand with a U (or less preferably, an A) at position one at the 5′ end. Hence, the ideal passenger strand should consist of C or G at the 5′ end to reduce the risk of selection, whereas the guide strand should contain a U or A at the 5′ end [157]. Furthermore, since there are reports that siRNAs with a rich G/C content are less potent, due to their increased overall duplex thermodynamic stability [147, 161], it is generally accepted as optimal that the G/C content of siRNA is between 30 and 64% [162]. Moreover, sequences with G/C stretches of nine or more nucleotides may reduce the gene silencing efficiency of siRNA and thus should be avoided [163].
Mammalian cells recognize dsRNAs by dsRNA-binding proteins and Toll-like receptors, which results in overall stoppage of protein synthesis and activation of the interferon response [164]. Despite the fact that initial studies assigned activation of the immune response to dsRNAs longer than 30 bp [116], some shorter siRNAs and miRNAs analogs have also been shown to activate innate immunity in a sequence-dependent manner [165, 166]. Notably, the length of the dsRNA threshold may vary among cell types, and even 23 bp siRNAs have been shown to induce interferon responses in some cell lines [167]. To date, several immune-related sequence motifs have been reported to activate Toll-like receptor (TRL) signaling. Unfortunately, these motifs are usually U-rich (e.g. 5′GUCCUUCAA3′, 5′UGUGU3′, 5′UGU3′, or 5′UGGC3′), and thus are hard to eliminate from an RNAi drug candidate sequence [168,169,170,171]. Alternate solutions to this problem focus on use of chemical modifications and non-endosomal delivery routes (e.g., electroporation), to prevent TRL activation [172, 173].
Taken together, the development of bioinformatics tools accompanied by better understanding of a drug candidate sequence’s relationship to its potency and specificity has facilitated efficient design of RNAi drug candidates. Nevertheless, such bioinformatic tools depend on the quality of the data deposited in sequence databases (often updated, and problematic in regard to non-coding regions and longer sequence repeats [174]). It should be noted that numerous miRNAs have been recently recognized as sequencing artifacts [175,176,177]. Furthermore, rules allowing motifs inducing stress response pathways to be avoided are still poorly understood. Therefore, extensive experimental validation of RNAi drug candidate sequence specificity and related off-targets as well as for any possible immunostimulatory adverse effects seems absolutely mandatory. Especially, some off-target siRNA effects can be reduced at concentrations that match the individual potency of these RNAs [178]. However, the recent rapid development and decreasing costs of next generation sequencing, and thus the ability to access entire transcriptome changes upon RNAi drug candidate administration, should facilitate the process of identification and selection of the best candidates with minimal adverse effects.
Chemical modifications
The vulnerability of RNAs to degradation by endogenous and exogenous nucleases [179, 180], resulting in poor pharmacokinetics, is another obstacle to RNAi therapy. Furthermore, although the right sequence optimization of RNAi drug candidates can greatly improve their specificity and potency and minimize the risk of adverse effects, it cannot completely eliminate the risk of immune response activation [25, 27]. Addressing these issues had resulted in development of numerous chemical modifications that, besides increasing RNAs stability and attenuating immune responses, can also enhance guide strand selection and delivery, as well as reduce RNAi off-target activity [25, 27, 181]. Finally, chemical modifications can be used to facilitate RNAi drug delivery [25, 27, 182]. Importantly, modifications to siRNAs and miRNAs analogs cannot impair their ability to effectively enter and function in the RNAi pathway, and thus prevent their interaction with Dicer and Ago proteins or compromise their silencing efficiency. Since the 5′ phosphate, the 5′ proximal part, and the central positions of the guide strand are crucial for interaction with the RISC, these sites cannot be easily modified [183]. On the other hand, alterations at the whole passenger strand and the 3′ proximal part and 3′ overhang of the guide strand are generally well tolerated [180]. Furthermore, in the case of pri-miRNA chemical modifications should allow nuclear processing [115]. As mentioned above, chemical modifications of antagomiRs determine microRNA fate by targeting it for degradation or accumulation in heteroduplexes [115, 138,139,140,141]. Interestingly, possibilities of piRNA chemical modifications are much less well explored, despite the fact that naturally existing piRNAs incorporate the 3′-end 2′-O-methyl modification that protects them from RNases [184].
Nowadays, numerous chemical strategies relying on base, sugar or backbone modifications of antisense strands are applied to improve RNAi drug candidate function and stability. Commonly used modifications employ ribose 2′-OH group substitution with other groups along with 2′-O-methyl (2′-O-Me), 2′-fluoro (2′-F) and 2′-methoxyethyl (2′-O-MOE) to increase RNAs nuclease resistance and reduce the risk of immune responses (by preventing TRL activation) [185,186,187,188]. However, these modifications may occasionally limit silencing efficiency [189,190,191].Similar advantages can be achieved by using locked nucleic acid (LNA) and unlocked nucleic acid (UNA) modifications that can also minimize the risk of off-target effects by ensuring proper guide strand selection. LNA creates a stable “locked” ring conformation by introducing into nucleic acid a methylene bridge between the 2′-O and the 4′-C of pentose [192]. Since LNA modification at the 5′ end of the passenger strand prevents incorporation into the RISC, it reduces the risk of off-target effects [193]. This modification also improves RNA stability and reduces its immunogenicity [165, 193]. However, LNA modifications are also reported to reduce siRNA potency [194].
UNA are based on removal of the C2′ and C3′-bond of the RNA ribose, which decreases modified RNA binding affinity to their target RNAs [195, 196]. Thus, UNA modifications in a seed region of the guide strand can be used to decrease sequence mismatch tolerance, and thus prevent miRNA-like off-target effects [195, 196]. Although single UNA modifications are generally well-tolerated in both the passenger and guide strands and improve RNA, UNA modifications of guide strands can also reduce silencing efficiency [195, 196].
Another chemical strategy relies on substituting the phosphodiester backbone linkages with other types of linkage. In the most common approach, the nonbridging phosphate oxygen atoms are substituted with a sulfur atom to create phosphorothioate (PS) [197]. PS significantly increases the stability of modified RNAs and enhances their pharmacokinetics via promotion of nonspecific binding to plasma proteins [198, 199]. However, in order to maintain RNAi drug candidate compatibility with the RISC pathway, only partial PS modification can be introduced, leaving the center region of the RNA duplex unmodified [200, 201]. Notably, PS modifications along with cholesterol conjugation improve systemic circulation of dsRNAs and stimulate their uptake by gymnosis [124, 202,203,204]. Furthermore, replacement of siRNA backbone phosphodiesters with the neutral phosphothioesters facilitates such an RNA cellular uptake, while cytosolic thioesterases revert this to native form (short interfering ribonucleic neutrals, siRNNs) [204]. Other chemical strategies are also reported to increase nuclease resistance and accordingly modulate the binding strength with target RNA by using peptide nucleic acids, (PNA) or morpholinos [205, 206].
Taken together, it is clear that optimal pharmacological results and potency of RNAi drug candidates can be obtained by combining the above-mentioned different chemical strategies [207, 208]. Although challenging, as in the classical drug development pipeline, sequential selection and optimization of differentially modified derivatives increases the chance of selecting the leader combination of chemical modifications in terms of stability, potency and specificity.
Targeted delivery
The cellular membrane constitutes a barrier preventing siRNA and miRNA analogs from entering the cytoplasm, due to their hydrophilic nature, size (~ 14-15 kDa) and negative charge. Moreover, naked nucleic acid molecules are the subject of rapid degradation in biological fluids and, following systemic administration, do not accumulate in target tissue. Hence, the efficient and targeted delivery of RNAi drug candidates still remains one of the major obstacles to the development of RNAi therapies [209]. It is also evident that an optimal delivery system cannot increase toxicity or induce immune response. Furthermore, an optimal carrier should also protect RNAi drugs from degradation in the circulation at physiological conditions and prevent their clearance by the mononuclear phagocytic system, and finally, it should allow their efficient endosomal escape into the cytosol [19, 182, 210].
Many initial clinical approaches and research reports have been based on DNA strategy and viral delivery in which RNAi drug candidates (including miRNA) are produced by intracellular processing of vectors encoding longer RNA hairpin transcripts [211,212,213,214,215,216,217,218]. Following transcription and processing, resultant short hairpin RNAs (shRNAs) and pre-miRNAs enter the RNAi pathway [219, 220]. While research use of this delivery strategy is relatively simple and efficient, and has a large potential for related gene therapy, in a clinical setting usage of viral-derived vectors raises serious concerns regarding their high immunogenicity and the risk of insertional mutagenesis [221,222,223,224,225,226,227,228,229].
Therefore, chemical excipients have become the leading strategy for delivering RNAi drugs, due to their better safety profile and lower production cost [25, 27, 40, 41, 115, 157, 219]. Furthermore, these chemical carriers/excipients can be modified to accomplish site-specific delivery (by incorporating targeting ligands, as summarized in [230]), or to enhance serum stability [209]. Polymer-based and lipid-based systems are the two main categories of RNA delivery systems.
In lipid-based systems several approaches are used. The simplest are so-called lipoplexes which are cationic lipid-RNA or DNA complexes and which, although successfully used in in vitro studies, appear toxic when administered into animals. More elaborated are long-circulating liposomes (~ 100 nm in diameter) containing either cationic lipid-nucleic acid (lipoplexes) or cationic polymer-nucleic acid complexes inside the liposome water space [231, 232]. RNA-containing lipoplexes or vesicles are taken up by cells mostly via endocytosis and released into the cytosol via the “endosomal escape” pathway [209, 232,233,234]. However, the lead technology for the non-viral delivery systems of genetic drugs is the so-called lipid nanoparticle system (LNPs) which is based on the method developed by Curtis et al. employing an ethanol-loading procedure, usage of ionizable cationic lipids and rapid mixing [235]. The resulting structure is a ~ 100 nm diameter particle covered with a PEG-lipid monolayer interacting with other constituent lipids in which water-filled cavities containing nucleic acid molecules can be seen [235]. This system has been found to be very efficient in hepatocyte transfection due to liver accumulation and interaction with ApoE. The efficiency of such a construct is high (0.005 mg siRNA/kg body weight in mice); the authors suggest that this is due to “the combination of the optimized cationic lipid MC3, cholesterol and DSPC, together with the rapidly dissociating PEGC14-lipid” [236] The above-mentioned properties and in addition tolerability led to the development the recently approved first RNAi drug, patisiran, directed against transthyretin-induced amyloidosis [23, 24].
In polymer-based delivery systems, cationic polymers are used to establish electrostatic polyplexes with the negatively charged RNA; for example synthetic polyethylenimine (PEI), cyclodextrins, Poly(lactic-co-glycolic acid) (PLGA) and Silica-based nanoparticles) [237,238,239,240,241,242,243,244,245,246,247,248,249,250,251,252]. Also, cationic proteins such as protamine and peptides, such as nona-arginine (9R) peptide can be used [253]. An interesting possibility is offered by the application of recombinant protamine as a fusion protein with an scFv antibody fragment which assures targeting of a protamine-nucleic acid complex against cells exposing particular marker molecules to the potential to be bound [254].
All the above-mentioned nanosized particles can enter cells via endocytosis and often promote endosomal escape. However, as mentioned above, because of their high charge density, some cationic nanoparticles are often toxic [27, 255]. Recently, natural cationic polymers such as chitosan, (derived from chitin), and atelocollagen, which is a protein obtained from calf dermis, have been proposed as dependable options for RNA delivery [255,256,257,258].
Finally, lipolyplexes consisting of both polymers and lipids are currently being developed to overcome the restraints of the exclusive polymer-based or lipid-based delivery system [249, 259,260,261].
Importantly, the success of therapeutic RNAi is also often highly dependent on tissue or cell type specific targeting, and thus avoiding unwanted on-target activity in non-target tissues. Target gene expression may be deregulated in target tissue (e.g., cancer cells), but at the same time at the correct levels in healthy non-target tissue (e.g., normal cells). Hence, modulating target gene expression in order to obtain therapeutic benefits in target tissue may be accompanied by deregulation of this gene expression in non-target tissues, leading to toxicity. This is especially important during systemic RNAi drug delivery, since its accumulation in tissues not intended for its activity may be toxic [262]. Furthermore, since miRNA expression is very often tissue and cell type specific [263, 264], targeted delivery is the key to the best potency and minimal off-target effects of related drugs. Hence, the development of targeting ligands for RNAi drugs (e.g., antibodies, aptamers, or small molecules, N-Acetylgalactosamine-GalNAc), as well as methods for their systemic and local administration create another major bottleneck in the further expansion of RNAi therapies [25, 26, 265,266,267,268,269,270].
Concluding notes and future prospects
It is clear that current progress in the RNAi therapy field provides an opportunity to deliver novel drugs that could change patients’ lives. However, despite the success story of Partisiran and multiple other RNAi drug candidates currently progressing through clinical trials, several technical barriers and hazards (Fig. 1) need to be overcome so such therapies could become common clinical treatment; that is, also accessible for orphan diseases.

Schematic representation of the RNAi drug discovery and development process. Full mechanistic understanding of the disease allows selection of highly disease specific therapy targets, and thus early elimination of off-targets. In the first phase, candidate sequence design and optimization allows early hazard identification and elimination, whereas chemical modifications can be applied to design out potential hazards and limitations. Furthermore, in later phases potential liabilities regarding delivery system choice should be assessed. Finally, broad pharmacological profiles of the lead drug candidates should be obtained, before drug candidates undergo further clinical development
From the clinical point of view, finding novel effective methods for systemic delivery of RNAi drugs to non-liver and non-kidney tissues, along with dedicated improvement of their pharmacokinetic and pharmacodynamics, remains one of the key challenges in achieving this goal [25, 182, 271]. Hopefully, further development of chemical modifications, as well as better understanding of cellular pathways governing endosomal escape and endocytosis [272,273,274], will eventually address this issue [275,276,277,278,279]. Finally, although the first RNAi drug is approved, we are very far from understanding the long-term effects of siRNA and miRNA therapy in vivo in human subjects.
Another main challenge is reducing the risks of RNAi drug candidate off-target effects. The initial hazards related to RNAs chemical modifications, non-viral delivery systems and immunogenicity can often be identified, tested in animal models and finally eliminated through the classical drug development pipelines that include in vitro pharmacology profiling [45, 280,281,282,283].
Nevertheless, the specific RNA sequences remain the main components and sources of hazards for the drug candidates. Bioinformatics tools try to prevent the design of siRNA with a seed region that is partially complementary with off-target transcripts, but they cannot fully eliminate the risks of all off-target interactions. Although small activating RNAs (ssRNAs) that are structurally identical to siRNA and that can mediate promoter sequence specific activation of some gene expression are also considered in therapy [284, 285], they also represent clear proof of siRNA related hazards. Furthermore, these prediction algorithms are based on consensus genome sequences, and do not eliminate the potential complications related to the occurrence of single nucleotide polymorphisms (SNPs) [67, 68, 286, 287]. As estimated for the human genome, SNP can occur once in every 300 bp in both coding and non-coding regions of genes [288], resulting in synonymous and non-synonymous changes that are often reflected in RNA sequences [289]. One such nucleotide change in the human genome may eliminate siRNA or miRNA seed region interaction with target RNA, or result in off-target degradations, as well as disturb miRNA biogenesis [67, 68, 286]. Hence, early detection of SNP-related off-target effects as well as paying attention to population stratification [290,291,292,293,294] are crucial to prevent RNAi drug candidate halt during clinical trials or even its market withdrawal.
For similar reasons to the siRNA/miRNA target sequence specificity, the use of in vivo translational models is very limited and does not allow fair assessment of such a drug candidate toxicity or off-target effects [295]. Furthermore, mRNAs and ncRNAs expression is often sex, age, organ or tissue specific, and thus preclinical development of RNAi drugs requires wide-ranging in vitro studies in different models to prevent both its off-target and on-target activities in non-target tissues [296,297,298,299,300]. Fortunately, recent development and decreasing costs of high-throughput genotyping technologies such as deep sequencing and single cell sequencing [301,302,303,304] should allow development of RNAi sequence design and related in vitro pharmacological profiling. Notably, these technologies should propel development of miRNA therapeutics, by advancing understanding of the mechanisms by which these RNAs modulate complex physiological [49, 98, 305,306,307,308,309,310,311,312,313,314,315,316,317,318,319,320] and pathological molecular networks [24, 43, 44, 91, 310, 321,322,323,324,325,326,327,328,329,330,331,332,333,334,335,336,337,338,339,340,341,342,343,344,345,346,347,348,349,350,351,352,353,354,355,356,357,358,359].
Furthermore, long non-coding RNA (lncRNA)-dependent modulation of miRNA levels may become a promising siRNA therapy target [360,361,362,363,364,365,366,367,368,369,370]. However, the biological roles of these ncRNAs, and thus potential off-target effects of lncRNA related therapies, require better understanding [310, 371,372,373,374,375,376,377,378,379,380,381,382,383,384].
Taken together, the critical challenge in the RNAi therapeutics field is the development of highly efficient pipelines for cost-effective selection of RNAi drug candidates that will also allow reduction of safety-related drug attrition. However, overcoming this challenge requires better understanding and more open cooperation between both drug developers and academic researchers. Although basic research studies commonly utilize siRNAs and miRNA analogs to increase our understanding of molecular mechanisms governing human health, they often focus on simplified (single pathway limited) models and thus are difficult to transfer into drug development processes [48, 148, 271, 310, 385,386,387,388,389,390,391,392,393,394,395,396,397]. It also has to be stressed that the bioinformatics databases used to predict siRNA/miRNA consequences are generally solely based on scientific literature, and thus are only as valid and efficient as the research underpinning them. However, the scientific literature lacks negative data on ncRNAs function (due to publishing limitations); while the related high scale of comprehensive analysis of publicly deposited genome-wide transcriptomics data is very challenging due to the need for harmonization of transcriptomic approaches and statistical analyses [398,399,400,401]. Effectively, the general knowledge obtained by pharmaceutical companies during unsuccessful clinical trials or during general RNAi drug design processes is rarely shared with academics [25, 182, 281,282,283, 402].
Obviously, closer cooperation between the academic research and pharmacy business realms would help RNAi therapy to realize its full potential to benefit patients.
Availability of data and materials
Not applicable.
Abbreviations
- Ago2:
-
Argonaute 2
- dsRNA:
-
double stranded RNA
- EMA:
-
European Medicine Agency
- FDA:
-
US Food and Drug Administration
- hATTR:
-
hereditary transthyretin amyloidosis
- LNA:
-
locked nucleic acid
- LNP:
-
lipid nanoparticle system
- miRNA:
-
microRNA
- ncRNA:
-
non-coding RNA
- nt:
-
nucleotide
- piRNA:
-
piwi-interacting RNA
- PNA:
-
peptide nucleic acids
- RNAi:
-
RNA interference
- shRNA:
-
short hairpin RNA
- siRNA:
-
small interfering RNA
- SNP:
-
single nucleotide polymorphism
- ssRNA:
-
single-stranded RNA
- TRL:
-
Toll-like receptor
- TTR:
-
transthyretin
- UNA:
-
unlocked nucleic acid
References
Crick FH. On protein synthesis. Symp Soc Exp Biol. 1958;12:138–63.
Crick F. Central dogma of molecular biology. Nature. 1970;227:561–3.
Cobb M. 60 years ago, Francis crick changed the logic of biology. PLoS Biol. 2017;15:e2003243.
Hambly K, Danzer J, Muskal S, Debe DA. Interrogating the druggable genome with structural informatics. Mol Divers. 2006;10:273–81.
Russ AP, Lampel S. The druggable genome: an update. Drug Discov Today. 2005;10:1607–10.
Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–30.
Dimitrov DS. Therapeutic proteins. Methods Mol Biol. 2012;899:1–26.
Dimitrov DS, Marks JD. Therapeutic antibodies: current state and future trends--is a paradigm change coming soon? Methods Mol Biol. 2009;525:1–27 xiii.
Franco-Serrano L, Huerta M, Hernandez S, Cedano J, Perez-Pons J, Pinol J, et al. Multifunctional proteins: involvement in human diseases and targets of current drugs. Protein J. 2018;37:444–53.
Bumcrot D, Manoharan M, Koteliansky V, Sah DW. RNAi therapeutics: a potential new class of pharmaceutical drugs. Nat Chem Biol. 2006;2:711–9.
Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–11.
Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–8.
Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15:188–200.
Caplen NJ, Parrish S, Imani F, Fire A, Morgan RA. Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proc Natl Acad Sci U S A. 2001;98:9742–7.
Kleinman ME, Yamada K, Takeda A, Chandrasekaran V, Nozaki M, Baffi JZ, et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008;452:591–7.
DeVincenzo JP, Wilkinson T, Vaishnaw A, Cehelsky J, Meyers R, Nochur S, et al. Viral load drives disease in humans experimentally infected with respiratory syncytial virus. Am J Respir Crit Care Med. 2010;182:1305–14.
DeVincenzo J, Lambkin-Williams R, Wilkinson T, Cehelsky J, Nochur S, Walsh E, et al. A randomized, double-blind, placebo-controlled study of an RNAi-based therapy directed against respiratory syncytial virus. Proc Natl Acad Sci U S A. 2010;107:8800–5.
Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA, et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067–70.
Zuckerman JE, Davis ME. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat Rev Drug Discov. 2015;14:843–56.
Ackley KL. Are we there yet? An update on oligonucleotide drug development. Chim Oggi Chem Today. 2016;34:Xxxv–Xxxviii.
Haussecker D. The business of RNAi therapeutics in 2012. Mol Ther Nucleic Acids. 2012;1:e8.
Conde J, Artzi N. Are RNAi and miRNA therapeutics truly dead? Trends Biotechnol. 2015;33:141–4.
Kristen AV, Ajroud-Driss S, Conceicao I, Gorevic P, Kyriakides T, Obici L. Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegener Dis Manag. 2019;9:5–23.
Adams D, Gonzalez-Duarte A, O'Riordan WD, Yang CC, Ueda M, Kristen AV, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379:11–21.
Setten RL, Rossi JJ, Han SP. The current state and future directions of RNAi-based therapeutics. Nat Rev Drug Discov. 2019;18:421–46.
Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203–22.
Lam JK, Chow MY, Zhang Y, Leung SW. siRNA versus miRNA as therapeutics for gene silencing. Mol Ther Nucleic Acids. 2015;4:e252.
Bartoszewska S, Cabaj A, Dabrowski M, Collawn JF, Bartoszewski R. miR-34c-5p modulates X-box-binding protein 1 (XBP1) expression during the adaptive phase of the unfolded protein response. FASEB J. 2019;33:11541–54.
Zhao Z, Lin CY, Cheng K. siRNA- and miRNA-based therapeutics for liver fibrosis. Transl Res. 2019. https://doi.org/10.1016/j.trsl.2019.07.007.
Miroshnichenko S, Patutina O. Enhanced inhibition of tumorigenesis using combinations of miRNA-targeted therapeutics. Front Pharmacol. 2019;10:488.
Bansal P, Kumar A, Chandna S, Arora M, Bansal R. Targeting miRNA for therapeutics using a Micronome based method for identification of miRNA-mRNA pairs and validation of key regulator miRNA. Methods Mol Biol. 1823;2018:185–95.
Li DJ, Sun CC. Editorial: towards MiRNA based therapeutics for lung Cancer. Curr Pharm Des. 2018;23:5971–2.
Wu M, Wang G, Tian W, Deng Y, Xu Y. MiRNA-based therapeutics for lung Cancer. Curr Pharm Des. 2018;23:5989–96.
Wen MM. Getting miRNA therapeutics into the target cells for neurodegenerative diseases: a mini-review. Front Mol Neurosci. 2016;9:129.
Zhu K, Liu D, Lai H, Li J, Wang C. Developing miRNA therapeutics for cardiac repair in ischemic heart disease. J Thorac Dis. 2016;8:E918–E27.
Kouri FM, Ritner C, Stegh AH. miRNA-182 and the regulation of the glioblastoma phenotype - toward miRNA-based precision therapeutics. Cell Cycle. 2015;14:3794–800.
Mishra S, Yadav T, Rani V. Exploring miRNA based approaches in cancer diagnostics and therapeutics. Crit Rev Oncol Hematol. 2016;98:12–23.
Bernardo BC, Ooi JY, Lin RC, McMullen JR. miRNA therapeutics: a new class of drugs with potential therapeutic applications in the heart. Future Med Chem. 2015;7:1771–92.
Nouraee N, Mowla SJ. miRNA therapeutics in cardiovascular diseases: promises and problems. Front Genet. 2015;6:232.
Kwekkeboom RF, Lei Z, Doevendans PA, Musters RJ, Sluijter JP. Targeted delivery of miRNA therapeutics for cardiovascular diseases: opportunities and challenges. Clin Sci (Lond). 2014;127:351–65.
Pereira DM, Rodrigues PM, Borralho PM, Rodrigues CM. Delivering the promise of miRNA cancer therapeutics. Drug Discov Today. 2013;18:282–9.
Ford LP. Using synthetic miRNA mimics for diverting cell fate: a possibility of miRNA-based therapeutics? Leuk Res. 2006;30:511–3.
Bartoszewska S, Kamysz W, Jakiela B, Sanak M, Kroliczewski J, Bebok Z, et al. miR-200b downregulates CFTR during hypoxia in human lung epithelial cells. Cell Mol Biol Lett. 2017;22:23.
Kalinowski L, Janaszak-Jasiecka A, Siekierzycka A, Bartoszewska S, Wozniak M, Lejnowski D, et al. Posttranscriptional and transcriptional regulation of endothelial nitric-oxide synthase during hypoxia: the role of microRNAs. Cell Mol Biol Lett. 2016;21:16.
Bowes J, Brown AJ, Hamon J, Jarolimek W, Sridhar A, Waldron G, et al. Reducing safety-related drug attrition: the use of in vitro pharmacological profiling. Nat Rev Drug Discov. 2012;11:909–22.
Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet. 2006;15 Spec No 1:R17–29.
Almeida R, Allshire RC. RNA silencing and genome regulation. Trends Cell Biol. 2005;15:251–8.
Bartoszewski R, Sikorski AF. Editorial focus: entering into the non-coding RNA era. Cell Mol Biol Lett. 2018;23:45.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97.
Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee JK, et al. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell. 2006;125:887–901.
Wang X, Xu X, Ma Z, Huo Y, Xiao Z, Li Y, et al. Dynamic mechanisms for pre-miRNA binding and export by Exportin-5. RNA. 2011;17:1511–28.
Treiber T, Treiber N, Meister G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat Rev Mol Cell Biol. 2019;20:5–20.
Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15:509–24.
Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597–610.
Siomi H, Siomi MC. Posttranscriptional regulation of microRNA biogenesis in animals. Mol Cell. 2010;38:323–32.
Davis BN, Hata A. Regulation of MicroRNA biogenesis: a miRiad of mechanisms. Cell Commun Signal. 2009;7:18.
Cheloufi S, Dos Santos CO, Chong MM, Hannon GJ. A dicer-independent miRNA biogenesis pathway that requires ago catalysis. Nature. 2010;465:584–9.
Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009;11:228–34.
Shin C, Nam JW, Farh KK, Chiang HR, Shkumatava A, Bartel DP. Expanding the microRNA targeting code: functional sites with centered pairing. Mol Cell. 2010;38:789–802.
Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9:102–14.
Eystathioy T, Chan EK, Tenenbaum SA, Keene JD, Griffith K, Fritzler MJ. A phosphorylated cytoplasmic autoantigen, GW182, associates with a unique population of human mRNAs within novel cytoplasmic speckles. Mol Biol Cell. 2002;13:1338–51.
Gibbings DJ, Ciaudo C, Erhardt M, Voinnet O. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat Cell Biol. 2009;11:1143–9.
van Dijk E, Cougot N, Meyer S, Babajko S, Wahle E, Seraphin B. Human Dcp2: a catalytically active mRNA decapping enzyme located in specific cytoplasmic structures. EMBO J. 2002;21:6915–24.
Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song JJ, et al. Argonaute2 is the catalytic engine of mammalian RNAi. Science. 2004;305:1437–41.
Meister G, Landthaler M, Patkaniowska A, Dorsett Y, Teng G, Tuschl T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol Cell. 2004;15:185–97.
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33.
Kroliczewski J, Sobolewska A, Lejnowski D, Collawn JF, Bartoszewski R. microRNA single polynucleotide polymorphism influences on microRNA biogenesis and mRNA target specificity. Gene. 2018;640:66–72.
Moszyńska A, Gebert M, Collawn JF, Bartoszewski R. SNPs in microRNA target sites and their potential role in human disease. Open Biol. 2017;7(4):170019. https://doi.org/10.1098/rsob.170019.
Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–73.
Benhamed M, Herbig U, Ye T, Dejean A, Bischof O. Senescence is an endogenous trigger for microRNA-directed transcriptional gene silencing in human cells. Nat Cell Biol. 2012;14:266–75.
Hwang HW, Wentzel EA, Mendell JT. A hexanucleotide element directs microRNA nuclear import. Science. 2007;315:97–100.
Catalanotto C, Cogoni C, Zardo G. MicroRNA in control of gene expression: an overview of nuclear functions. Int J Mol Sci. 2016;17(10):1712.
Agrawal N, Dasaradhi PV, Mohmmed A, Malhotra P, Bhatnagar RK, Mukherjee SK. RNA interference: biology, mechanism, and applications. Microbiol Mol Biol Rev. 2003;67:657–85.
Tang G. siRNA and miRNA: an insight into RISCs. Trends Biochem Sci. 2005;30:106–14.
Valencia-Sanchez MA, Liu J, Hannon GJ, Parker R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 2006;20:515–24.
Doench JG, Petersen CP, Sharp PA. siRNAs can function as miRNAs. Genes Dev. 2003;17:438–42.
Lippman Z, May B, Yordan C, Singer T, Martienssen R. Distinct mechanisms determine transposon inheritance and methylation via small interfering RNA and histone modification. PLoS Biol. 2003;1:E67.
Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–55.
Czech B, Hannon GJ. One loop to rule them all: the ping-pong cycle and piRNA-guided silencing. Trends Biochem Sci. 2016;41:324–37.
Rojas-Rios P, Simonelig M. piRNAs and PIWI proteins: regulators of gene expression in development and stem cells. Development. 2018;145:dev161786.
Rouget C, Papin C, Boureux A, Meunier AC, Franco B, Robine N, et al. Maternal mRNA deadenylation and decay by the piRNA pathway in the early Drosophila embryo. Nature. 2010;467:1128–32.
Barckmann B, Pierson S, Dufourt J, Papin C, Armenise C, Port F, et al. Aubergine iCLIP reveals piRNA-dependent decay of mRNAs involved in germ cell development in the early embryo. Cell Rep. 2015;12:1205–16.
Sivagurunathan S, Arunachalam JP, Chidambaram S. PIWI-like protein, HIWI2 is aberrantly expressed in retinoblastoma cells and affects cell-cycle potentially through OTX2. Cell Mol Biol Lett. 2017;22:17.
Ponnusamy M, Yan KW, Liu CY, Li PF, Wang K. PIWI family emerging as a decisive factor of cell fate: an overview. Eur J Cell Biol. 2017;96:746–57.
Burroughs AM, Iyer LM, Aravind L. Two novel PIWI families: roles in inter-genomic conflicts in bacteria and mediator-dependent modulation of transcription in eukaryotes. Biol Direct. 2013;8:13.
Kwon C, Tak H, Rho M, Chang HR, Kim YH, Kim KT, et al. Detection of PIWI and piRNAs in the mitochondria of mammalian cancer cells. Biochem Biophys Res Commun. 2014;446:218–23.
Suzuki R, Honda S, Kirino Y. PIWI expression and function in Cancer. Front Genet. 2012;3:204.
Martinez VD, Vucic EA, Thu KL, Hubaux R, Enfield KS, Pikor LA, et al. Unique somatic and malignant expression patterns implicate PIWI-interacting RNAs in cancer-type specific biology. Sci Rep. 2015;5:10423.
Mei Y, Wang Y, Kumari P, Shetty AC, Clark D, Gable T, et al. A piRNA-like small RNA interacts with and modulates p-ERM proteins in human somatic cells. Nat Commun. 2015;6:7316.
Mei Y, Clark D, Mao L. Novel dimensions of piRNAs in cancer. Cancer Lett. 2013;336:46–52.
Gebert M, Bartoszewska S, Janaszak-Jasiecka A, Moszynska A, Cabaj A, Kroliczewski J, et al. PIWI proteins contribute to apoptosis during the UPR in human airway epithelial cells. Sci Rep. 2018;8:16431.
Saberi F, Kamali M, Najafi A, Yazdanparast A, Moghaddam MM. Natural antisense RNAs as mRNA regulatory elements in bacteria: a review on function and applications. Cell Mol Biol Lett. 2016;21:6.
Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014;157:77–94.
Ishizu H, Siomi H, Siomi MC. Biology of PIWI-interacting RNAs: new insights into biogenesis and function inside and outside of germlines. Genes Dev. 2012;26:2361–73.
Erhard F, Zimmer R. Classification of ncRNAs using position and size information in deep sequencing data. Bioinformatics. 2010;26:i426–32.
Szell M, Bata-Csorgo Z, Kemeny L. The enigmatic world of mRNA-like ncRNAs: their role in human evolution and in human diseases. Semin Cancer Biol. 2008;18:141–8.
Huang R, Jaritz M, Guenzl P, Vlatkovic I, Sommer A, Tamir IM, et al. An RNA-Seq strategy to detect the complete coding and non-coding transcriptome including full-length imprinted macro ncRNAs. PLoS One. 2011;6:e27288.
Gurtan AM, Sharp PA. The role of miRNAs in regulating gene expression networks. J Mol Biol. 2013;425:3582–600.
Xu H, Zhang Y, Xie SJ, Xu SJ, Zhou H, Qu LH. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps during heart development. Cardiology. 2013;126:62.
Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–86.
Olejniczak M, Kotowska-Zimmer A, Krzyzosiak W. Stress-induced changes in miRNA biogenesis and functioning. Cell Mol Life Sci. 2018;75:177–91.
van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–9.
Leung AK, Sharp PA. MicroRNA functions in stress responses. Mol Cell. 2010;40:205–15.
Mendell JT, Olson EN. MicroRNAs in stress signaling and human disease. Cell. 2012;148:1172–87.
Ge XL, Wang JL, Liu X, Zhang J, Liu C, Guo L. Inhibition of miR-19a protects neurons against ischemic stroke through modulating glucose metabolism and neuronal apoptosis. Cell Mol Biol Lett. 2019;24:37.
Lu G, Zhang Y. MicroRNA-340-5p suppresses non-small cell lung cancer cell growth and metastasis by targeting ZNF503. Cell Mol Biol Lett. 2019;24:34.
Fu Y, Lin L, Xia L. MiR-107 function as a tumor suppressor gene in colorectal cancer by targeting transferrin receptor 1. Cell Mol Biol Lett. 2019;24:31.
Liu L, Zuo Y, Xu Y, Zhang Z, Li Y, Pang J. MiR-613 inhibits proliferation and invasion and induces apoptosis of rheumatoid arthritis synovial fibroblasts by direct down-regulation of DKK1. Cell Mol Biol Lett. 2019;24:8.
Cai Y, Li Y. Upregulation of miR-29b-3p protects cardiomyocytes from hypoxia-induced apoptosis by targeting TRAF5. Cell Mol Biol Lett. 2019;24:27.
Li S, Sun Z, Chen T, Pan J, Shen Y, Chen X, et al. The role of miR-431-5p in regulating pulmonary surfactant expression in vitro. Cell Mol Biol Lett. 2019;24:25.
Sun L, Lian JX, Meng S. MiR-125a-5p promotes osteoclastogenesis by targeting TNFRSF1B. Cell Mol Biol Lett. 2019;24:23.
Zeng Q, Wang Y, Gao J, Yan Z, Li Z, Zou X, et al. miR-29b-3p regulated osteoblast differentiation via regulating IGF-1 secretion of mechanically stimulated osteocytes. Cell Mol Biol Lett. 2019;24:11.
Yang Y, Bao Y, Yang GK, Wan J, Du LJ, Ma ZH. MiR-214 sensitizes human colon cancer cells to 5-FU by targeting Hsp27. Cell Mol Biol Lett. 2019;24:22.
Ma F, Lin P, Chen Q, Lu X, Zhang YE, Wu CI. Direct measurement of pervasive weak repression by microRNAs and their role at the network level. BMC Genomics. 2018;19:362.
van Rooij E, Kauppinen S. Development of microRNA therapeutics is coming of age. Embo Molec Med. 2014;6:851–64.
Gantier MP, Williams BR. The response of mammalian cells to double-stranded RNA. Cytokine Growth Factor Rev. 2007;18:363–71.
Lee HY, Zhou K, Smith AM, Noland CL, Doudna JA. Differential roles of human dicer-binding proteins TRBP and PACT in small RNA processing. Nucleic Acids Res. 2013;41:6568–76.
Cifuentes D, Xue H, Taylor DW, Patnode H, Mishima Y, Cheloufi S, et al. A novel miRNA processing pathway independent of dicer requires Argonaute2 catalytic activity. Science. 2010;328:1694–8.
Kim DH, Behlke MA, Rose SD, Chang MS, Choi S, Rossi JJ. Synthetic dsRNA dicer substrates enhance RNAi potency and efficacy. Nat Biotechnol. 2005;23:222–6.
Siolas D, Lerner C, Burchard J, Ge W, Linsley PS, Paddison PJ, et al. Synthetic shRNAs as potent RNAi triggers. Nat Biotechnol. 2005;23:227–31.
Hefner E, Clark K, Whitman C, Behlke MA, Rose SD, Peek AS, et al. Increased potency and longevity of gene silencing using validated dicer substrates. J Biomol Tech. 2008;19:231–7.
Snead NM, Wu X, Li A, Cui Q, Sakurai K, Burnett JC, et al. Molecular basis for improved gene silencing by dicer substrate interfering RNA compared with other siRNA variants. Nucleic Acids Res. 2013;41:6209–21.
Parmar R, Willoughby JL, Liu J, Foster DJ, Brigham B, Theile CS, et al. 5′-(E)-Vinylphosphonate: a stable phosphate mimic can improve the RNAi activity of siRNA-GalNAc conjugates. Chembiochem. 2016;17:985–9.
Ly S, Navaroli DM, Didiot MC, Cardia J, Pandarinathan L, Alterman JF, et al. Visualization of self-delivering hydrophobically modified siRNA cellular internalization. Nucleic Acids Res. 2017;45:15–25.
Yu D, Pendergraff H, Liu J, Kordasiewicz HB, Cleveland DW, Swayze EE, et al. Single-stranded RNAs use RNAi to potently and allele-selectively inhibit mutant huntingtin expression. Cell. 2012;150:895–908.
Lima WF, Prakash TP, Murray HM, Kinberger GA, Li W, Chappell AE, et al. Single-stranded siRNAs activate RNAi in animals. Cell. 2012;150:883–94.
Stein CA, Hansen JB, Lai J, Wu S, Voskresenskiy A, Hog A, et al. Efficient gene silencing by delivery of locked nucleic acid antisense oligonucleotides, unassisted by transfection reagents. Nucleic Acids Res. 2010;38:e3.
Shi B, Ma C, Liu G, Guo Y. MiR-106a directly targets LIMK1 to inhibit proliferation and EMT of oral carcinoma cells. Cell Mol Biol Lett. 2019;24:1.
Terasawa K, Shimizu K, Tsujimoto G. Synthetic pre-miRNA-based shRNA as potent RNAi triggers. J Nucleic Acids. 2011;2011:131579.
Wang H, Jiang Y, Peng H, Chen Y, Zhu P, Huang Y. Recent progress in microRNA delivery for cancer therapy by non-viral synthetic vectors. Adv Drug Deliv Rev. 2015;81:142–60.
Zhang Y, Wang Z, Gemeinhart RA. Progress in microRNA delivery. J Control Release. 2013;172:962–74.
Stenvang J, Petri A, Lindow M, Obad S, Kauppinen S. Inhibition of microRNA function by antimiR oligonucleotides. Silence. 2012;3:1.
Velu CS, Grimes HL. Utilizing antagomiR (antisense microRNA) to knock down microRNA in murine bone marrow cells. Methods Mol Biol. 2012;928:185–95.
Esau CC. Inhibition of microRNA with antisense oligonucleotides. Methods. 2008;44:55–60.
Wang JY, Cheng H, Zhang HY, Ye YQ, Feng Q, Chen ZM, et al. Suppressing microRNA-29c promotes biliary atresia-related fibrosis by targeting DNMT3A and DNMT3B. Cell Mol Biol Lett. 2019;24:10.
Gong ZH, Zhou F, Shi C, Xiang T, Zhou CK, Wang QQ, et al. miRNA-221 promotes cutaneous squamous cell carcinoma progression by targeting PTEN. Cell Mol Biol Lett. 2019;24:9.
Chen L, Hu W, Li G, Guo Y, Wan Z, Yu J. Inhibition of miR-9-5p suppresses prostate cancer progress by targeting StarD13. Cell Mol Biol Lett. 2019;24:20.
Davis S, Propp S, Freier SM, Jones LE, Serra MJ, Kinberger G, et al. Potent inhibition of microRNA in vivo without degradation. Nucleic Acids Res. 2009;37:70–7.
Elmen J, Lindow M, Schutz S, Lawrence M, Petri A, Obad S, et al. LNA-mediated microRNA silencing in non-human primates. Nature. 2008;452:896–9.
Torres AG, Fabani MM, Vigorito E, Gait MJ. MicroRNA fate upon targeting with anti-miRNA oligonucleotides as revealed by an improved northern-blot-based method for miRNA detection. RNA. 2011;17:933–43.
Krutzfeldt J, Kuwajima S, Braich R, Rajeev KG, Pena J, Tuschl T, et al. Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res. 2007;35:2885–92.
Staton AA, Giraldez AJ. Use of target protector morpholinos to analyze the physiological roles of specific miRNA-mRNA pairs in vivo. Nat Protoc. 2011;6:2035–49.
Summerton JE. Morpholino, siRNA, and S-DNA compared: impact of structure and mechanism of action on off-target effects and sequence specificity. Curr Top Med Chem. 2007;7:651–60.
Hudziak RM, Barofsky E, Barofsky DF, Weller DL, Huang SB, Weller DD. Resistance of morpholino phosphorodiamidate oligomers to enzymatic degradation. Antisense Nucleic Acid Drug Dev. 1996;6:267–72.
Summerton J. Morpholino antisense oligomers: the case for an RNase H-independent structural type. Biochim Biophys Acta. 1999;1489:141–58.
Arora V, Devi GR, Iversen PL. Neutrally charged phosphorodiamidate morpholino antisense oligomers: uptake, efficacy and pharmacokinetics. Curr Pharm Biotechnol. 2004;5:431–9.
Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. Rational siRNA design for RNA interference. Nat Biotechnol. 2004;22:326–30.
Fakhr E, Zare F, Teimoori-Toolabi L. Precise and efficient siRNA design: a key point in competent gene silencing. Cancer Gene Ther. 2016;23:73–82.
Chaudhary A, Srivastava S, Garg S. Development of a software tool and criteria evaluation for efficient design of small interfering RNA. Biochem Biophys Res Commun. 2011;404:313–20.
Zhong R, Kim J, Kim HS, Kim M, Lum L, Levine B, et al. Computational detection and suppression of sequence-specific off-target phenotypes from whole genome RNAi screens. Nucleic Acids Res. 2014;42:8214–22.
Hajalirezay Yazdi S, Paryan M, Mohammadi-Yeganeh S. An integrated approach of bioinformatic prediction and in vitro analysis identified that miR-34a targets MET and AXL in triple-negative breast cancer. Cell Mol Biol Lett. 2018;23:51.
Saxena S, Jonsson ZO, Dutta A. Small RNAs with imperfect match to endogenous mRNA repress translation. Implications for off-target activity of small inhibitory RNA in mammalian cells. J Biol Chem. 2003;278:44312–9.
Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, et al. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol. 2003;21:635–7.
Scacheri PC, Rozenblatt-Rosen O, Caplen NJ, Wolfsberg TG, Umayam L, Lee JC, et al. Short interfering RNAs can induce unexpected and divergent changes in the levels of untargeted proteins in mammalian cells. Proc Natl Acad Sci U S A. 2004;101:1892–7.
Holen T, Moe SE, Sorbo JG, Meza TJ, Ottersen OP, Klungland A. Tolerated wobble mutations in siRNAs decrease specificity, but can enhance activity in vivo. Nucleic Acids Res. 2005;33:4704–10.
Eastman P, Shi J, Ramsundar B, Pande VS. Solving the RNA design problem with reinforcement learning. PLoS Comput Biol. 2018;14:e1006176.
Petri S, Meister G. siRNA design principles and off-target effects. Methods Mol Biol. 2013;986:59–71.
Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias (vol 115, pg 209, 2003). Cell. 2003;115:505.
Angart P, Vocelle D, Chan C, Walton SP. Design of siRNA therapeutics from the molecular scale. Pharmaceuticals (Basel). 2013;6:440–68.
Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199–208.
Wang X, Varma RK, Beauchamp L, Magdaleno S, Sendera TJ. Selection of hyperfunctional siRNAs with improved potency and specificity. Nucleic Acids Res. 2009;37:e152.
Birmingham A, Anderson E, Sullivan K, Reynolds A, Boese Q, Leake D, et al. A protocol for designing siRNAs with high functionality and specificity. Nat Protoc. 2007;2:2068–78.
Ui-Tei K, Naito Y, Nishi K, Juni A, Saigo K. Thermodynamic stability and Watson-Crick base pairing in the seed duplex are major determinants of the efficiency of the siRNA-based off-target effect. Nucleic Acids Res. 2008;36:7100–9.
Kawasaki T, Kawai T. Chapter one - discrimination between self and non-self-nucleic acids by the innate immune system. In: Vanpouille-Box C, Galluzzi L, editors. International review of cell and molecular biology. Volume 344. Cambridge: Academic Press; 2019. p. 1–30.
Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, et al. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med. 2005;11:263–70.
Judge AD, Sood V, Shaw JR, Fang D, McClintock K, MacLachlan I. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat Biotechnol. 2005;23:457–62.
Reynolds A, Anderson EM, Vermeulen A, Fedorov Y, Robinson K, Leake D, et al. Induction of the interferon response by siRNA is cell type- and duplex length-dependent. RNA. 2006;12:988–93.
Fedorov Y, Anderson EM, Birmingham A, Reynolds A, Karpilow J, Robinson K, et al. Off-target effects by siRNA can induce toxic phenotype. RNA. 2006;12:1188–96.
Sioud M. Overcoming the challenges of siRNA activation of innate immunity: design better therapeutic siRNAs. Methods Mol Biol. 2015;1218:301–19.
Robbins M, Judge A, MacLachlan I. siRNA and innate immunity. Oligonucleotides. 2009;19:89–102.
Goodchild A, Nopper N, King A, Doan T, Tanudji M, Arndt GM, et al. Sequence determinants of innate immune activation by short interfering RNAs. BMC Immunol. 2009;10:40.
Bramsen JB, Kjems J. Engineering small interfering RNAs by strategic chemical modification. Methods Mol Biol. 2013;942:87–109.
Marques JT, Williams BR. Activation of the mammalian immune system by siRNAs. Nat Biotechnol. 2005;23:1399–405.
Chen L, Liu P, Evans TC Jr, Ettwiller LM. DNA damage is a pervasive cause of sequencing errors, directly confounding variant identification. Science. 2017;355:752–6.
Chugh P, Dittmer DP. Potential pitfalls in microRNA profiling. Wiley Interdiscip Rev RNA. 2012;3:601–16.
Van Peer G, Lefever S, Anckaert J, Beckers A, Rihani A, Van Goethem A, et al. miRBase Tracker: keeping track of microRNA annotation changes. Database (Oxford). 2014;2014:bau080.
Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019;47:D155–D62.
Caffrey DR, Zhao J, Song Z, Schaffer ME, Haney SA, Subramanian RR, et al. SiRNA off-target effects can be reduced at concentrations that match their individual potency. PLoS One. 2011;6:e21503.
Layzer JM, McCaffrey AP, Tanner AK, Huang Z, Kay MA, Sullenger BA. In vivo activity of nuclease-resistant siRNAs. RNA. 2004;10:766–71.
Bramsen JB, Laursen MB, Nielsen AF, Hansen TB, Bus C, Langkjaer N, et al. A large-scale chemical modification screen identifies design rules to generate siRNAs with high activity, high stability and low toxicity. Nucleic Acids Res. 2009;37:2867–81.
Behlke MA. Chemical modification of siRNAs for in vivo use. Oligonucleotides. 2008;18:305–19.
Haussecker D. Current issues of RNAi therapeutics delivery and development. J Control Release. 2014;195:49–54.
Wang Y, Sheng G, Juranek S, Tuschl T, Patel DJ. Structure of the guide-strand-containing argonaute silencing complex. Nature. 2008;456:209–13.
Balaratnam S, West N, Basu S. A piRNA utilizes HILI and HIWI2 mediated pathway to down-regulate ferritin heavy chain 1 mRNA in human somatic cells. Nucleic Acids Res. 2018;46:10635–48.
Chiu YL, Rana TM. siRNA function in RNAi: a chemical modification analysis. RNA. 2003;9:1034–48.
Judge AD, Bola G, Lee AC, MacLachlan I. Design of noninflammatory synthetic siRNA mediating potent gene silencing in vivo. Mol Ther. 2006;13:494–505.
Cekaite L, Furset G, Hovig E, Sioud M. Gene expression analysis in blood cells in response to unmodified and 2′-modified siRNAs reveals TLR-dependent and independent effects. J Mol Biol. 2007;365:90–108.
Hamm S, Latz E, Hangel D, Muller T, Yu P, Golenbock D, et al. Alternating 2′-O-ribose methylation is a universal approach for generating non-stimulatory siRNA by acting as TLR7 antagonist. Immunobiology. 2010;215:559–69.
Odadzic D, Bramsen JB, Smicius R, Bus C, Kjems J, Engels JW. Synthesis of 2′-O-modified adenosine building blocks and application for RNA interference. Bioorg Med Chem. 2008;16:518–29.
Amarzguioui M, Holen T, Babaie E, Prydz H. Tolerance for mutations and chemical modifications in a siRNA. Nucleic Acids Res. 2003;31:589–95.
Prakash TP, Allerson CR, Dande P, Vickers TA, Sioufi N, Jarres R, et al. Positional effect of chemical modifications on short interference RNA activity in mammalian cells. J Med Chem. 2005;48:4247–53.
Veedu RN, Wengel J. Locked nucleic acids: promising nucleic acid analogs for therapeutic applications. Chem Biodivers. 2010;7:536–42.
Elmen J, Thonberg H, Ljungberg K, Frieden M, Westergaard M, Xu Y, et al. Locked nucleic acid (LNA) mediated improvements in siRNA stability and functionality. Nucleic Acids Res. 2005;33:439–47.
Grunweller A, Wyszko E, Bieber B, Jahnel R, Erdmann VA, Kurreck J. Comparison of different antisense strategies in mammalian cells using locked nucleic acids, 2′-O-methyl RNA, phosphorothioates and small interfering RNA. Nucleic Acids Res. 2003;31:3185–93.
Laursen MB, Pakula MM, Gao S, Fluiter K, Mook OR, Baas F, et al. Utilization of unlocked nucleic acid (UNA) to enhance siRNA performance in vitro and in vivo. Mol BioSyst. 2010;6:862–70.
Vaish N, Chen F, Seth S, Fosnaugh K, Liu Y, Adami R, et al. Improved specificity of gene silencing by siRNAs containing unlocked nucleobase analogs. Nucleic Acids Res. 2011;39:1823–32.
Campbell JM, Bacon TA, Wickstrom E. Oligodeoxynucleoside phosphorothioate stability in subcellular extracts, culture media, sera and cerebrospinal fluid. J Biochem Biophys Methods. 1990;20:259–67.
Eckstein F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 2014;24:374–87.
Braasch DA, Paroo Z, Constantinescu A, Ren G, Oz OK, Mason RP, et al. Biodistribution of phosphodiester and phosphorothioate siRNA. Bioorg Med Chem Lett. 2004;14:1139–43.
Schwarz DS, Tomari Y, Zamore PD. The RNA-induced silencing complex is a Mg2+−dependent endonuclease. Curr Biol. 2004;14:787–91.
Choung S, Kim YJ, Kim S, Park HO, Choi YC. Chemical modification of siRNAs to improve serum stability without loss of efficacy. Biochem Biophys Res Commun. 2006;342:919–27.
Shmushkovich T, Monopoli KR, Homsy D, Leyfer D, Betancur-Boissel M, Khvorova A, et al. Functional features defining the efficacy of cholesterol-conjugated, self-deliverable, chemically modified siRNAs. Nucleic Acids Res. 2018;46:10905–16.
Ligtenberg MA. Pico de Coana Y, Shmushkovich T, Yoshimoto Y, Truxova I, Yang Y, et al. self-delivering RNAi targeting PD-1 improves tumor-specific T cell functionality for adoptive cell therapy of malignant melanoma. Mol Ther. 2018;26:1482–93.
Meade BR, Gogoi K, Hamil AS, Palm-Apergi C, van den Berg A, Hagopian JC, et al. Efficient delivery of RNAi prodrugs containing reversible charge-neutralizing phosphotriester backbone modifications. Nat Biotechnol. 2014;32:1256–61.
Nielsen PE. Peptide nucleic acid (PNA) a structural DNA mimic. Prog Biophys Mol Biol. 1996;65:Sb103.
Nielsen PE, Egholm M, Buchardt O. Peptide nucleic acid (PNA). A DNA mimic with a peptide backbone. Bioconjug Chem. 1994;5:3–7.
Willoughby JLS, Chan A, Sehgal A, Butler JS, Nair JK, Racie T, et al. Evaluation of GalNAc-siRNA conjugate activity in pre-clinical animal models with reduced Asialoglycoprotein receptor expression. Mol Ther. 2018;26:105–14.
Nair JK, Attarwala H, Sehgal A, Wang Q, Aluri K, Zhang X, et al. Impact of enhanced metabolic stability on pharmacokinetics and pharmacodynamics of GalNAc-siRNA conjugates. Nucleic Acids Res. 2017;45:10969–77.
Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non-viral vectors for gene-based therapy. Nat Rev Genet. 2014;15:541–55.
Dowdy SF. Overcoming cellular barriers for RNA therapeutics. Nat Biotechnol. 2017;35:222–9.
Chung KH, Hart CC, Al-Bassam S, Avery A, Taylor J, Patel PD, et al. Polycistronic RNA polymerase II expression vectors for RNA interference based on BIC/miR-155. Nucleic Acids Res. 2006;34:e53.
Xia XG, Zhou H, Xu Z. Multiple shRNAs expressed by an inducible pol II promoter can knock down the expression of multiple target genes. Biotechniques. 2006;41:64–8.
Sun D, Melegari M, Sridhar S, Rogler CE, Zhu L. Multi-miRNA hairpin method that improves gene knockdown efficiency and provides linked multi-gene knockdown. Biotechniques. 2006;41:59–63.
Sui G, Soohoo C, Affar el B, Gay F, Shi Y, Forrester WC. A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc Natl Acad Sci U S A. 2002;99:5515–20.
Gupta S, Schoer RA, Egan JE, Hannon GJ, Mittal V. Inducible, reversible, and stable RNA interference in mammalian cells. Proc Natl Acad Sci U S A. 2004;101:1927–32.
Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–3.
Couto LB, High KA. Viral vector-mediated RNA interference. Curr Opin Pharmacol. 2010;10:534–42.
Brandt MR, Kirste AG, Pozzuto T, Schubert S, Kandolf R, Fechner H, et al. Adenovirus vector-mediated RNA interference for the inhibition of human parvovirus B19 replication. Virus Res. 2013;176:155–60.
Aagaard L, Rossi JJ. RNAi therapeutics: principles, prospects and challenges. Adv Drug Deliv Rev. 2007;59:75–86.
Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 2002;16:948–58.
Ehlert EM, Eggers R, Niclou SP, Verhaagen J. Cellular toxicity following application of adeno-associated viral vector-mediated RNA interference in the nervous system. BMC Neurosci. 2010;11:20.
Davidson BL, McCray PB Jr. Current prospects for RNA interference-based therapies. Nat Rev Genet. 2011;12:329–40.
Carlin CR. New Insights to Adenovirus-Directed Innate Immunity in Respiratory Epithelial Cells. Microorganisms. 2019;7(8):216.
Hendrickx R, Stichling N, Koelen J, Kuryk L, Lipiec A, Greber UF. Innate immunity to adenovirus. Hum Gene Ther. 2014;25:265–84.
Baden LR, Liu J, Li H, Walsh S, Johnson J, Milner D, et al. A phase1 clinical trial to evaluate the safety, mucosal and innate immunity of adenovirus type 26 HIV-1 vaccine in healthy, HIV-1 uninfected adults. AIDS Res Hum Retrovir. 2011;27:A124.
Baum C, Kustikova O, Modlich U, Li Z, Fehse B. Mutagenesis and oncogenesis by chromosomal insertion of gene transfer vectors. Hum Gene Ther. 2006;17:253–63.
Pauwels K, Gijsbers R, Toelen J, Schambach A, Willard-Gallo K, Verheust C, et al. State-of-the-art lentiviral vectors for research use: risk assessment and biosafety recommendations. Curr Gene Ther. 2009;9:459–74.
Seow Y, Wood MJ. Biological gene delivery vehicles: beyond viral vectors. Mol Ther. 2009;17:767–77.
Polansky H, Schwab H. Latent viruses can cause disease by disrupting the competition for the limiting factor p300/CBP. Cell Mol Biol Lett. 2018;23:56.
Toporkiewicz M, Meissner J, Matusewicz L, Czogalla A, Sikorski AF. Toward a magic or imaginary bullet? Ligands for drug targeting to cancer cells: principles, hopes, and challenges. Int J Nanomedicine. 2015;10:1399–414.
Wyrozumska P, Meissner J, Toporkiewicz M, Szarawarska M, Kuliczkowski K, Ugorski M, et al. Liposome-coated lipoplex-based carrier for antisense oligonucleotides. Cancer Biol Ther. 2015;16:66–76.
Meissner JM, Toporkiewicz M, Czogalla A, Matusewicz L, Kuliczkowski K, Sikorski AF. Novel antisense therapeutics delivery systems: in vitro and in vivo studies of liposomes targeted with anti-CD20 antibody. J Control Release. 2015;220:515–28.
Wyrozumska P, Stebelska K, Grzybek M, Sikorski AF. Synthetic vectors for genetic drug delivery. In: Mozafari MR, editor. Nanocarrier technologies: frontiers of nanotherapy. Dordrecht: Springer Netherlands; 2006. p. 139–74. https://doi.org/10.1007/978-1-4020-5041-1_9.
Tseng YC, Mozumdar S, Huang L. Lipid-based systemic delivery of siRNA. Adv Drug Deliv Rev. 2009;61:721–31.
Cullis PR, Hope MJ. Lipid nanoparticle Systems for Enabling Gene Therapies. Mol Ther. 2017;25:1467–75.
Jayaraman M, Ansell SM, Mui BL, Tam YK, Chen J, Du X, et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew Chem Int Ed Eng. 2012;51:8529–33.
Neuberg P, Kichler A. Recent developments in nucleic acid delivery with polyethylenimines. Adv Genet. 2014;88:263–88.
Urban-Klein B, Werth S, Abuharbeid S, Czubayko F, Aigner A. RNAi-mediated gene-targeting through systemic application of polyethylenimine (PEI)-complexed siRNA in vivo. Gene Ther. 2005;12:461–6.
Somani S, Laskar P, Altwaijry N, Kewcharoenvong P, Irving C, Robb G, et al. PEGylation of polypropylenimine dendrimers: effects on cytotoxicity, DNA condensation, gene delivery and expression in cancer cells. Sci Rep. 2018;8:9410.
Peng L. Dendrimers as nanovectors for Rna delivery in gene therapy. Eur J Pharm Sci. 2013;50:E31.
Dufes C, Uchegbu IF, Schatzlein AG. Dendrimers in gene delivery. Adv Drug Deliv Rev. 2005;57:2177–202.
Shcharbin D, Shakhbazau A, Bryszewska M. Poly(amidoamine) dendrimer complexes as a platform for gene delivery. Expert Opin Drug Deliv. 2013;10:1687–98.
Liu X, Li G, Su Z, Jiang Z, Chen L, Wang J, et al. Poly(amido amine) is an ideal carrier of miR-7 for enhancing gene silencing effects on the EGFR pathway in U251 glioma cells. Oncol Rep. 2013;29:1387–94.
Shah V, Taratula O, Garbuzenko OB, Taratula OR, Rodriguez-Rodriguez L, Minko T. Targeted nanomedicine for suppression of CD44 and simultaneous cell death induction in ovarian cancer: an optimal delivery of siRNA and anticancer drug. Clin Cancer Res. 2013;19:6193–204.
Siu KS, Chen D, Zheng X, Zhang X, Johnston N, Liu Y, et al. Non-covalently functionalized single-walled carbon nanotube for topical siRNA delivery into melanoma. Biomaterials. 2014;35:3435–42.
Chien C-S, Chiou S-H. Abstract 3321: Cationic polyurethanes-short branch PEI-mediated delivery of Mir145 inhibited epithelial-mesenchymal transdifferentiation and cancer stem-like properties and in lung adenocarcinoma. Cancer Res. 2012;72:3321.
Davis ME, Brewster ME. Cyclodextrin-based pharmaceutics: past, present and future. Nat Rev Drug Discov. 2004;3:1023–35.
Bartlett DW, Davis ME. Impact of tumor-specific targeting and dosing schedule on tumor growth inhibition after intravenous administration of siRNA-containing nanoparticles. Biotechnol Bioeng. 2008;99:975–85.
Videira M, Arranja A, Rafael D, Gaspar R. Preclinical development of siRNA therapeutics: towards the match between fundamental science and engineered systems. Nanomedicine. 2014;10:689–702.
Bitar A, Ahmad NM, Fessi H, Elaissari A. Silica-based nanoparticles for biomedical applications. Drug Discov Today. 2012;17:1147–54.
Chen Y, Wang X, Liu T, Zhang DS, Wang Y, Gu H, et al. Highly effective antiangiogenesis via magnetic mesoporous silica-based siRNA vehicle targeting the VEGF gene for orthotopic ovarian cancer therapy. Int J Nanomedicine. 2015;10:2579–94.
Chen Y, Gu H, Zhang DS, Li F, Liu T, Xia W. Highly effective inhibition of lung cancer growth and metastasis by systemic delivery of siRNA via multimodal mesoporous silica-based nanocarrier. Biomaterials. 2014;35:10058–69.
Kumar P, Ban HS, Kim SS, Wu H, Pearson T, Greiner DL, et al. T cell-specific siRNA delivery suppresses HIV-1 infection in humanized mice. Cell. 2008;134:577–86.
Peer D, Zhu P, Carman CV, Lieberman J, Shimaoka M. Selective gene silencing in activated leukocytes by targeting siRNAs to the integrin lymphocyte function-associated antigen-1. Proc Natl Acad Sci U S A. 2007;104:4095–100.
Serrano-Sevilla I, Artiga Á, Mitchell SG, De Matteis L, de la Fuente JM. Natural polysaccharides for siRNA delivery: Nanocarriers based on chitosan, hyaluronic acid, and their derivatives. Molecules. 2019;24(14):2570.
Deng X, Cao M, Zhang J, Hu K, Yin Z, Zhou Z, et al. Hyaluronic acid-chitosan nanoparticles for co-delivery of MiR-34a and doxorubicin in therapy against triple negative breast cancer. Biomaterials. 2014;35:4333–44.
Kawakami E, Kawai N, Kinouchi N, Mori H, Ohsawa Y, Ishimaru N, et al. Local applications of myostatin-siRNA with atelocollagen increase skeletal muscle mass and recovery of muscle function. PLoS One. 2013;8:e64719.
Yhee JY, Song S, Lee SJ, Park SG, Kim KS, Kim MG, et al. Cancer-targeted MDR-1 siRNA delivery using self-cross-linked glycol chitosan nanoparticles to overcome drug resistance. J Control Release. 2015;198:1–9.
Schafer J, Hobel S, Bakowsky U, Aigner A. Liposome-polyethylenimine complexes for enhanced DNA and siRNA delivery. Biomaterials. 2010;31:6892–900.
Jang YL, Yun UJ, Lee MS, Kim MG, Son S, Lee K, et al. Cell-penetrating peptide mimicking polymer-based combined delivery of paclitaxel and siRNA for enhanced tumor growth suppression. Int J Pharm. 2012;434:488–93.
Alshamsan A, Hamdy S, Samuel J, El-Kadi AO, Lavasanifar A, Uludag H. The induction of tumor apoptosis in B16 melanoma following STAT3 siRNA delivery with a lipid-substituted polyethylenimine. Biomaterials. 2010;31:1420–8.
Soucek L, Whitfield JR, Sodir NM, Masso-Valles D, Serrano E, Karnezis AN, et al. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013;27:504–13.
Sood P, Krek A, Zavolan M, Macino G, Rajewsky N. Cell-type-specific signatures of microRNAs on target mRNA expression. Proc Natl Acad Sci U S A. 2006;103:2746–51.
Nowakowski TJ, Rani N, Golkaram M, Zhou HR, Alvarado B, Huch K, et al. Regulation of cell-type-specific transcriptomes by microRNA networks during human brain development. Nat Neurosci. 2018;21:1784–92.
Song E, Zhu P, Lee SK, Chowdhury D, Kussman S, Dykxhoorn DM, et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotechnol. 2005;23:709–17.
McNamara JO 2nd, Andrechek ER, Wang Y, Viles KD, Rempel RE, Gilboa E, et al. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat Biotechnol. 2006;24:1005–15.
Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M, et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432:173–8.
Zhou JH, Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat Rev Drug Discov. 2017;16:181–202.
Cuellar TL, Barnes D, Nelson C, Tanguay J, Yu SF, Wen X, et al. Systematic evaluation of antibody-mediated siRNA delivery using an industrial platform of THIOMAB-siRNA conjugates. Nucleic Acids Res. 2015;43:1189–203.
Kim SW, Kim NY, Choi YB, Park SH, Yang JM, Shin S. RNA interference in vitro and in vivo using an arginine peptide/siRNA complex system. J Control Release. 2010;143:335–43.
Behlke MA. Progress towards in vivo use of siRNAs. Mol Ther. 2006;13:644–70.
Xu C, Haque F, Jasinski DL, Binzel DW, Shu D, Guo P. Favorable biodistribution, specific targeting and conditional endosomal escape of RNA nanoparticles in cancer therapy. Cancer Lett. 2018;414:57–70.
Yang X, Fan B, Gao W, Li L, Li T, Sun J, et al. Enhanced endosomal escape by photothermal activation for improved small interfering RNA delivery and antitumor effect. Int J Nanomedicine. 2018;13:4333–44.
Durak-Kozica M, Baster Z, Kubat K, Stepien E. 3D visualization of extracellular vesicle uptake by endothelial cells. Cell Mol Biol Lett. 2018;23:57.
Dowdy SF, Levy M. RNA therapeutics (almost) comes of age: targeting, delivery and endosomal escape. Nucleic Acid Ther. 2018;28:107–8.
Smith SA, Selby LI, Johnston APR, Such GK. The endosomal escape of nanoparticles: toward more efficient cellular delivery. Bioconjug Chem. 2019;30:263–72.
Cupic KI, Rennick JJ, Johnston AP, Such GK. Controlling endosomal escape using nanoparticle composition: current progress and future perspectives. Nanomedicine (London). 2019;14:215–23.
Ichimizu S, Watanabe H, Maeda H, Hamasaki K, Ikegami K, Chuang VTG, et al. Cell-penetrating mechanism of intracellular targeting albumin: contribution of macropinocytosis induction and endosomal escape. J Control Release. 2019;304:156–63.
Degors IMS, Wang C, Rehman ZU, Zuhorn IS. Carriers break barriers in drug delivery: endocytosis and endosomal escape of gene delivery vectors. Acc Chem Res. 2019;52:1750–60.
Stevens JL. Future of toxicology--mechanisms of toxicity and drug safety: where do we go from here? Chem Res Toxicol. 2006;19:1393–401.
Pentin J, Smith J. Drug calculations: are they safer with or without a calculator? Br J Nurs. 2006;15:778–81.
Smith DA, Schmid EF. Drug withdrawals and the lessons within. Curr Opin Drug Discov Devel. 2006;9:38–46.
Visconti JA, Smith MC. The economics of adverse drug reactions--case studies. Ann Pharmacother. 2006;40:529–36.
Li LC, Okino ST, Zhao H, Pookot D, Place RF, Urakami S, et al. Small dsRNAs induce transcriptional activation in human cells. Proc Natl Acad Sci U S A. 2006;103:17337–42.
Setten RL, Lightfoot HL, Habib NA, Rossi JJ. Development of MTL-CEBPA: small activating RNA drug for hepatocellular carcinoma. Curr Pharm Biotechnol. 2018;19:611–21.
Sethupathy P, Collins FS. MicroRNA target site polymorphisms and human disease. Trends Genet. 2008;24:489–97.
Bartoszewski R, Kroliczewski J, Piotrowski A, Jasiecka AJ, Bartoszewska S, Vecchio-Pagan B, et al. Codon bias and the folding dynamics of the cystic fibrosis transmembrane conductance regulator. Cell Mol Biol Lett. 2016;21:23.
Wu J, Jiang R. Prediction of deleterious nonsynonymous single-nucleotide polymorphism for human diseases. Sci World J. 2013;2013:675851.
Shastry BS. SNPs in disease gene mapping, medicinal drug development and evolution. J Hum Genet. 2007;52:871–80.
Brinster R, Scherer D, Lorenzo BJ. Optimal selection of genetic variants for adjustment of population stratification in European association studies. Brief Bioinform. 2019. https://doi.org/10.1093/bib/bbz023.
Hellwege JN, Keaton JM, Giri A, Gao X, Velez Edwards DR, Edwards TL. Population stratification in genetic association studies. Curr Protoc Hum Genet. 2017;95:1.22.1–1.22.23.
Lacour A, Schuller V, Drichel D, Herold C, Jessen F, Leber M, et al. Novel genetic matching methods for handling population stratification in genome-wide association studies. BMC Bioinformatics. 2015;16:84.
Guan W, Liang L, Boehnke M, Abecasis GR. Genotype-based matching to correct for population stratification in large-scale case-control genetic association studies. Genet Epidemiol. 2009;33:508–17.
Tebbutt SJ, He JQ, Burkett KM, Ruan J, Opushnyev IV, Tripp BW, et al. Microarray genotyping resource to determine population stratification in genetic association studies of complex disease. Biotechniques. 2004;37:977–85.
Pushparaj PN, Aarthi JJ, Manikandan J, Kumar SD. siRNA, miRNA, and shRNA: in vivo applications. J Dent Res. 2008;87:992–1003.
Zhu J, Chen G, Zhu S, Li S, Wen Z, Bin L, et al. Identification of tissue-specific protein-coding and noncoding transcripts across 14 human tissues using RNA-seq. Sci Rep. 2016;6:28400.
Ludwig N, Leidinger P, Becker K, Backes C, Fehlmann T, Pallasch C, et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016;44:3865–77.
Kwekel JC, Vijay V, Desai VG, Moland CL, Fuscoe JC. Age and sex differences in kidney microRNA expression during the life span of F344 rats. Biol Sex Differ. 2015;6:1.
Shu Y, Zhang H, Cai Q, Tang D, Wang G, Liu T, et al. Integrated mRNA and miRNA expression profile analyses reveal the potential roles of sex-biased miRNA-mRNA pairs in gonad tissues of the Chinese concave-eared torrent frog (Odorrana tormota). J Exp Zool B Mol Dev Evol. 2019;332:69–80.
Guo L, Zhang Q, Ma X, Wang J, Liang T. miRNA and mRNA expression analysis reveals potential sex-biased miRNA expression. Sci Rep. 2017;7:39812.
Shapiro E, Biezuner T, Linnarsson S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat Rev Genet. 2013;14:618–30.
Venema WTU, Voskuil MD, Vila AV, Graham DB, Weersma RK, Festen EA. Single cell Rna sequencing of T cells in crohn’s disease identifies tissue specific drug targets. Gastroenterology. 2018;154:S189.
Schmidt F, Efferth T. Tumor heterogeneity, single-cell sequencing, and drug resistance. Pharmaceuticals (Basel). 2016;9(2):33. https://doi.org/10.3390/ph9020033.
Wu H, Wang C, Wu S. Single-cell sequencing for drug discovery and drug development. Curr Top Med Chem. 2017;17:1769–77.
Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3:87–98.
Cai Y, Yu X, Hu S, Yu J. A brief review on the mechanisms of miRNA regulation. Genomics Proteomics Bioinformatics. 2009;7:147–54.
Listowski MA, Heger E, Boguslawska DM, Machnicka B, Kuliczkowski K, Leluk J, et al. microRNAs: fine tuning of erythropoiesis. Cell Mol Biol Lett. 2013;18:34–46.
Zhang WW, Sun XF, Tong HL, Wang YH, Li SF, Yan YQ, et al. Effect of differentiation on microRNA expression in bovine skeletal muscle satellite cells by deep sequencing. Cell Mol Biol Lett. 2016;21:8.
Alvarez-Garcia I, Miska EA. MicroRNA functions in animal development and human disease. Development. 2005;132:4653–62.
Wang X, Jin H, Jiang S, Xu Y. MicroRNA-495 inhibits the high glucose-induced inflammation, differentiation and extracellular matrix accumulation of cardiac fibroblasts through downregulation of NOD1. Cell Mol Biol Lett. 2018;23:23.
Wang Y, Zou X, Guo Y, Wang L, Liu Y, Zeng Q, et al. Mechanical strain affects some Microrna profiles in pre-Oeteoblasts. Cell Mol Biol Lett. 2015;20:586–96.
Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med. 2014;20:460–9.
Ardekani AM, Naeini MM. The role of MicroRNAs in human diseases. Avicenna J Med Biotechnol. 2010;2:161–79.
Li Y, Kowdley KV. MicroRNAs in common human diseases. Genomics Proteomics Bioinformatics. 2012;10:246–53.
Das J, Podder S, Ghosh TC. Insights into the miRNA regulations in human disease genes. BMC Genomics. 2014;15:1010.
Yang H, Wu D, Li H, Chen N, Shang Y. Downregulation of microRNA-448 inhibits IL-1beta-induced cartilage degradation in human chondrocytes via upregulation of matrilin-3. Cell Mol Biol Lett. 2018;23:7.
Chen Q, Li L, Tu Y, Zheng LL, Liu W, Zuo XY, et al. MiR-34a regulates apoptosis in liver cells by targeting the KLF4 gene. Cell Mol Biol Lett. 2014;19:52–64.
Vucicevic D, Schrewe H, Orom UA. Molecular mechanisms of long ncRNAs in neurological disorders. Front Genet. 2014;5:48.
Wang W, Kwon EJ, Tsai LH. MicroRNAs in learning, memory, and neurological diseases. Learn Mem. 2012;19:359–68.
Luck ME, Muljo SA, Collins CB. Prospects for therapeutic targeting of MicroRNAs in human immunological diseases. J Immunol. 2015;194:5047–52.
Peng Y, Croce CM. The role of MicroRNAs in human cancer. Signal Transduct Target Ther. 2016;1:15004.
Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–69.
Sun J, Yan J, Yuan X, Yang R, Dan T, Wang X, et al. A computationally constructed ceRNA interaction network based on a comparison of the SHEE and SHEEC cell lines. Cell Mol Biol Lett. 2016;21:21.
Patel M, Antala B, Shrivastava N. In silico screening of alleged miRNAs associated with cell competition: an emerging cellular event in cancer. Cell Mol Biol Lett. 2015;20:798–815.
Fomeshi MR, Ebrahimi M, Mowla SJ, Khosravani P, Firouzi J, Khayatzadeh H. Evaluation of the expressions pattern of miR-10b, 21, 200c, 373 and 520c to find the correlation between epithelial-to-mesenchymal transition and melanoma stem cell potential in isolated cancer stem cells. Cell Mol Biol Lett. 2015;20:448–65.
Liu Y, Wang J. Therapeutic potentials of noncoding RNAs: targeted delivery of ncRNAs in Cancer cells. Adv Exp Med Biol. 2016;927:429–58.
de Almeida RA, Fraczek MG, Parker S, Delneri D, O'Keefe RT. Non-coding RNAs and disease: the classical ncRNAs make a comeback. Biochem Soc Trans. 2016;44:1073–8.
Gong R, Lv X, Liu F. MiRNA-17 encoded by the miR-17-92 cluster increases the potential for steatosis in hepatoma cells by targeting CYP7A1. Cell Mol Biol Lett. 2018;23:16.
Kong R, Ma Y, Feng J, Li S, Zhang W, Jiang J, et al. The crucial role of miR-126 on suppressing progression of esophageal cancer by targeting VEGF-A. Cell Mol Biol Lett. 2016;21:3.
Viallard C, Larrivee B. Tumor angiogenesis and vascular normalization: alternative therapeutic targets. Angiogenesis. 2017;20:409–26.
van Beijnum JR, Giovannetti E, Poel D, Nowak-Sliwinska P, Griffioen AW. miRNAs: micro-managers of anticancer combination therapies. Angiogenesis. 2017;20:269–85.
Arabzadeh S, Hossein G, Salehi-Dulabi Z, Zarnani AH. WNT5A-ROR2 is induced by inflammatory mediators and is involved in the migration of human ovarian cancer cell line SKOV-3. Cell Mol Biol Lett. 2016;21:9.
Wang H, Wu Q, Zhang Y, Zhang HN, Wang YB, Wang W. TGF-beta1-induced epithelial-mesenchymal transition in lung cancer cells involves upregulation of miR-9 and downregulation of its target, E-cadherin. Cell Mol Biol Lett. 2017;22:22.
Zeng M, Zhu L, Li L, Kang C. miR-378 suppresses the proliferation, migration and invasion of colon cancer cells by inhibiting SDAD1. Cell Mol Biol Lett. 2017;22:12.
Liu H, Zhang N, Tian D. MiR-30b is involved in methylglyoxal-induced epithelial-mesenchymal transition of peritoneal mesothelial cells in rats. Cell Mol Biol Lett. 2014;19:315–29.
Nicolas FE. Role of ncRNAs in development, diagnosis and treatment of human Cancer. Recent Pat Anticancer Drug Discov. 2017;12:128–35.
Ivan M, Harris AL, Martelli F, Kulshreshtha R. Hypoxia response and microRNAs: no longer two separate worlds. J Cell Mol Med. 2008;12:1426–31.
Ali N, Mah N, McLoughlin P, Costello CM. Identification of a hypoxia-responsive MicroRNA signature in lung endothelial cells. Ir J Med Sci. 2012;181:S414.
Kulshreshtha R, Ferracin M, Wojcik SE, Garzon R, Alder H, Agosto-Perez FJ, et al. A microRNA signature of hypoxia. Mol Cell Biol. 2007;27:1859–67.
Bandara V, Michael MZ, Gleadle JM. Hypoxia represses microRNA biogenesis proteins in breast cancer cells. BMC Cancer. 2014;14:533.
Janaszak-Jasiecka A, Bartoszewska S, Kochan K, Piotrowski A, Kalinowski L, Kamysz W, et al. miR-429 regulates the transition between Hypoxia-Inducible Factor (HIF)1A and HIF3A expression in human endothelial cells. Sci Rep. 2016;6:22775.
Zhang Y, Zhang W, Zha C, Liu Y. Platelets activated by the anti-beta2GPI/beta2GPI complex release microRNAs to inhibit migration and tube formation of human umbilical vein endothelial cells. Cell Mol Biol Lett. 2018;23:24.
Hassan G, Bahjat M, Kasem I, Soukkarieh C, Aljamali M. Platelet lysate induces chondrogenic differentiation of umbilical cord-derived mesenchymal stem cells. Cell Mol Biol Lett. 2018;23:11.
Chamorro-Jorganes A, Araldi E, Suarez Y. MicroRNAs as pharmacological targets in endothelial cell function and dysfunction. Pharmacol Res. 2013;75:15–27.
Njock MS, Fish JE. Endothelial miRNAs as cellular messengers in Cardiometabolic diseases. Trends Endocrinol Metab. 2017;28:237–46.
Kulshreshtha R, Davuluri RV, Calin GA, Ivan M. A microRNA component of the hypoxic response. Cell Death Differ. 2008;15:667–71.
Crosby ME, Devlin CM, Glazer PM, Calin GA, Ivan M. Emerging roles of microRNAs in the molecular responses to hypoxia. Curr Pharm Des. 2009;15:3861–6.
Chen Y, Zhang L, Liu WX, Wang K. VEGF and SEMA4D have synergistic effects on the promotion of angiogenesis in epithelial ovarian cancer. Cell Mol Biol Lett. 2018;23:2.
Feng W, Xue T, Huang S, Shi Q, Tang C, Cui G, et al. HIF-1alpha promotes the migration and invasion of hepatocellular carcinoma cells via the IL-8-NF-kappaB axis. Cell Mol Biol Lett. 2018;23:26.
Liyanage KIP, Ganegoda GU. Therapeutic approaches and role of ncRNAs in cardiovascular disorders and insulin resistance. Biomed Res Int. 2017;2017:4078346.
Thum T. MicroRNAs in cardiovascular disease - from disease mechanisms to therapeutic approaches. Vasc Pharmacol. 2012;56:329–30.
Siasos G, Tousoulis D, Tourikis P, Mazaris S, Zakynthinos G, Oikonomou E, et al. MicroRNAs in cardiovascular therapeutics. Curr Top Med Chem. 2013;13:1605–18.
Karapetsas A, Tokamani M, Kolettas E, Sandaltzopoulos R. Novel microRNAs as putative therapeutic targets in cardiovascular diseases. Curr Vasc Pharmacol. 2015;13:564–5.
Vogel B, Katus HA, Meder B. MicroRNAs as novel biomarkers in cardiovascular medicine. MicroRNAs as Novel Biomarkers in Cardiovascular Medicine. In: Keller A, Meese E, editors Nucleic Acids as Molecular Diagnostics. 2014. https://doi.org/10.1002/9783527672165.ch02.
Papageorgiou N, Tslamandris S, Giolis A, Tousoulis D. MicroRNAs in cardiovascular disease: perspectives and reality. Cardiol Rev. 2016;24:110–8.
Tian ZQ, Jiang H, Lu ZB. MiR-320 regulates cardiomyocyte apoptosis induced by ischemia-reperfusion injury by targeting AKIP1. Cell Mol Biol Lett. 2018;23:41.
Li Y, Yang C, Zhang L, Yang P. MicroRNA-210 induces endothelial cell apoptosis by directly targeting PDK1 in the setting of atherosclerosis. Cell Mol Biol Lett. 2017;22:3.
Zou Y, Kong M. Tetrahydroxy stilbene glucoside alleviates palmitic acid-induced inflammation and apoptosis in cardiomyocytes by regulating miR-129-3p/Smad3 signaling. Cell Mol Biol Lett. 2019;24:5.
Zhang Z, Han Y, Sun G, Liu X, Jia X, Yu X. MicroRNA-325-3p inhibits cell proliferation and induces apoptosis in hepatitis B virus-related hepatocellular carcinoma by down-regulation of aquaporin 5. Cell Mol Biol Lett. 2019;24:13.
Pickard MR, Williams GT. Targeting long non-coding RNAs (lncRNAs) with oligonucleotides in cancer therapy. Transl Cancer Res. 2016;5:S926–S7.
Engreitz JM, Haines JE, Perez EM, Munson G, Chen J, Kane M, et al. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature. 2016;539:452–5.
Wu Z, Liu X, Liu L, Deng H, Zhang J, Xu Q, et al. Regulation of lncRNA expression. Cell Mol Biol Lett. 2014;19:561–75.
Si Y, Yang Z, Ge Q, Yu L, Yao M, Sun X, et al. Long non-coding RNA Malat1 activated autophagy, hence promoting cell proliferation and inhibiting apoptosis by sponging miR-101 in colorectal cancer. Cell Mol Biol Lett. 2019;24:50.
Pei Y, Yao Q, Li Y, Zhang X, Xie B. microRNA-211 regulates cell proliferation, apoptosis and migration/invasion in human osteosarcoma via targeting EZRIN. Cell Mol Biol Lett. 2019;24:48.
Liu M, Gong C, Xu R, Chen Y, Wang X. MicroRNA-5195-3p enhances the chemosensitivity of triple-negative breast cancer to paclitaxel by downregulating EIF4A2. Cell Mol Biol Lett. 2019;24:47.
Chai L, Yang G. MiR-216a-5p targets TCTN1 to inhibit cell proliferation and induce apoptosis in esophageal squamous cell carcinoma. Cell Mol Biol Lett. 2019;24:46.
Deng H, Ouyang W, Zhang L, Xiao X, Huang Z, Zhu W. LncRNA GASL1 is downregulated in chronic heart failure and regulates cardiomyocyte apoptosis. Cell Mol Biol Lett. 2019;24:41.
Kong Q, Liang C, Jin Y, Pan Y, Tong D, Zhou J. The lncRNA MIR4435-2HG is upregulated in hepatocellular carcinoma and promotes cancer cell proliferation by upregulating miRNA-487a. Cell Mol Biol Lett. 2019;24:26.
Su H, Zou D, Sun Y, Dai Y. Hypoxia-associated circDENND2A promotes glioma aggressiveness by sponging miR-625-5p. Cell Mol Biol Lett. 2019;24:24.
Lu T, Wang Y, Chen D, Liu J, Jiao W. Potential clinical application of lncRNAs in non-small cell lung cancer. Onco Targets Ther. 2018;11:8045–52.
Kung JT, Colognori D, Lee JT. Long noncoding RNAs: past, present, and future. Genetics. 2013;193:651–69.
Fang Y, Fullwood MJ. Roles, functions, and mechanisms of long non-coding RNAs in Cancer. Genomics Proteomics Bioinformatics. 2016;14:42–54.
Qin D, Xu C. Study strategies for long non-coding RNAs and their roles in regulating gene expression. Cell Mol Biol Lett. 2015;20:323–49.
Xu X, Ji S, Li W, Yi B, Li H, Zhang H, et al. LncRNA H19 promotes the differentiation of bovine skeletal muscle satellite cells by suppressing Sirt1/FoxO1. Cell Mol Biol Lett. 2017;22:10.
Liu J, Yang C, Gu Y, Li C, Zhang H, Zhang W, et al. Knockdown of the lncRNA SNHG8 inhibits cell growth in Epstein-Barr virus-associated gastric carcinoma. Cell Mol Biol Lett. 2018;23:17.
Heilmann K, Toth R, Bossmann C, Klimo K, Plass C, Gerhauser C. Genome-wide screen for differentially methylated long noncoding RNAs identifies Esrp2 and lncRNA Esrp2-as regulated by enhancer DNA methylation with prognostic relevance for human breast cancer. Oncogene. 2017;36:6446–61.
Martens JA, Wu PY, Winston F. Regulation of an intergenic transcript controls adjacent gene transcription in Saccharomyces cerevisiae. Genes Dev. 2005;19:2695–704.
Bumgarner SL, Dowell RD, Grisafi P, Gifford DK, Fink GR. Toggle involving cis-interfering noncoding RNAs controls variegated gene expression in yeast. Proc Natl Acad Sci U S A. 2009;106:18321–6.
Latos PA, Pauler FM, Koerner MV, Senergin HB, Hudson QJ, Stocsits RR, et al. Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science. 2012;338:1469–72.
Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, Feil R, et al. The air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science. 2008;322:1717–20.
Wang X, Arai S, Song X, Reichart D, Du K, Pascual G, et al. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature. 2008;454:126–30.
Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–4.
Dang X, Lian L, Wu D. The diagnostic value and pathogenetic role of lncRNA-ATB in patients with osteoarthritis. Cell Mol Biol Lett. 2018;23:55.
Zhuang L, Tian J, Zhang X, Wang H, Huang C. Lnc-DC regulates cellular turnover and the HBV-induced immune response by TLR9/STAT3 signaling in dendritic cells. Cell Mol Biol Lett. 2018;23:43.
Nikitakis NG, Gkouveris I, Aseervatham J, Barahona K, Ogbureke KUE. DSPP-MMP20 gene silencing downregulates cancer stem cell markers in human oral cancer cells. Cell Mol Biol Lett. 2018;23:30.
Lu J, Xu F, Zhang Y, Lu H, Zhang J. ClC-2 knockdown prevents cerebrovascular remodeling via inhibition of the Wnt/beta-catenin signaling pathway. Cell Mol Biol Lett. 2018;23:29.
Shi B, Yan W, Liu G, Guo Y. MicroRNA-488 inhibits tongue squamous carcinoma cell invasion and EMT by directly targeting ATF3. Cell Mol Biol Lett. 2018;23:28.
Ge J, Chen Q, Liu B, Wang L, Zhang S, Ji B. Knockdown of Rab21 inhibits proliferation and induces apoptosis in human glioma cells. Cell Mol Biol Lett. 2017;22:30.
Xu C, Zheng J. siRNA against TSG101 reduces proliferation and induces G0/G1 arrest in renal cell carcinoma - involvement of c-myc, cyclin E1, and CDK2. Cell Mol Biol Lett. 2019;24:7.
Wang R, Wen B, Sun D. miR-573 regulates cell proliferation and apoptosis by targeting Bax in nucleus pulposus cells. Cell Mol Biol Lett. 2019;24:2.
Tokatlian T, Segura T. siRNA applications in nanomedicine. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2010;2:305–15.
Dorsett Y, Tuschl T. siRNAs: applications in functional genomics and potential as therapeutics. Nat Rev Drug Discov. 2004;3:318–29.
Fuchs U, Borkhardt A. The application of siRNA technology to cancer biology discovery. Adv Cancer Res. 2007;96:75–102.
Cejka D, Losert D, Wacheck V. Short interfering RNA (siRNA): tool or therapeutic? Clin Sci (Lond). 2006;110:47–58.
Lee W-C, Berry R, Hohenstein P, Davies J. siRNA as a tool for investigating organogenesis: the pitfalls and the promises. Organogenesis. 2008;4:176–81.
Dykxhoorn DM, Lieberman J. The silent revolution: RNA interference as basic biology, research tool, and therapeutic. Annu Rev Med. 2005;56:401–23.
Bhindi R, Fahmy RG, Lowe HC, Chesterman CN, Dass CR, Cairns MJ, et al. Brothers in arms: DNA enzymes, short interfering RNA, and the emerging wave of small-molecule nucleic acid-based gene-silencing strategies. Am J Pathol. 2007;171:1079–88.
Borrageiro G, Haylett W, Seedat S, Kuivaniemi H, Bardien S. A review of genome-wide transcriptomics studies in Parkinson's disease. Eur J Neurosci. 2018;47:1–16.
Sahraeian SME, Mohiyuddin M, Sebra R, Tilgner H, Afshar PT, Au KF, et al. Gaining comprehensive biological insight into the transcriptome by performing a broad-spectrum RNA-seq analysis. Nat Commun. 2017;8:59.
Ward RM, Schmieder R, Highnam G, Mittelman D. Big data challenges and opportunities in high-throughput sequencing. Syst Biomed. 2014;1:29–34.
Yin N, Zhu L, Ding L, Yuan J, Du L, Pan M, et al. MiR-135-5p promotes osteoblast differentiation by targeting HIF1AN in MC3T3-E1 cells. Cell Mol Biol Lett. 2019;24:51.
Burnett JC, Rossi JJ, Tiemann K. Current progress of siRNA/shRNA therapeutics in clinical trials. Biotechnol J. 2011;6:1130–46.
Acknowledgments
Not applicable.
Competing financial interests
The authors declare no competing financial and non-financial interests.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
RB and AFS wrote, read and revised the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bartoszewski, R., Sikorski, A.F. Editorial focus: understanding off-target effects as the key to successful RNAi therapy. Cell Mol Biol Lett 24, 69 (2019). https://doi.org/10.1186/s11658-019-0196-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11658-019-0196-3