
Abstract
Antibody-based therapeutics currently enjoy unprecedented success, growth in research and revenues, and recognition of their potential. It appears that the promise of the “magic bullet” has largely been realized. There are currently 22 monoclonal antibodies (mAbs) approved by the United States Food and Drug Administration (FDA) for clinical use and hundreds are in clinical trials for treatment of various diseases including cancers, immune disorders, and infections. The revenues from the top five therapeutic antibodies (Rituxan, Remicade, Herceptin, Humira, and Avastin) nearly doubled from $6.4 billion in 2004 to $11.7 billion in 2006. During the last several years major pharmaceutical companies raced to acquire antibody companies, with a recent example of MedImmune being purchased for $15.6 billion by AstraZeneca. These therapeutic and business successes reflect the major advances in antibody engineering which have resulted in the generation of safe, specific, high-affinity, and non-immunogenic antibodies during the last three decades. Currently, second and third generations of antibodies are under development, mostly to improve already existing antibody specificities. However, although the refinement of already known methodologies is certainly of great importance for potential clinical use, there are no conceptually new developments in the last decade comparable, for example, to the development of antibody libraries, phage display, domain antibodies (dAbs), and antibody humanization to name a few. A fundamental question is then whether there will be another change in the paradigm of research as happened 1–2 decades ago or the current trend of gradual improvement of already developed methodologies and therapeutic antibodies will continue. Although any prediction could prove incorrect, it appears that conceptually new methodologies are needed to overcome the fundamental problems of drug (antibody) resistance due to genetic or/and epigenetic alterations in cancer and chronic infections, as well as problems related to access to targets and complexity of biological systems. If new methodologies are not developed, it is likely that gradual saturation will occur in the pipeline of conceptually new antibody therapeutics. In this scenario we will witness an increase in combination of targets and antibodies, and further attempts to personalize targeted treatments by using appropriate biomarkers as well as to develop novel scaffolds with properties that are superior to those of the antibodies now in clinical use.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
- Antibody therapy
- Rituxan
- Herceptin
- Remicade
- Synagis
- Humira
- Avastin
- IgG1
- domain antibodies
- antibody-derived scaffold
1 Introduction
Antibody therapy has its roots thousands of years ago; early forms of vaccination against infectious diseases were developed in China as early as 200 BC. However, the history of true antibody therapy began about a century ago with the discovery that serum from animals immunized with toxins, for example, diphtheria toxin or viruses, is an effective therapeutic against the disease caused by the same agent in humans. In the 1880s von Behring developed an antitoxin that did not kill the bacteria, but neutralized the toxin that the bacteria released into the body. Von Behring was awarded the first Nobel Prize in Medicine in 1901 for his role in the discovery and development of a serum therapy for diphtheria. As he emphasized in his Nobel lecture, the serum therapy would not be possible without prior work mostly of Loffler (who discovered the diphtheria bacilli) and Roux who reasoned that the disease (diphtheria) is caused by the toxin and not by the bacteria (http://nobelprize.org/nobel_prizes/medicine/laureates/1901/behring-lecture.html The birth of the therapeutic antibodies would not have been possible without the paradigm change at the end of the past century – understanding that microorganisms and toxins they produce do exist and they can cause diseases. This new knowledge combined with the development at that time of a number of new methodologies for the study and manipulation of microorganisms and better understanding of cell and human physiology all were critically important for the discovery of the first antibody-based therapy. It was called serum therapy because whole serum from the blood of immunized animals was used for treatment. However, the existence of antibodies was anticipated and von Behring specifically used the term anti-bodies although antibodies were not isolated or characterized until decades later.
Following the initial successes in the late 1800s, sera from humans or animals containing antibodies were widely used for prophylaxis and therapy of viral and bacterial diseases (1–4). Serum therapy of most bacterial infections was abandoned in the 1940s after antibiotics became widely available (3). However, polyclonal antibody preparations are being used for some toxin-mediated infectious diseases and venomous bites (1). Serum immunoglobulin is also being used for viral diseases where there are few treatments available, although immunoglobulin is largely used for pre- or post-exposure prophylaxis (5–7). Antibody products licensed in the USA for prevention or treatment of viral diseases include human immunoglobulin for use against hepatitis A and measles, virus-specific polyclonal human immunoglobulin against cytomegalovirus, hepatitis B, rabies, respiratory syncytial virus (RSV), vaccinia, and varicella-zoster, and the humanized monoclonal antibody (mAb) Synagis (5) (see also Table 1.1 ). Polyclonal immunoglobulin has also been used with various success for diseases caused by other human viruses including parvovirus B19 (PV B19) (8–11), Lassa virus (12, 13), West Nile virus (14, 15), some enteroviruses (16, 17), herpes simplex virus (18), Crimean-Congo hemorrhagic fever virus (CCHFV) (19), Junin virus (20), SARS-CoV (21, 22), and HIV (23–28), and for treatment of some diseases of the immune system, for example, for treatment of primary immunodeficiency disorders associated with defects in humoral immunity (see also GAMMAGARD® in Table 1.1 ).
Although serum polyclonal antibody preparations have been clinically effective in many cases, problems related to toxicity including a risk for allergic reactions, lot-to-lot variation, and uncertain dosing have limited their use (1). In addition, the active antigen-specific antibodies in a polyclonal preparation typically represent a relatively small portion of the total antibodies (1%); the rest of the antibodies are not only ineffective but could be even toxic or immunogenic. However, until the 1970s it was not possible to produce large amounts of antibodies with the desired specificity.
The beginning of the paradigm change for antibodies began in 1975 with the publication of the seminal article (29) describing hybridoma technology which can provide unlimited quantities of mAbs with predefined specificity. In addition, this technology was not patented and could be used freely. A major limitation of the hybridoma technology has been the inability to produce human mAbs. Administration of murine mAbs in humans resulted in immune responses against the foreign proteins with the generation of human anti-mouse antibodies (HAMAs). However, the advent of a number of molecular biology techniques, mostly recombinant DNA technology, and the increased understanding of the antibody structure and function led to the development of chimeric and humanized mAbs. Finally, phage-display techniques and other techniques based on the progress of molecular biology, including the generation of transgenic animals, allowed the development of fully human antibodies; these methodologies have been extensively reviewed (30–58). This completed the paradigm change which occurred mostly during a period of 2–3 decades beginning in the 1970s and ending in the 1990s. We are witnessing the fruits of this paradigm change which have resulted in a number of useful therapeutic antibodies approved for clinical use during the last decade.
However, during the last decade the basic concepts and methodologies for antibody generation have not changed significantly but have been applied to numerous new targets. Do we expect another paradigm change in the near future? Are the currently used methodologies and antibodies developed based on these methodologies reaching their limit? Is it possible to produce conceptually new antibodies that are able to resolve long-standing problems including efficient oral delivery, penetration into solid tumors, and low cost of production which are the major drawbacks of antibodies in comparison to small molecules? Or perhaps, increasing the complexity by making multifunctional antibody-based drugs including nanoparticle conjugates with antibodies in various formats could result in novel therapeutics with unique and useful properties. Here we briefly overview the current state of antibody therapeutics and try to answer these and other questions related to the directions which this field may follow in the future. More indepth analysis and details can be found in the excellent reviews (56, 59–61).
2 Lessons from mAbs Currently in Clinical Use
A total of 22 mAbs are currently approved by the US FDA for clinical use; almost all of them are for treatment of cancer and diseases related to the immune system (Tables 1.1 and 1.2 ). (In April 2008 FDA approved Cimzia for treatment of Crohn’s disease) Many more mAbs are in clinical trials (1373 entries for ongoing or completed clinical trials were retrieved from (http://www.clinicaltrials.gov) by searching with “therapy and mAbs” as of March 2008). During the last decade and especially in the last years the number of clinical trials with therapeutic antibodies has increased dramatically (Table 1.3 presents a snapshot of clinical trials up to the year 2000). However, this increase has been largely due to an increase in the number of targets and indications for the same antibodies especially in combination with other therapeutics. The number of targets and corresponding antibodies in preclinical development and in the discovery phase has also increased significantly during the past decade (see, e.g., the latest and largest meeting on Molecular Targets and Cancer Therapeutics, October 22–26, 2007, San Francisco, http://www.aacr.org, where the proportion of presentations related to mAbs has increased significantly compared to previous years). Therefore, currently research and development of mAbs as potential therapeutics is growing.
The mAb market ushered into a “take-off” phase by the 1997 launch of Rituxan (rituximab) (marketed as MabThera in Europe) for non-Hodgkin’s lymphoma (NHL). Rituxan represented the first mAb product to succeed commercially in a high-revenue/high-growth market (oncology) and to provide significant enhancements in the efficacy of treatment versus existing non-mAb therapies. As a result, Rituxan rapidly became established as the gold-standard therapy for NHL and the first-launched mAb product which went on to achieve blockbuster status (revenues above $1 billion per year).
Several mAbs launched in subsequent years also became blockbusters: Herceptin (1998), Remicade (1998), Synagis (1998), Humira (2002), and Avastin (2004); the six mAbs generated total revenues of more than $12 billion in 2006 (Table 1.4 ). The other 15 mAbs generated about 10% (about $1 billion) of the total revenues from mAbs for 2006. The revenues from blockbuster mAbs have been steadily increasing typically with double-digit percentage growth each year (Table 1.4 ) and are projected to continue to increase. They are mostly products from four established companies at the top end of the market: Genentech, Roche, Abbott, and Johnson & Johnson each of which generated mAb revenues in excess of $2 billion in 2006. MedImmune (now part of AstraZeneca) follows closely. An additional tier of four companies, Biogen Idec, Amgen, Novartis, and UCB Pharma, is also evident with each forecast to record absolute annual mAb sales growth in excess of $1 billion over the period 2006–2012. The dramatic increase in revenues for the last decade and the forecast for even larger revenues in the next decade has prompted the major pharmaceutical companies to acquire a number of antibody companies in an equally dramatic race during the last several years (Table 1.5 ) and/or create their own antibody or biologicals departments. Today all major pharmaceutical companies and a still increasing number of smaller biotech companies identify and develop novel antibody-based therapeutics. This completes the paradigm change resulting in the conversion of mAbs from promising therapeutics, being developed mostly by biotech companies, into “regular” therapeutics about as important as, or perhaps in some cases more important than, small-molecule drugs. Thus most of the new antibody therapeutics or improvements in existing ones which could be clinically used are expected to be developed at large companies in their biologicals departments.
Currently about 200 different antibody-based candidate therapeutics are in clinical trials targeting about 70 different molecules (see, e.g., www.phrma.org where 418 biomedicines in clinical trials, including mAbs, are listed for 2006). At least 1–3 different antibodies are being developed at different companies for each relevant therapeutic target, with a notable exception, the IGF-IR, which is being targeted by more than ten different mAbs (62). Second- and third-generation mAbs are being developed against already validated targets. For example, based on Synagis, an antibody (motavizumab (MEDI-524; NuMax)) was developed with much higher affinity to the F protein of the RSV (63); it is expected to be approved by FDA this year and ultimately replace Synagis for which the patent expires in 2015. The improvement of already existing antibodies also includes an increase (to a certain extent) in their binding to Fc receptors for enhancement of ADCC and half-life, selection of appropriate frameworks to increase stability and yield, decrease of immunogenicity by using in silico and in vitro methods, and conjugation to small molecules and various fusion proteins to enhance cytotoxicity. A major lesson from the current state of antibody-based therapeutics is that gradual improvement in the properties of existing antibodies and identification of novel antibodies and novel targets is likely to continue in the foreseeable future. This is likely to be a major driving force of the field until saturation is reached presumably in the next decade or two, and various combinations of antibodies and other drugs may dominate unless a major change in the current paradigm occurs.
3 Beyond Antibodies as an Alternative to a Paradigm Change
The rapid expansion in mAb revenues in the next decade is likely to be driven by a number of key individual products recording peak sales growth and the launch of new products. Furthermore, a number of key mAb products are the subject of horizontal indication broadening strategies. This trend is expected to further enhance revenue growth. The most notable example of this strategy is Genentech and Roche’s Avastin (bevacizumab). Given its broad-spectrum mode of action (it targets angiogenesis) it can be used across a wide range of tumor types. Although by no means representing an end of mAb market sales expansion, revenue growth may begin to slow by 2012. Competition between rival mAb products will begin to slow sales growth for some franchises (Humira sales growth at the expense of Remicade for example), while some second-generation product launches (such as MedImmune’s Numax (motavizumab, MEDI-524) which is expected to be launched this (2008) year for the 2008/2009 RSV season) will cannibalize sales of first-generation mAb products (MedImmune’s Synagis). Ultimately, organic revenue expansion in any market is finite and this will prove the case in the mAb segment, despite the indication broadening opportunities available for many brands. However, revenues from mAbs will still grow faster than those from small molecules which face an unattractive combination of high exposure to generic competition, no major focus on areas of highest unmet need, and little access to novel target space – all conspiring to make this product set the slowest growing to 2012.
The question then is whether a new paradigm change could trigger a new dramatic expansion of some novel, still unknown, types of therapeutics. We do not know the answer to this question and surprises are always possible but currently there are no indications that another paradigm change in the discovery of biological therapeutics is coming anytime soon. It rather appears that there will be gradual improvements in existing antibodies and identification of antibodies to novel targets using currently available methodologies. However, one area where one could expect conceptually novel antibody-based candidate therapeutics, although within the current paradigm, is going beyond traditional antibody structures (see, e.g., the latest (2008) meeting Beyond Antibodies http://www.ibclifesciences.com/beyond/overview.xml)
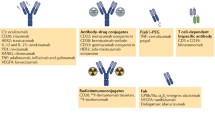
Currently, almost all FDA-approved therapeutic antibodies (Tables 1.1 , 1.2 , and 1.3 ) (except ReoPro, Lucentis, and Cimzia which are Fabs) and the vast majority of those in clinical trials are full-size antibodies mostly in IgG1 format of about 150-kDa size. A fundamental problem for such large molecules is their poor penetration into tissues (e.g., solid tumors) and poor or absent binding to regions on the surface of some molecules (e.g., on the HIV envelope glycoprotein) which are accessible by molecules of smaller size. Therefore, a large amount of work especially during the last decade has been aimed at developing novel scaffolds of much smaller size and higher stability (see, e.g., a recent review (54)). Such scaffolds are based on various human and non-human molecules of high stability and could be divided into two major groups for the purposes of this review – antibody-derived and others. Here we will briefly discuss antibody-derived scaffolds, specifically those derived from antibody domains, as an example of potentially useful candidate therapeutics; an excellent recent review describes the second group (54).
The first two domain antibodies (dAbs) entered clinical trials (phase I) last year. One of them, ALX-0081, is a camelid dAb targeting the von Willebrand factor (vWF). (Because of their small size the camelid dAbs are also termed nanobodies by the company, Ablynx, which develops them). The neutralization of the vWF could reduce the risk of thrombosis in patients with acute coronary syndrome (ACS) and thrombotic thrombocytopenic purpura (TTP). Ablynx reported the results from its phase I study in December 2007 (http://www.ablynx.com). The other dAb, ART621, is a human protein targeting TNFα. In preclinical studies, it demonstrated potency levels at least equivalent to a market-leading anti-TNF drug in an animal model of rheumatoid arthritis. The phase I clinical trial which showed that the drug was well-tolerated in healthy volunteers was successfully completed in November 2007. In March 2008, the biotechnology company Arana Therapeutics Limited (http://www.arana.com) which develops this antibody announced that it has commenced recruitment for a phase II trial in psoriasis. If successful, the company plans to initiate a phase III trial in 2009. This antibody was licensed from Domantis (http://www.domantis.com) which is now a wholly owned subsidiary of GSK. Many additional dAbs are in early stages of development.
What are the features of the dAbs which make them attractive as candidate therapeutics? Firstly, their size (12–15 kDa) is about an order of magnitude smaller than the size of an IgG1 (about 150 kDa). The small size leads to relatively good penetration into tissues and the ability to bind into cavities or active sites of protein targets which may not be accessible to full-size antibodies. This could be particularly important for the development of therapeutics against rapidly mutating viruses, for example, HIV. Because these viruses have evolved in humans to escape naturally occurring antibodies of large size, some of their surface regions which are critical for the viral life cycle may be vulnerable to targeting by molecules of smaller size including dAbs. Secondly, dAbs may be more stable than full-size antibodies in the circulation and can be relatively easily engineered to further increase their stability. For example, some dAbs with increased stability could be taken orally or delivered via the pulmonary route or may even penetrate the blood–brain barrier, and retain activity even after being subjected to harsh conditions, such as freeze-drying or heat denaturation. In addition, dAbs are typically monomeric, of high solubility, and do not significantly aggregate or can be engineered to reduce aggregation. Their half-life in the circulation can be relatively easily adjusted from minutes or hours to weeks. In contrast to conventional antibodies, dAbs are well expressed in bacterial, yeast, and mammalian cell systems. Finally, the small size of dAbs allows for higher molar quantities per gram of product, which should provide a significant increase in potency per dose and reduction in overall manufacturing cost (http://www.domantis.com).
Research on novel antibody-derived scaffold continues. We have identified a VH-based scaffold which is stable and highly soluble (64, 65). It was used for the construction of a large-size (20-billion clone) dAb phage library by grafting CDR3s and CDR2s from five of our other Fab libraries and randomly mutagenizing CDR1. Panning of this library with an HIV Env complexed with CD4 resulted in the identification of a very potent broadly cross-reactive dAb against HIV, m36, which neutralized primary HIV isolates from different clades with IC50s and IC90s in the low µg/ml range. Very recently one of the authors (DSD) has proposed to use engineered antibody constant domains (CH2 of IgG, IgA, and IgD, and CH3 of IgE and IgM) as scaffolds for construction of libraries. Because of their small size and the domains role in antibody effector functions, these have been termed nano-antibodies, the smallest fragments that could be engineered to exhibit simultaneously antigen-binding and effector functions. Several large libraries (up to 50-billion clones) were constructed and antigen-specific binders successfully identified (Xiao, Vu, Dimitrov et al., in preparation). It is possible that these and other novel scaffolds under development could provide new opportunities for identification of potentially useful therapeutics.
4 Conclusions
The rapid progress made in the last few decades toward the development of potent therapeutic antibodies mostly against cancer and immune diseases raises a number of questions for the future directions of this field. A key question is whether there are any indications of a paradigm change that could lead to radically different therapeutics as occurred 2–3 decades ago and which resulted in an explosion of antibody therapeutics approved for clinical use during the last decade. If history provides an answer and such a paradigm shift occurs, it will probably take decades before we witness the fruition of such a shift in terms of new licensed antibody therapeutics. Meanwhile, gradual improvements in the characteristics of existing antibodies, discovery of novel antibodies and novel targets, combining antibodies, conjugating them with drugs, nanoparticles, and other reagents, and going beyond antibodies by developing novel antibody-based scaffolds with superior properties to those already in use will be major areas of research and development in the coming decades. A decade from now it is likely that we will see many antibody-based therapeutics based on different scaffolds than the IgG1 approved for clinical use and hundreds more in preclinical and clinical development.
References
Casadevall, A. (1999). Passive antibody therapies: progress and continuing challenges. Clin. Immunol. 93, 5–15.
Casadevall, A. and Scharff, M. D. (1995). Return to the past: the case for antibody-based therapies in infectious diseases. Clin. Infect. Dis. 21, 150–161.
Casadevall, A. and Scharff, M. D. (1994). Serum therapy revisited: animal models of infection and development of passive antibody therapy. Antimicrob. Agents Chemother. 38, 1695–1702.
Zeitlin, L., Cone, R. A., Moench, T. R., and Whaley, K. J. (2000). Preventing infectious disease with passive immunization. Microbes Infect. 2, 701–708.
Sawyer, L. A. (2000). Antibodies for the prevention and treatment of viral diseases. Antiviral Res. 47, 57–77.
Bayry, J., Lacroix-Desmazes, S., Kazatchkine, M. D., and Kaveri, S. V. (2004). Intravenous immunoglobulin for infectious diseases: back to the pre-antibiotic and passive prophylaxis era? Trends Pharmacol. Sci. 25, 306–310.
DesJardin, J. A. and Snydman, D. R. (1998). Antiviral immunotherapy – a review of current status. Biodrugs 9, 487–507.
Moudgil, A., Shidban, H., Nast, C. C., Bagga, A., Aswad, S., Graham, S. L., Mendez, R., and Jordan, S. C. (1997). Parvovirus B19 infection-related complications in renal transplant recipients: treatment with intravenous immunoglobulin. Transplantation 64, 1847–1850.
Kurtzman, G., Frickhofen, N., Kimball, J., Jenkins, D. W., Nienhuis, A. W., and Young, N. S. (1989). Pure red-cell aplasia of 10 years’ duration due to persistent parvovirus B19 infection and its cure with immunoglobulin therapy. N. Engl. J Med. 321, 519–523.
Kerr, J. R., Cunniffe, V. S., Kelleher, P., Bernstein, R. M., and Bruce, I. N. (2003). Successful intravenous immunoglobulin therapy in 3 cases of parvovirus B19-associated chronic fatigue syndrome. Clin. Infect. Dis. 36, e100–e106.
Koduri, P. R., Kumapley, R., Valladares, J., and Teter, C. (1999). Chronic pure red cell aplasia caused by parvovirus B19 in AIDS: use of intravenous immunoglobulin – a report of eight patients. Am. J Hematol. 61, 16–20.
Clayton, A. J. (1977). Lassa immune serum. Bull. World Health Organ. 55, 435–439.
Krasnianskii, V. P., Gradoboev, V. N., Borisevich, I. V., Potryvaeva, N. V., Lebedinskaia, E. V., Chernikova, N. K., and Timan'kova, G. D. (1997). Development and study of properties of immunoglobulins against Lassa fever. Vopr. Virusol. 42, 168–171.
Shimoni, Z., Niven, M. J., Pitlick, S., and Bulvik, S. (2001). Treatment of West Nile virus encephalitis with intravenous immunoglobulin. Emerg. Infect. Dis. 7, 759.
Hamdan, A., Green, P., Mendelson, E., Kramer, M. R., Pitlik, S., and Weinberger, M. (2002). Possible benefit of intravenous immunoglobulin therapy in a lung transplant recipient with West Nile virus encephalitis. Transpl. Infect. Dis. 4, 160–162.
Pasic, S., Jankovic, B., Abinun, M., and Kanjuh, B. (1997). Intravenous immunoglobulin prophylaxis in an echovirus 6 and echovirus 4 nursery outbreak. Pediatr. Infect. Dis. J. 16, 718–720.
Rotbart, H. A., O’Connell, J. F., and McKinlay, M. A. (1998). Treatment of human enterovirus infections. Antiviral Res. 38, 1–14.
Masci, S., De Simone, C., Famularo, G., Gravante, M., Ciancarelli, M., Andreassi, M., Amerio, P., and Santini, G. (1995). Intravenous immunoglobulins suppress the recurrences of genital herpes simplex virus: a clinical and immunological study. Immunopharmacol. Immunotoxicol. 17, 33–47.
Vassilenko, S. M., Vassilev, T. L., Bozadjiev, L. G., Bineva, I. L., and Kazarov, G. Z. (1990). Specific intravenous immunoglobulin for Crimean-Congo haemorrhagic fever. Lancet 335, 791–792.
Enria, D. A., Briggiler, A. M., Fernandez, N. J., Levis, S. C., and Maiztegui, J. I. (1984). Importance of dose of neutralising antibodies in treatment of Argentine haemorrhagic fever with immune plasma. Lancet 2, 255–256.
Ali, M. B. (2003). Treating severe acute respiratory syndrome with hyperimmune globulins. Hong. Kong. Med. J. 9, 391–392.
Burnouf, T. and Radosevich, M. (2003). Treatment of severe acute respiratory syndrome with convalescent plasma. Hong. Kong. Med. J. 9, 309.
Vittecoq, D., Chevret, S., Morand-Joubert, L., Heshmati, F., Audat, F., Bary, M., Dusautoir, T., Bismuth, A., Viard, J. P., and Barre-Sinoussi, F. (1995). Passive immunotherapy in AIDS: a double-blind randomized study based on transfusions of plasma rich in anti-human immunodeficiency virus 1 antibodies vs. transfusions of seronegative plasma. Proc. Natl. Acad. Sci. USA 92, 1195–1199.
Jablonowski, H., Sander, O., Willers, R., Adams, O., Bartmann, P., and Wahn, V. (1994). The use of intravenous immunoglobulins in symptomatic HIV infection. Results of a randomized study. Clin. Investig. 72, 220–224.
Olopoenia, L., Young, M., White, D., Barnes, S., Rahbar, F., and Fomufod, A. (1997). Intravenous immunoglobulin in symptomatic and asymptomatic children with perinatal HIV infection. J. Natl. Med. Assoc. 89, 543–547.
Guay, L. A., Musoke, P., Hom, D. L., Nakabiito, C., Bagenda, D., Fletcher, C. V., Marum, L. H., Fowler, M. G., Falksveden, L. G., Wahren, B., Kataaha, P., Wigzell, H., Mmiro, F. A., and Jackson, J. B. (2002). Phase I/II trial of HIV-1 hyperimmune globulin for the prevention of HIV-1 vertical transmission in Uganda. AIDS 16, 1391–1400.
Dezube, B. J., Proper, J., Zhang, J., Choy, V. J., Weeden, W., Morrissey, J., Burns, E. M., Dixon, J. D., O’Loughlin, C., Williams, L. A., Pickering, P. J., Crumpacker, C. S., and Gelder, F. B. (2003). A passive immunotherapy, (PE)HRG214, in patients infected with human immunodeficiency virus: a phase I study. J. Infect. Dis. 187, 500–503.
Zolla-Pazner, S. and Gorny, M. K. (1992). Passive immunization for the prevention and treatment of HIV infection. AIDS 6, 1235–1247.
Kohler, G. and Milstein, C. (1975). Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497.
Hoogenboom, H. R., Marks, J. D., Griffiths, A. D., and Winter, G. (1992). Building antibodies from their genes. Immunol. Rev. 130, 41–68.
Marks, J. D., Hoogenboom, H. R., Griffiths, A. D., and Winter, G. (1992). Molecular evolution of proteins on filamentous phage. Mimicking the strategy of the immune system. J. Biol. Chem. 267, 16007–16010.
Barbas, C. F., Burton, D. R., Scott, J. K., and Silverman, G. J. (2001). Phage Display: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor.
Bradbury, A. R. and Marks, J. D. (2004). Antibodies from phage antibody libraries. J. Immunol. Methods 290, 29–49.
Soderlind, E., Simonsson, A. C., and Borrebaeck, C. A. (1992). Phage display technology in antibody engineering: design of phagemid vectors and in vitro maturation systems. Immunol. Rev. 130, 109–124.
Winter, G., Griffiths, A. D., Hawkins, R. E., and Hoogenboom, H. R. (1994). Making antibodies by phage display technology. Annu. Rev. Immunol. 12, 433–455.
Rader, C. and Barbas, C. F., III (1997). Phage display of combinatorial antibody libraries. Curr. Opin. Biotechnol. 8, 503–508.
Hoogenboom, H. R., de Bruine, A. P., Hufton, S. E., Hoet, R. M., Arends, J. W., and Roovers, R. C. (1998). Antibody phage display technology and its applications. Immunotechnology 4, 1–20.
Griffiths, A. D. and Duncan, A. R. (1998). Strategies for selection of antibodies by phage display. Curr. Opin. Biotechnol. 9, 102–108.
Pini, A. and Bracci, L. (2000). Phage display of antibody fragments. Curr. Protein Pept. Sci. 1, 155–169.
Schmitz, U., Versmold, A., Kaufmann, P. and Frank, H. G. (2000). Phage display: a molecular tool for the generation of antibodies – a review. Placenta 21 Suppl A, S106–S112.
Watkins, N. A. and Ouwehand, W. H. (2000). Introduction to antibody engineering and phage display. Vox Sang. 78, 72–79.
Jarolim, P. (2001). The phage display technique and transfusion medicine. Transfusion 41, 1–3.
Siegel, D. L. (2001). Research and clinical applications of antibody phage display in transfusion medicine. Transfus. Med. Rev. 15, 35–52.
Kretzschmar, T. and von, R. T. (2002). Antibody discovery: phage display. Curr. Opin. Biotechnol. 13, 598–602.
Hoogenboom, H. R. (2002). Overview of antibody phage-display technology and its applications. Methods Mol. Biol. 178, 1–37.
He, M. and Khan, F. (2005). Ribosome display: next-generation display technologies for production of antibodies in vitro. Expert. Rev. Proteomics. 2, 421–430.
Conrad, U. and Scheller, J. (2005). Considerations on antibody-phage display methodology. Comb. Chem. High Throughput Screen. 8, 117–126.
Smith, J., Kontermann, R. E., Embleton, J. and Kumar, S. (2005). Antibody phage display technologies with special reference to angiogenesis. FASEB J. 19, 331–341.
Hust, M. and Dubel, S. (2005). Phage display vectors for the in vitro generation of human antibody fragments. Methods Mol. Biol. 295, 71–96.
Lonberg, N. (2008). Human monoclonal antibodies from transgenic mice. Handb. Exp. Pharmacol. 69–97.
Almagro, J. C. and Fransson, J. (2008). Humanization of antibodies. Front. Biosci. 13, 1619–1633.
Mondon, P., Dubreuil, O., Bouayadi, K., and Kharrat, H. (2008). Human antibody libraries: a race to engineer and explore a larger diversity. Front Biosci. 13, 1117–1129.
Harmsen, M. M. and de Haard, H. J. (2007). Properties, production, and applications of camelid single-domain antibody fragments. Appl. Microbiol. Biotechnol. 77, 13–22.
Skerra, A. (2007). Alternative non-antibody scaffolds for molecular recognition. Curr. Opin. Biotechnol. 18, 295–304.
Filpula, D. (2007). Antibody engineering and modification technologies. Biomol. Eng. 24, 201–215.
Carter, P. J. (2006). Potent antibody therapeutics by design. Nat. Rev. Immunol. 6, 343–357.
Dufner, P., Jermutus, L., and Minter, R. R. (2006). Harnessing phage and ribosome display for antibody optimisation. Trends Biotechnol. 24, 523–529.
Denkberg, G. and Reiter, Y. (2006). Recombinant antibodies with T-cell receptor-like specificity: novel tools to study MHC class I presentation. Autoimmun. Rev. 5, 252–257.
Schrama, D., Reisfeld, R. A., and Becker, J. C. (2006). Antibody targeted drugs as cancer therapeutics. Nat. Rev. Drug Discov. 5, 147–159.
Casadevall, A., Dadachova, E., and Pirofski, L. A. (2004). Passive antibody therapy for infectious diseases. Nat. Rev. Microbiol. 2, 695–703.
Waldmann, T. A. (2003). Immunotherapy: past, present and future. Nat. Med. 9, 269–277.
Feng, Y. and Dimitrov, D. S. (2008). Monoclonal antibodies against components of the IGF system for cancer treatment. Curr. Opin. Drug Discov. Devel. 11, 178–185.
Wu, H., Pfarr, D. S., Tang, Y., An, L. L., Patel, N. K., Watkins, J. D., Huse, W. D., Kiener, P. A., and Young, J. F. (2005). Ultra-potent antibodies against respiratory syncytial virus: effects of binding kinetics and binding valence on viral neutralization. J. Mol. Biol. 350, 126–144.
Chen, W. Zhu, Z. Feng, Y. Dimitrov, D. S. (2008). Human domain antibodies to conserved sterically restricted regions on gp120 as exceptionally potent cross-reactive HIV-1 neutralizers. Proc. Natl. Acad. Sci. USA 105, 17121–17126.
Chen, W. Zhu, Z. Feng, Y. Xiao, X. Dimitrov, D. S. (2008). Construction of a large phage-displayed human antibody domain library with a scaffold based on a newly identified highly soluble, stable heavy chain variable domain. J. Mol. Biol. 382, 779–789.
Acknowledgments
This study was supported by the NIH NCI CCR intramural program, the NIH intramural AIDS program (IATAP), and the NIH intramural biodefense program to DSD.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Humana Press, a part of Springer Science+Business Media, LLC
About this protocol
Cite this protocol
Dimitrov, D.S., Marks, J.D. (2009). Therapeutic Antibodies: Current State and Future Trends – Is a Paradigm Change Coming Soon?. In: Dimitrov, A. (eds) Therapeutic Antibodies. Methods in Molecular Biology™, vol 525. Humana Press. https://doi.org/10.1007/978-1-59745-554-1_1
Download citation
DOI: https://doi.org/10.1007/978-1-59745-554-1_1
Published:
Publisher Name: Humana Press
Print ISBN: 978-1-934115-92-3
Online ISBN: 978-1-59745-554-1
eBook Packages: Springer Protocols