
Abstract
Introduction
Migraine has long been known as a common complex disease caused by genetic and environmental factors. The pathophysiology and the specific genetic susceptibility are poorly understood. Common variants only explain a small part of the heritability of migraine. It is thought that rare genetic variants with bigger effect size may be involved in the disease. Since migraine has a tendency to cluster in families, a family approach might be the way to find these variants. This is also indicated by identification of migraine-associated loci in classical linkage-analyses in migraine families. A single migraine study using a candidate-gene approach was performed in 2010 identifying a rare mutation in the TRESK potassium channel segregating in a large family with migraine with aura, but this finding has later become questioned. The technologies of next-generation sequencing (NGS) now provides an affordable tool to investigate the genetic variation in the entire exome or genome. The family-based study design using NGS is described in this paper. We also review family studies using NGS that have been successful in finding rare variants in other common complex diseases in order to argue the promising application of a family approach to migraine.
Method
PubMed was searched to find studies that looked for rare genetic variants in common complex diseases through a family-based design using NGS, excluding studies looking for de-novo mutations, or using a candidate-gene approach and studies on cancer. All issues from Nature Genetics and PLOS genetics 2014, 2015 and 2016 (UTAI June) were screened for relevant papers. Reference lists from included and other relevant papers were also searched. For the description of the family-based study design using NGS an in-house protocol was used.
Results
Thirty-two successful studies, which covered 16 different common complex diseases, were included in this paper. We also found a single migraine study. Twenty-three studies found one or a few family specific variants (less than five), while other studies found several possible variants. Not all of them were genome wide significant. Four studies performed follow-up analyses in unrelated cases and controls and calculated odds ratios that supported an association between detected variants and risk of disease. Studies of 11 diseases identified rare variants that segregated fully or to a large degree with the disease in the pedigrees.
Conclusion
It is possible to find rare high risk variants for common complex diseases through a family-based approach. One study using a family approach and NGS to find rare variants in migraine has already been published but with strong limitations. More studies are under way.
Similar content being viewed by others
Background
With a lifetime prevalence of 16%, migraine affects 75 million Europeans. It can be very disabling for the individual and is a large economic burden to society [1]. Unraveling the genetics of migraine is therefore highly relevant. Migraine is a complex disorder caused by several genes and environmental factors [2, 3]. A higher concordance of migraine in monozygotic than in dizygotic twins, and the 1.9-3.8 fold higher risk of migraine among first degree relatives of affected individuals, indicates an important genetic component [2–5] Twin studies show a heritability of 34%–65% [5, 6]. Heritability of migraine with typical aura (MA) that affects approximately one third of migraineurs is higher than for migraine without aura (MO), and evidence supports that MA and MO have different, though somewhat overlapping, etiology [7, 8]. The diagnosis of migraine is based solely on patients’ history in the absence of validated biomarkers.
Causal mutations in three genes have been identified for familial hemiplegic migraine (FHM), a rare and severe, autosomal dominantly inherited subtype of migraine with aura [9–11]. However, these genes are apparently not involved in the prevalent types of migraine [12, 13]. Several linkage studies and association studies using a candidate-gene approach have failed to identify any robust association between genetic variants and the prevalent types of migraine (see Table 1 for explanation of terms and methods mentioned) [14]. Recently, a large meta-analysis including almost 60,000 affected subjects demonstrated 44 independent common single nucleotide polymorphisms (SNPs) associated with migraine [15]. Odds ratios (ORs) ranged between 0.85 and 1.11, and the mechanisms of action in migraine are unknown. It is now clear that no common variants are associated with a medium or high risk of migraine. Thus, common variants cannot explain much of the observed heritability of migraine. In general, only 1.5%–50% of heritability of common complex diseases and traits can be explained by common variants [16]. In other words, variants with medium or high risk must be rare and therefore not possible to capture by genome-wide association studies (GWAS) in unrelated case–control samples [17]. Migraine sometimes clusters in families with an inheritance pattern that often looks dominant. MA in particular seems to aggregate in large families [2, 3]. It is reasonable to think that rare genetic variants or mutations with medium to high effect size play an important role in these large families [18]. The same could be the case for families with multiple individuals affected by MO. A search for rare variants conferring a medium to high risk of migraine should therefore focus on families with many affected with migraine. Thus, it is hypothesized that the prevalent types of migraine can be oligogenic or even monogenic inherited, as is also hypothesized for many other common complex diseases [18]. If oligogenetic inherited, susceptibility is due to a specific combination of rare variants and unrelated cases represent a wide range of different combinations. These would be impossible to capture by GWAS alone. In a family, the combination of variants is probably specific to that one family, and because of this clustering of variants it will increase the chance to find it. Linkage analysis is the method of choice when studying monogenic disorders, but linkage studies are difficult in case of oligogenic inheritance. Previous linkage analyses in migraine families have shown association between several loci and MA and MO with LOD > 3 [19–25]. None of these associations have been consistently replicated and the causal genetic variants remain to be identified, but it shows that the family approach is promising for migraine. Based on an a-priori hypothesis about the involvement of the TRESK potassium channel (encoded by the KCNK18 gene) in MA, Lafreniére et al. 2010 sequenced the entire gene region (i.e. a candidate-gene approach) and identified five variants in a case–control sample, of which one rare variant was subsequently shown to segregate perfectly with MA in a large multigenerational family (eight affected and 8 unaffected members) [26]. However, this association has later become questioned [26–28]. The technology of next-generation sequencing (NGS) now provides an affordable tool to investigate genetic variation in the entire exome or genome [29]. In theory, a family based study design and genome sequencing can embrace the search for rare variants with both high and medium effect size, whether monogenic or oligogenic. The family approach using NGS has not yet been used in migraine genetic research on a larger scale, but we and probably several other groups now have ongoing studies. We therefore judge it timely to investigate how the family approach and NGS has been used successfully in other common complex disorders, and to deduce from that the possibilities for its application in migraine.
Method
This is a focused review and by no means exhaustive on the topic of NGS in common complex diseases.
PubMed was searched to find studies on rare genetic variants using NGS in common complex diseases with positive findings.. There is no clear definition of the term “common disease”. The definition of a “rare disease” is not clear either, but it is defined by the European Commission as “prevalence of less than five per 10,000 in the Community” [30]. We therefore defined diseases not fitting this definition as common. Successful studies with a family-based design using a NGS method, not looking for de novo mutations or already known candidate genes were included. Studies focusing on cancer were also excluded. We searched the following terms: “Exome AND sequencing AND pedigree AND rare AND variants” which resulted in 192 articles of which 29 abstracts were read, 15 were read in full and 14 were included. Also the terms “family-based AND “exome-sequencing” AND rare AND variants” were searched and yielded 17 articles (three already included) out of which nine abstracts were read and eight articles were read in full resulting in three included studies.
“Migraine AND “whole genome sequencing ”and “migraine AND” “whole exome sequencing” only yielded three and nine hits respectively and one article were read in full and included.
We especially wanted to include studies of bipolar disorder and schizophrenia, because the diagnosis, like for migraine, relies on clinical characteristics in lack of a biomarker. Therefore, we also searched the terms “Schizophrenia AND “whole genome sequencing” AND families” yielding six articles whereof two were included, “Schizophrenia AND “whole exome sequencing” AND families” yielding 13 articles (one already included) of which 8 abstracts were read, 5 of them in full resulting in 3 included. The terms ““bipolar disorder” AND “whole exome sequencing” AND families” resulted in one article which was not included and ““bipolar disorder” AND “whole genome sequencing” AND families” resulted in two articles of which one was included.
The last search was performed the 12th of June 2016.
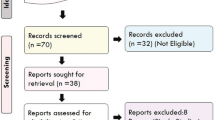
We expected highly relevant articles on the topic would be published in Nature Genetics and PLOS genetics. Therefore every issue of the two journals published in 2014, 2015 and 2016 (UTAI June), were screened for relevant papers. This and reading through reference lists of reviews and other relevant papers lead to inclusion of another 10 studies. A total of 32 studies were included (Fig. 1).

Flowchart of the searching process
Methodology of the family approach
In the following we shall describe a promising design for a family-study to find rare high risk variants in complex diseases used in our ongoing studies. For explanation of the different methods and terms mentioned, see table 1.
Inclusion
Multigenerational pedigrees with as many affected and unaffected individuals as possible will optimize the chance to find causal variants. The affected family members have to be in direct bloodline, with a minimum of affected spouses. There must be a trio of an affected child and an affected as well as a non-affected parent and also an affected relative in direct blood line as many meioses away as possible to narrow down the number of shared variants.
Diagnosis
To distinguish affected individuals from unaffected, the diagnostic process is of crucial importance. A single wrongly diagnosed individual in a family, whether affected or unaffected, will diminish the chance to identify the causal genetic variant(s) in the segregation analysis. For migraine this is difficult as no diagnostic biomarker exists. The diagnosis relies on a detailed recording of symptoms and unambiguous diagnostic criteria of the International Headache Society (international classification of headache disorders third edition (ICHD 3-beta) [31]). A validated semi-structured interview based on the diagnostic criteria, allows a reliable diagnosis. The interview should be performed by a specially trained physician or senior medical student. An example is the validated, semi-structured migraine interview used at the Danish Headache Center [32].
Molecular genetics
Whole exome sequencing (WES) or preferably whole genome sequencing (WGS) can be performed on DNA from blood samples or cells from other biological materials like mucosal cells from a buccal swab. The analysis will be a combination of an association- and a segregation analysis which will require a combination of several sequencing and genotyping methods. Based on an in-house protocol and literature review we propose the following.
A so called family trio is selected from each pedigree for sequencing (in some cases it can be preferable to only sequence the proband). WGS is optimal because causal genetic variants can be localized in non-protein coding regions. WES may be chosen because of the lower cost. A trio composed of a nuclear family with an affected child, an affected parent, a non-affected parent (Fig. 2) and a distantly related affected family member from the same pedigree as many meioses from the trio as possible are sequenced.

Example of a MA family suited for a family approach analysis. A trio and a distantly related, affected family member are marked with dashed lines. The arrow marks the proband
Several hundred variants are usually shared between the affected individuals. The disease-causing variants will often not be present in DNA from the unaffected relative. The use of a more distantly related affected individual in the analysis will decrease the number of possible causal variants because a distantly related person shares less DNA with the two affected individuals in the trio. Quality testing of the sequencing like depth of coverage (the a verage number of times a nucleotide is expected and observed to be sequenced) will not be explained here in details, but the importance of this step should be noted.
Filtering
To narrow down the number of variants, a filtering process is hereafter necessary. This can be done in several ways. They all depend on several existing databases with genetic information. These databases consist of collections of structural variants like SNPs and copy number variants (CNVs) detected by different studies. All SNPs known to date are for example gathered in public databases like dbSNP [33]. Other examples are the 1000 genomes project data, the Database of Genomic Variants [34], and more local databases like LuCamp containing 700.000 SNPS from 2000 Danish individuals [35]. The strategy is to filter out and exclude all variants known in all available genetic databases as the causal variants are expected to be private for a family. Some studies choose to include rare variants present in databases with minor allele frequency (MAF) <0.5% or <0.1% [36]. A third strategy is to filter out variants with e.g. MAF >0,1%, thereby excluding variants common in the population from the study data [37, 38].
Co-segregation studies
The remaining family specific variants then have to be tested for segregation with disease in the pedigree. This can be done by a classical Sanger sequencing of the relevant genes in all the non-sequenced family members or by SNP genotyping. Ideally the causal variant is present only in affected individuals, but the possibility of unaffected carriers has to be taken into account because of incomplete penetrance, or later debut of the disease. Calculating a so called LOD score (LOD = logarithm of the odds), can help as a measure of probability of linkage between a variant and a disease.
Oligogenic inheritance where more than one variant are found to segregate in a pedigree is very likely in common diseases. Therefore, it is possible that this approach will result in multiple variants segregating in one family, all playing a role for the pathogenicity in that family.
Further analyses
Further analysis, like testing whether some of the segregating variants are shared between different families within the same study is interesting. The likelihood of a variant to be disease-causing can be tested by screening a sample of unrelated cases or additional sets of affected families for the mutation [36]. Screening for other mutations in the involved genes is also relevant. A so called burden test can be done. The load of rare mutations in a gene or gene family present in cases vs. controls reflects the probability of a gene to be involved in disease pathogenesis. Afterwards, functional studies involving laboratory tests are crucial to investigate how detected variants are involved in pathogenic mechanisms of the disease of interest. Different kinds of software like polyphen-2 [39] and SIFT [40] have been developed to predict to which extent a variant or mutation is functional. This prediction is based on the possibility of a variant to be e.g. deleterious or in-frame. Laboratory tests may show whether the mutation resides in a gene (this will always be the case when using WES) or in a regulatory area. Other relevant questions to ask are: in which processes or pathways is the variant involved? Does this affect gene or protein expression or function? Can this explain development of symptoms of the investigated disease? Functional laboratory studies in cells from humans or animals can help answer these questions (Fig. 3).

Description of the family approach in summary
Successful use of the family approach in diseases other than migraine
Family studies using different variants of the approach described above or some of its elements have already provided interesting results in common complex diseases other than migraine.
Thirty-two studies that fulfilled the criteria were included in this review [36–38, 41–69]. Six of them studied bipolar disorder, five of them studied schizophrenia (SCZ), three of them autism spectrum disorder (ASD) and two of them type II diabetes, while four studies focused on late onset Alzheimer’s disease (LOAD). There was one study focusing on each of the following: Parkinson’s disease, age-related macular degeneration (AMD), adiponectin level, atrial fibrillation, intracranial aneurysms, nonsyndromic cleft lip and palate (NSCLP), nonsyndromic hearing impairment, otitis media, preeclampsia, primary open angle glaucoma, inflammatory bowel disease and reumathoid arthritis. The study groups ranged from one to eighty families of varying size. They all used a combination of genetic methods like WGS, WES, sanger sequncing, GWAS and linkage-analysis. Some also used identity by descent analysis (IBD). Only three studies did WGS and three studies made use of family trios. One study searched for rare CNVs instead of SNPs [41]. 23 of the studies found one or a few variants (<5) specific for a family and the rest found several variants without finding one causal variant. Study details on disease, sample, techniques and findings are listed in Additional file 1: Table S1.
Four studies performed a follow-up analysis in unrelated cases and controls and presented an OR. Wetzel-smith et al. detected a missense variant (rs137875858 in UNC5C) in a large family with 8 LOAD cases using WGS, WES, linkage-analysis and assay genotyping. The same variant was found in four other pedigrees. Further genotyping of LOAD cases (8,050) and controls (98,194) resulted in an OR = 2.15 (95% CI = [1.21; 3.84], P = 0.0095) [42]. By WES and genotyping Cruchaga et al. detected a rare variant (rs145999145 in PLD3) segregating in 2 pedigrees with multiple LOAD cases. When testing independent cohorts of sporadic cases (4,998) and controls (6,356) they found an OR = 2.10 (95% CI = [1.47; 2,99] P=2.39×10−10-). For familial LOAD cases (1,106) vs. unrelated controls (6,356) an OR of 3.39 (95% CI = [2.14; 5.39] P = 1.18×10−6) was calculated [36]. Kohli et al. made use of WES, linkage analysis, genotyping and Sanger sequencing to study one pedigree counting 15 individuals. They found a rare variant (rs377155188 in TTC3) which segregated perfectly with LOAD in the pedigree. They calculated an OR for LOAD of 3.35 for sporadic cases (6,669) vs. controls (5,585) [43]. The result did not reach statistical significance (CI not specified). Goes et al. exome sequenced 36 individuals from 8 families and calculated ORs for variants in three genes with association to bipolar disorder in a case–control follow-up study (3,541 cases, 4,774 controls). It resulted in ORs of 2.73 (P = 0.016), 6.7 (P = 0.0039) and 2.78 (P = 0.045) for variants in MLK4, APPL2 and HSP90AA1 respectively [44].
Some studies found complete segregation between a variant and the studied disease and a few of them will be highlighted in the following. Cruceanu et al. exome-sequenced DNA from caucasian individuals affected with a highly heritable subtype of bipolar disorder in multigenerational families with three to seven affected individuals. Focus was on variants with MAF<1% in the general population. A missense variant was detected on chromosome 11 (position116652892) that leads to substitution of an aminoacid in the encoded protein. The variant segregated with affected family members in a family (five individuals) and was not present in unaffected family members selected as controls (six individuals). Whether this variant contributes to bipolar disorder is not known because of lack of knowledge about the involved gene [37].
Nyegaard et al. studied a five generational family containing 17 individuals with nonsyndromic hearing impairment (two of them dead). Seven already known hearing loss genes were not involved. 11 individuals were selected for SNP genotyping. 11.034 detected SNPs were included in a parametric linkage analysis which resulted in a significant linkage peak at chromosome six. To narrow down the size of the detected locus, 26 family members were genotyped for seven microsatellite markers. Then DNA from an affected individual was selected to undergo next generation sequencing (NGS) at the locus site by a costume designed sequence array. This identified 28,300 variants. One variant found in a coding region and predicted to have a functional effect was identified after excluding common variants. The mutation c.574C>T in CD164 was found in all affected individuals including a young girl with signs of a beginning hearing loss. The variant was absent in 12 unaffected family members, in one with unknown phenotype and in 1200 unrelated controls [45].
Suggested application of the family approach to migraine
The studies reviewed here largely followed the steps we have described, but with many variations. In our description of a family based study design, we suggest to use family trios for WGS. Only three studies [46–48], one of them still ongoing, did this. Also, only three studies [42, 46, 49] made use of WGS, which probably reflects that the cost of WGS has only been manageable very recently. It is highly likely that the non-coding regulatory areas play an important role in common migraine [70]. Georgi et al. chose to study a pedigree in an isolated population which has also been suggested as a possible approach to find rare variants [16, 46, 71, 72]. 23 studies [36–38, 41–45, 48–62] reported the finding of less than five family specific variants (not all significant). Some studies could not find one specific causal variant probably because disease susceptibility is caused by more than one rare variant specific to a family (oligogenic inheritance). Detection of variants only makes sense if it is followed by further studies to clarify the causality of the variant. An et al. found several variants associated with autism spectrum disorder. They found enrichment of rare causal variants in key neurobiological processes, and overrepresentation of the rare causal variants in functions involving neuronal development, signal transduction and synapse development [48].
In the future, combinations of gene variants might be analyzed by “omics” approach, where bioinformatics integrate genomics, epigenomics, transcriptomics, proteomics and metabolomics [73, 74]. We excluded Ratnapriya et al. [75] from the reviewing part of this study because they studied a rare subtype of macular degeneration. They found a rare variant in a family with early onset macular degeneration in FBN2, but also a common variant in the same gene with a modest association to AMD cases. It is an excellent example of how both rare and common variants in a single gene can contribute to complex forms of a disease phenotype and the understanding of its pathophysiology.
Few studies focused on CNV’s, and only Van Den Bossche et al. [41], studying schizophrenia, succeeded in finding a CNV associated with disease. Rare inherited CNVs were more frequent in familial schizophrenia than in an unaffected control cohort [76]. This supports CNVs as an area of interest when searching for rare disease variants in migraine. In the future, NGS methods will be able to capture CNVs [77].
As mentioned, ORs for SNPs associated with migraine found through a GWAS ranged between 0.85 and 1.24. In the studies using a family approach reviewed here the ORs in follow-up case–control studies ranged between 2.10 and 6.7, the last one for the association between a variant in APPL2 and bipolar disorder. Like migraine, bipolar disorder and schizophrenia are common and complex neurological disorders with a clearly heritable factor and a diagnosis based on history in the absence of a biomarker. Success in these disorders raises hope to find specific rare variants with high relative risk in migraine families. Jiang et al. reported the preliminary finding of six novel rare non-synonymous mutations in a Chinese family with clustering of migraine without aura using WES. They included four cases (a father and three children) and four unrelated controls. However, the study had several limitations [78]. As far as we know, Jiang et al. is the only study using a family approach and NGS in migraine that has been published. F. Michael Cutrer, Mayo Clinic, Rochester has carried out WES in two large migraine families, according to a published grant description [79]. Five candidate genes were found to segregate with MA in one family. In the other family, including individuals with varying phenotypes, a single variant was detected. Whether the variants are rare was not stated. These results have unfortunately not been published. A similar study is ongoing on at the Danish Headache Center. This project aims to find rare genetic variants conferring a high risk of migraine using a family approach exactly as described previously. Extended Danish families with MA or MO are included. The study is still collecting data and biological material for sequencing and genotyping. A family approach in migraine will encounter obstacles. Correct phenotyping cannot avoid that unaffected controls may develop migraine later. The probability that some affected family members do not carry a family specific variant is high due to the high prevalence of migraine in the general population. Also, unaffected carriers are a possibility due to low level of penetrance. This will complicate the analysis. Many families contain a mix of individuals with MO, MA or so called MAMO (co-occurring MA and MO). It is not known whether the two phenotypes are part of a spectrum of the same disease or different diseases. Taking these problems into account, we still believe that a family approach is the best way to find variants with a high relative risk. Such variants can be the key to understand the pathophysiological mechanisms of migraine, and much more so than the common variants discovered by GWAS. It is obvious that migraine is highly genetically heterogeneous. Pathophysiology as well as response to prophylactic drugs vary considerably [80, 81]. On the other hand, 80% of patients respond to injection of Sumatriptan suggesting the existence of a final common pathway [82, 83]. If the etiology of just a few sub-phenotypes can be identified with certainty, it seems possible to identify one or more migraine pathwaysthat may be relevant for many patients, even if the genetic cause that lead to the discovery is rare. New targets for better and more specific treatments may then be discovered.
Review and conclusions
It has proven possible to find rare high risk variants for common complex diseases through a family based approach. One study using a family approach and NGS to find rare variants in migraine has already been published but it has strong limitations. More studies are under way.
Future family approach studies could be advanced by choosing isolated populations or individuals with severe phenotypes as study groups and include analysis of mitochondrial DNA and “omics”.
Abbreviations
- AMD:
-
Age-related macular degeneration
- ASD:
-
Autism spectrum disorder
- CNV:
-
Copy number variant
- GWAS:
-
Genome wide associations study
- IBD:
-
Identity by descent
- LOAD:
-
Late onset Alzheimer’s disease
- LOD:
-
Logarithm of the odds
- MA:
-
Migraine with aura
- MAF:
-
Minor allele frequency
- MAMO:
-
Migraine with and without aura
- MO:
-
Migraine without aura
- NGS:
-
Next generation sequencing
- NSCLP:
-
Non syndromic cleft lip palate
- SCZ:
-
Schizophrenia
- SNP:
-
Single nucleotide polymorphism
- WES:
-
Whole exome sequencing
- WGS:
-
Whole genome sequencing
References
Gustavsson A, Svensson M, Jacobi F et al (2011) Cost of disorders of the brain in Europe 2010. Eur Neuropsychopharmacol 21:718–779. doi:10.1016/j.euroneuro.2011.08.008
Russell MB, Iselius L, Olesen J (1996) Migraine without aura and migraine with aura are inherited disorders. Cephalalgia 16:305–309
Russell MB, Olesen J (2014) Increased familial risk and evidence of genetic factor in migraine. Br Med J 311:541–544
Ulrich V, Gervil M, Kyvik KO et al (1999) Evidence of a genetic factor in migraine with aura: a population-based danish twin study. Ann Neurol 45:242–246. doi:10.1002/1531-8249(199902)45:2<242::AID-ANA15>3.0.CO;2-1
Mulder EJ, van Baal C, Gaist D et al (2012) Genetic and environmental influences on migraine: a twin study across Six countries. Twin Res 6:422–431. doi:10.1375/twin.6.5.422
Ulrich V, Gervil M, Kyvik KO et al (1999) The inheritance of migraine with aura estimated by means of structural equation modelling. J Med Genet 36:225–227
Russell MB, Ulrich V, Gervil M, Olesen J (2002) Migraine without aura and migraine with aura Are distinct disorders. A population-based twin survey. Headache 42:332–336
Ulrich V, Gervil M, Fenger K et al (1960) The prevalence and characteristics of migraine in twins from the general population. Headache 39:173–180
De Fusco M, Marconi R, Silvestri L et al (2003) Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump alpha2 subunit associated with familial hemiplegic migraine type 2. Nat Genet 33:192–196. doi:10.1038/ng1081
Dichgans M, Freilinger T, Eckstein G et al (2005) Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 366:371–377. doi:10.1016/S0140-6736(05)66786-4
Ophoff RA, Terwindt GM, Vergouwe MN et al (1996) Familial hemiplegic migraine and episodic ataxia type-2 Are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 87:543–552. doi:10.1016/S0092-8674(00)81373-2
Kirchmann M, Thomsen LL, Olesen J (2006) The CACNA1A and ATP1A2 genes are not involved in dominantly inherited migraine with aura. Am J Med Genet B Neuropsychiatr Genet 141B(3):250–256. doi:10.1002/ajmg.b.30277
Nyholt DR, Laforge KS, Kallela M et al (2008) A high-density association screen of 155 ion transport genes for involvement with common migraine. Hum Mol Genet 17:3318–3331. doi:10.1093/hmg/ddn227
de Vries B, Frants RR, Ferrari MD, van den Maagdenberg AMJM (2009) Molecular genetics of migraine. Hum Genet 126:115–132. doi:10.1007/s00439-009-0684-z
Gormley P, Anttila V, Winsvold BS et al (2016) Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat Genet 48:16–18. doi:10.1038/ng.3598
Manolio TA, Collins FS, Cox NJ et al (2009) Finding the missing heritability of complex diseases. Nature 461:747–753. doi:10.1038/nature08494
McCarthy MI, Abecasis GR, Cardon LR et al (2008) Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet 9:356–369. doi:10.1038/nrg2344
Schork NJ, Murray SS, Frazer KA, Topol EJ (2009) Common vs. rare allele hypotheses for complex diseases. Curr Opin Genet Dev 19:212–219. doi:10.1016/j.gde.2009.04.010
Nyholt DR, Lea RA, Goadsby PJ, Brimage PJGL (1998) Familial typical migraine. Neurology 50:1428–1432
Tikka-Kleemola P, Artto V, Vepsäläinen S et al (2010) A visual migraine aura locus maps to 9q21-q22. Neurology 74:1171–1177. doi:10.1212/WNL.0b013e3181d8ffcb
Oterino A, Toriello M, Castillo J et al (2012) Family-based association study of chromosome 6p12.2-p21.1 migraine locus. Headache 52:393–399. doi:10.1111/j.1526-4610.2011.02040.x
Anttila V, Nyholt DR, Kallela M, Artto V, Vepsäläinen S, Jakkula E, Wennerström A et al (1992) Consistently replicating locus linked to migraine on 10q22-q23. Am J Hum Genet 82:1051–1063, S0002-9297(08)00213-9
Russo L, Mariotti P, Sangiorgi E et al (2005) A New susceptibility locus for migraine with aura in the 15q11-q13 genomic region containing three GABA-a receptor genes. Am J Hum Genet 76:327–333. doi:10.1086/427521
Carlsson A, Forsgren L, Nylander P-O et al (2002) Identification of a susceptibility locus for migraine with and without aura on 6p12.2-p21.1. Neurology 59:1804–1807
Cader ZM, Noble-topham S, Dyment DA et al (2003) Significant linkage to migraine with aura on chromosome 11q24. Hum Mol Genet 12:2511–2517. doi:10.1093/hmg/ddg252
Lafrenière RG, Cader MZ, Poulin J-F et al (2010) A dominant-negative mutation in the TRESK potassium channel is linked to familial migraine with aura. Nat Med 16:1157–1160. doi:10.1038/nm.2216
Andres-Enguix I, Shang L, Stansfeld PJ et al (2012) Functional analysis of missense variants in the TRESK (KCNK18) K channel. Sci Rep 2:1–8. doi:10.1038/srep00237
Maher BH, Taylor M, Stuart S et al (2013) Analysis of 3 common polymorphisms in the KCNK18 gene in an Australian Migraine case-control cohort. Gene 528:343–346. doi:10.1016/j.gene.2013.07.030
Li B, Liu DJ, Leal SM (2013) Identifying rare variants associated with complex traits via sequencing. Curr Protoc Hum Genet 144:724–732. doi:10.1038/jid.2014.371
European Commission (2005) Useful Information on rare dieseases from an EU perspective., pp 2005–2006
Road C (2013) The international classification of headache disorders, 3rd edition (beta version). Cephalalgia 33:629–808. doi:10.1177/0333102413485658
Kirchmann M, Seven E, Björnsson Á et al (2006) Validation of the deCODE Migraine Questionnaire (DMQ3) for use in genetic studies. Eur J Neurol 13:1239–1244. doi:10.1111/j.1468-1331.2006.01491.x
dbSNP. https://www.ncbi.nlm.nih.gov/SNP/. Accessed 14 Mar 2016
Database of Genomic Variants. http://dgv.tcag.ca/dgv/app/about?ref. Accessed 14 Mar 2016
LuCamp. http://www.lucamp.org/#/172977/. Accessed 15 Mar 2016
Cruchaga C, Karch CM, Jin SC et al (2014) Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature 505:550–554. doi:10.1038/nature12825
Cruceanu C, Ambalavanan A, Spiegelman D et al (2013) Family-based exome-sequencing approach identifies rare susceptibility variants for lithium-responsive bipolar disorder. Genome 56:634–640
Yu Y, Triebwasser MP, Wong EKS et al (2014) Whole-exome sequencing identifies rare, functional CFH variants in families with macular degeneration. Hum Mol Genet 23:5283–5293. doi:10.1093/hmg/ddu226
Polyphen-2. http://genetics.bwh.harvard.edu/pph2/. Accessed 14 Mar 2016
SIFT. http://sift.jcvi.org/. Accessed 14 Mar 2016
Van Den Bossche MJ, Strazisar M, Cammaerts S et al (2013) Identification of rare copy number variants in high burden schizophrenia families. Am J Med Genet B Neuropsychiatr Genet 162:273–282. doi:10.1002/ajmg.b.32146
Wetzel-Smith MK, Hunkapiller J, Bhangale TR et al (2014) A rare mutation in UNC5C predisposes to late-onset Alzheimer’s disease and increases neuronal cell death. Nat Med 20:1452–1457. doi:10.1038/nm.3736
Kohli MA, Cukier HN, Hamilton-Nelson KL et al (2016) Segregation of a rare TTC3 variant in an extended family with late-onset Alzheimer disease. Neurol Genet 2:e41. doi:10.1212/NXG.0000000000000041
Goes FS, Pirooznia M, Parla JS et al (2016) Exome sequencing of familial bipolar disorder. JAMA Psychiat 13:537–551. doi:10.1001/jamapsychiatry.2016.0251
Nyegaard M, Rendtorff ND, Nielsen MS et al (2015) A novel locus harbouring a functional CD164 nonsense mutation identified in a large danish family with nonsyndromic hearing impairment. PLoS Genet 11:1–25. doi:10.1371/journal.pgen.1005386
Georgi B, Craig D, Kember RL et al (2014) Genomic view of bipolar disorder revealed by whole genome sequencing in a genetic isolate. PLoS Genet 10:e1004229. doi:10.1371/journal.pgen.1004229
N. Matoba, M. Kataoka, K. Fujii, Y. Suzuki, S. Sugano TK Trio-based pathway analysis of bipolar disorder. http://www.ashg.org/2013meeting/abstracts/fulltext/f130121585.htm.
An JY, Cristino AS, Zhao Q et al (2014) Towards a molecular characterization of autism spectrum disorders: an exome sequencing and systems approach. Transl Psychiatry 4:e394. doi:10.1038/tp.2014.38
Thygesen JH, Zambach SK, Ingason A, et al. (2015) Linkage and whole genome sequencing identify a locus on 6q25–26 for formal thought disorder and implicate MEF2A regulation. Schizophr Res 6–11. doi: 10.1016/j.schres.2015.08.037.
Strauss KA, Markx S, Georgi B et al (2014) A population-based study of KCNH7 p. Arg394His and bipolar spectrum disorder. Hum Mol Genet 23:1–30. doi:10.1093/hmg/ddu335
Timms AE, Dorschner MO, Wechsler J et al (2013) Support for the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia from exome sequencing in multiplex families. JAMA Psychiat 70:582–590. doi:10.1001/jamapsychiatry.2013.1195
Prudente S, Jungtrakoon P, Marucci A et al (2015) Loss-of-function mutations in APPL1 in familial diabetes mellitus. Am J Hum Genet 97:177–185. doi:10.1016/j.ajhg.2015.05.011
Bowden DW, An SS, Palmer ND et al (2010) Molecular basis of a linkage peak: exome sequencing and family-based analysis identify a rare genetic variant in the ADIPOQ gene in the IRAS Family Study. Hum Mol Genet 19:4112–4120. doi:10.1093/hmg/ddq327
Santos-Cortez RLP, Chiong CM, Reyes-Quintos MRT et al (2015) Rare A2ML1 variants confer susceptibility to otitis media. Nat Genet 47:917–920. doi:10.1038/ng.3347
Deng H-X, Shi Y, Yang Y, et al. (2016) Identification of TMEM230 mutations in familial Parkinson’s disease. Nat Genet advance on:733–739. doi: 10.1038/ng.3589.
Egawa J, Watanabe Y, Sugimoto A et al (2015) Whole-exome sequencing in a family with a monozygotic twin pair concordant for autism spectrum disorder and a follow-up study. Psychiatry Res 229:599–601. doi:10.1016/j.psychres.2015.07.018
Aylward A, Cai Y, Lee A et al (2016) Genetic epidemiology using whole exome sequencing to identify candidate genes with rare variants in nonsyndromic cleft lip and palate. Genet Epidemiol 40:432–441. doi:10.1002/gepi.21972
Johnson M, Loset M, Brennecke S et al (2012) OS049. Exome sequencing identifies likely functional variantsinfluencing preeclampsia and CVD risk. Pregnancy Hypertens 2:203–204. doi:10.1016/j.preghy.2012.04.050
Okada Y, Diogo D, Greenberg JD et al (2014) Integration of sequence data from a consanguineous family with genetic data from an outbred population identifies PLB1 as a candidate rheumatoid arthritis risk gene. PLoS One 9:1–12. doi:10.1371/journal.pone.0087645
Tanaka D, Nagashima K, Sasaki M et al (2013) Exome sequencing identifies a new candidate mutation for susceptibility to diabetes in a family with highly aggregated type 2 diabetes. Mol Genet Metab 109:112–117. doi:10.1016/j.ymgme.2013.02.010
Okou DT, Mondal K, Faubion WA et al (2014) Exome sequencing identifies a novel FOXP3 mutation in a 2-generation family with inflammatory bowel disease. J Pediatr Gastroenterol Nutr 58:561–568. doi:10.1097/MPG.0000000000000302
Zhou Z, Hu Z, Zhang L et al (2016) Identification of RELN variation p. Thr3192Ser in a Chinese family with schizophrenia. Sci Rep 6:24327. doi:10.1038/srep24327
Shi L, Zhang X, Golhar R et al (2013) Whole-genome sequencing in an autism multiplex family. Mol Autism 4:8. doi:10.1186/2040-2392-4-8
Weeke P, Muhammad R, Delaney JT, et al. (2014) Whole-exome sequencing in familial atrial fibrillation. Eur Heart J 1–7. doi: 10.1093/eurheartj/ehu156 .
Cukier HN, Dueker ND, Slifer SH et al (2014) Exome sequencing of extended families with autism reveals genes shared across neurodevelopmental and neuropsychiatric disorders. Mol Autism 5:1. doi:10.1186/2040-2392-5-1
Farlow JL, Lin H, Sauerbeck L et al (2015) Lessons learned from whole exome sequencing in multiplex families affected by a complex genetic disorder, intracranial aneurysm. PLoS One 10:1–25. doi:10.1371/journal.pone.0121104
Saad M, Brkanac Z, Wijsman EM (2015) Family-based genome scan for age at onset of late-onset Alzheimer’s disease in whole exome sequencing data. Genes Brain Behav 14:607–617. doi:10.1111/gbb.12250
Liu T, Xie L, Ye J, He X (2014) Family-based analysis identified CD2 as a susceptibility gene for primary open angle glaucoma in Chinese Han population. J Cell Mol Med 18:600–609. doi:10.1111/jcmm.12201
Homann OR, Misura K, Lamas E, et al. (2016) Whole-genome sequencing in multiplex families with psychoses reveals mutations in the SHANK2 and SMARCA1 genes segregating with illness. Mol Psychiatry 1–6. doi: 10.1038/mp.2016.24.
Xiong HY, Alipanahi B, Lee LJ et al (2015) The human splicing code reveals new insight into the genetic determinants of disease, vol 347., p 1254806. doi:10.1613/jair.301
Zuk O, Schaffner SF, Samocha K et al (2014) Searching for missing heritability: designing rare variant association studies. Proc Natl Acad Sci U S A 111:E455–E464. doi:10.1073/pnas.1322563111
Lettre G, Hirschhorn JN (2015) Small island, big genetic discoveries. Nat Genet 47:1224–1225. doi:10.1038/ng.3426
Zameel Cader M (2013) The molecular pathogenesis of migraine: New developments and opportunities. Hum Mol Genet 22:39–44. doi:10.1093/hmg/ddt364
Lionetto L, Gentile G, Bellei E et al (2013) The omics in migraine. J Headache Pain 14:55. doi:10.1186/1129-2377-14-55
Ratnapriya R, Zhan X, Fariss RN et al (2014) Rare and common variants in extracellular matrix gene Fibrillin 2 (FBN2) are associated with macular degeneration. Hum Mol Genet 23:5827–5837. doi:10.1093/hmg/ddu276
Xu B, Woodroffe A, Rodriguez-Murillo L et al (2009) Elucidating the genetic architecture of familial schizophrenia using rare copy number variant and linkage scans. Proc Natl Acad Sci U S A 106:16746–16751. doi:10.1073/pnas.0908584106
de Ligt J, Boone PM, Pfundt R et al (2013) Detection of clinically relevant copy number variants with whole-exome sequencing. Hum Mutat 34:1439–1448. doi:10.1002/humu.22387
Jiang Y, Wu R, Chen C et al (2015) Six novel rare non-synonymous mutations for migraine without aura identified by exome sequencing. J Neurogenet 29:188–194. doi:10.3109/01677063.2015.1122787
Cutrer F Whole Exome Sequencing as a Strategy for Gene Discovery in a Large Well Characterized Family with Migraine. http://www.migraineresearchfoundation.org/completed-research.html. Accessed 14 Mar 2016
Tfelt-Hansen P, Brøsen K (2008) Pharmacogenomics and migraine: possible implications. J Headache Pain 9:13–18. doi:10.1007/s10194-008-0009-y
Ophoff RA, Van Den Maagdenberg AMJM, Roon KI et al (2001) The impact of pharmacogenetics for migraine. Eur J Pharmacol 413:1–10
The Subcutaneous Sumatriptan International Study Group (1991) Treatment of migraine attacks with sumatriptan. N Engl J Med 325:316–321
Cady RK, Wendt JK, Kirchner JR, Sargent JD, Rothrock JF, Skaggs H (1991) Treatment of acute migraine with subcutaneous sumatriptan. JAMA 265:2831–2856
Feero WG, Guttmache AE, Collins FS (2010) Genomic medicine - An updated primer. N Engl J Med 362:2001–2011
Guttmacher ALEG, Collins AFSC (2002) Genomic medicine - A primer. N Engl J Med 347:1512–1520
The International SNP Map Working Group (2001) A map of human genome sequence variation containing 1. 42 million single nucleotide polymorphisms. Nature 409:928–933
Frazer KA, Ballinger DG, Cox DR et al (2007) A second generation human haplotype map of over 3.1 million SNPs. Nature 449:851–861. doi:10.1038/nature06258
Bernstein BE, Birney E, Dunham I et al (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489:57–74. doi:10.1038/nature11247
Venter JC, Adams MD, Myers EW et al (2001) The sequence of the human genome. Science 291(80-):1304–1351
The International HapMap Consortium (2005) A haplotype map of the human genome. Nature 437:1299–1320. doi:10.1038/nature04226
Abecasis GR, Auton A, Brooks LD et al (2012) An integrated map of genetic variation from 1,092 human genomes. Nature 491:56–65. doi:10.1038/nature11632
Abecasis GR, Altshuler D, Auton A et al (2010) A map of human genome variation from population-scale sequencing. Nature 467:1061–1073. doi:10.1038/nature09534
Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247. doi:10.1038/ng1195-241
Altmu J, Palmer LJ, Fischer G et al (2001) Genomewide scans of complex human diseases : true linkage is hard to find. Am J Hum Genet 69:936–950
Sanger F, Nicklen S, Coulson A (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 74:5463–5467
Marchini J, Howie B (2010) Genotype imputation for genome-wide association studies. Nat Rev Genet 11:499–511. doi:10.1038/nrg2796
Hickey JM, Kinghorn BP, Tier B et al (2011) A combined long-range phasing and long haplotype imputation method to impute phase for SNP genotypes. Genet Sel Evol 43:12. doi:10.1186/1297-9686-43-12
Aknowledgements
We would like to thank Torben Hansen, Professor at Novo Nordisk Foundation Center for Basic Metabolic Research, University of Copenhagen, for comments that improved the manuscript.
Authors’contributions
JO and AC were involved in the conception and design of this work. RH collected litterature and in cooperation with AC, relevant studies where sorted out to be reviewed. The manuscript where mainly drafted by RH but with guidance and inputs from JO and AC who also revised it critically. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
Summary of family studies. Statistical measures are stated where possible. (DOCX 64 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hansen, R.D., Christensen, A.F. & Olesen, J. Family studies to find rare high risk variants in migraine. J Headache Pain 18, 32 (2017). https://doi.org/10.1186/s10194-017-0729-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10194-017-0729-y