
Abstract
Background
Autism spectrum disorders (ASDs) comprise a range of neurodevelopmental conditions of varying severity, characterized by marked qualitative difficulties in social relatedness, communication, and behavior. Despite overwhelming evidence of high heritability, results from genetic studies to date show that ASD etiology is extremely heterogeneous and only a fraction of autism genes have been discovered.
Methods
To help unravel this genetic complexity, we performed whole exome sequencing on 100 ASD individuals from 40 families with multiple distantly related affected individuals. All families contained a minimum of one pair of ASD cousins. Each individual was captured with the Agilent SureSelect Human All Exon kit, sequenced on the Illumina Hiseq 2000, and the resulting data processed and annotated with Burrows-Wheeler Aligner (BWA), Genome Analysis Toolkit (GATK), and SeattleSeq. Genotyping information on each family was utilized in order to determine genomic regions that were identical by descent (IBD). Variants identified by exome sequencing which occurred in IBD regions and present in all affected individuals within each family were then evaluated to determine which may potentially be disease related. Nucleotide alterations that were novel and rare (minor allele frequency, MAF, less than 0.05) and predicted to be detrimental, either by altering amino acids or splicing patterns, were prioritized.
Results
We identified numerous potentially damaging, ASD associated risk variants in genes previously unrelated to autism. A subset of these genes has been implicated in other neurobehavioral disorders including depression (SLIT3), epilepsy (CLCN2, PRICKLE1), intellectual disability (AP4M1), schizophrenia (WDR60), and Tourette syndrome (OFCC1). Additional alterations were found in previously reported autism candidate genes, including three genes with alterations in multiple families (CEP290, CSMD1, FAT1, and STXBP5). Compiling a list of ASD candidate genes from the literature, we determined that variants occurred in ASD candidate genes 1.65 times more frequently than in random genes captured by exome sequencing (P = 8.55 × 10-5).
Conclusions
By studying these unique pedigrees, we have identified novel DNA variations related to ASD, demonstrated that exome sequencing in extended families is a powerful tool for ASD candidate gene discovery, and provided further evidence of an underlying genetic component to a wide range of neurodevelopmental and neuropsychiatric diseases.
Similar content being viewed by others

Background
Autism spectrum disorders (ASDs) encompass a constellation of neurodevelopmental conditions characterized by three features: marked qualitative difficulties in social relatedness, communication, and behavior [1]. ASDs occur in approximately one of every 88 individuals and genetic studies to date demonstrate that the ASD etiology is highly complex with over 100 candidate genes being implicated in ASD etiology through linkage, association, and candidate gene studies [2, 3]. This genetic complexity is compounded by the fact that many families present with private and rare alterations and that known pathogenic alterations can result in a variety of clinical consequences [4–6]. Indeed, genetic overlap has been reported between ASDs and other neurodevelopmental and neuropsychiatric disorders including attention deficit hyperactivity disorder (ADHD), intellectual disability, schizophrenia, and Tourette syndrome [7–11].
With the advent of whole exome sequencing, studies have been rapidly identifying rare genetic variants and pinpointing the causes of classic Mendelian disorders [12]. However, exome sequencing of more complex disorders, such as autism, have primarily focused either on simplex families to discover de novo alterations [13–17] or consanguineous families that carry recessive mutations [18, 19]. In contrast, we designed a study to perform whole exome sequencing in extended, multiplex families with at least an affected cousin pair to identify potential new ASD loci. We hypothesized that identical by descent (IBD) filtering in these pedigrees would permit us to isolate genes contributing to ASD pathogenesis since these extended families are likely to carry novel ASD susceptibility loci of moderate to high effect. Our strategy discovered potentially damaging alterations in both known and novel ASD candidate genes, as well as in genes that carry variations known to be pathogenic in other neurological disorders.
Methods
Ethics statement
We ascertained individuals at the John P. Hussman Institute for Human Genomics (HIHG) at the University of Miami, Miller School of Medicine (Miami, FL, USA), the University of South Carolina (Columbia, SC, USA), and the Center for Human Genetics Research at Vanderbilt University (Nashville, TN, USA). Written informed consent was obtained from parents for all minor children and those who were unable to give consent. In addition, we obtained assent from all participants of the appropriate developmental and chronological age. All participants were ascertained using the protocol approved by the appropriate Institutional Review Boards. Patients were collected for this study for over a decade, with protocols and amendments being approved at each stage. Oversight of the study falls under the University of Miami (UM) Institutional Review Board (IRB). This study was approved by the UM Medical Sciences IRB Committee B members: Ofelia Alvarez MD, Abdul Mian PhD, Jose Castro MD, Rabbi Hector Epelbaum MA, Jean Jose DO, Howard Landy MD, Bruce Nolan MD FACP FAASM, Eric Zetka PharmD, and Liza Gordillo BA BS.
Sample selection
One hundred and sixty-four individuals (100 ASD patients: 90 males and 10 females; 5 relatives with ASD features: 2 males and 3 females; and 59 unaffected relatives: 27 males and 32 females) from 40 ASD extended families were used in this study [Additional file 1: Table S1]. We define extended families as multiplex families with at least one pair of ASD affected cousins. Each family has between two to five ASD individuals and relationships to each other range from first degree relatives (that is, parent–child and siblings) to distant relatives (that is, third cousins). Thirty-nine families were of European ancestry, while a single family (7606) was of African ancestry. All participants were enrolled using protocols approved by the appropriate Institutional Review Boards. Core inclusion criteria for ASD individuals included: (1) between 3 and 21 years of age, (2) a presumptive clinical diagnosis of ASD, (3) an expert clinical determination of an ASD diagnosis using DSM-IV criteria [1] supported by the Autism Diagnostic Interview-Revised (ADI-R) [20], and (4) an IQ equivalent >35 or developmental level >18 months as determined by the Vineland Adaptive Behavior Scale (VABS) [21]. Diagnostic determination was based on review by a panel consisting of experienced clinical psychologists and a pediatric medical geneticist. In those instances where an ADI-R was not available, a best-estimate diagnosis was assigned using all available clinical information including clinician summaries, a caregiver report, and medical records. IQ was obtained for the majority of individuals from administration of any of several measures (for example, age appropriate Wechsler scale, Leiter intelligence test, or Mullen Scales of Early Learning, MSEL) or from medical records. A summary of the sample is provided in Additional file 1: Table S2. DNA was isolated either from saliva (n = 2) or whole blood collected via venipuncture (n = 162).
Whole exome sequencing and variant detection
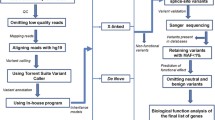
One hundred and sixty-four samples from extended ASD families were prepared following standard Agilent (Santa Clara, CA, USA) and Illumina (San Diego, CA, USA) protocols for whole exome sequencing (Figure 1). Briefly, 3 μg of genomic DNA was sheared to approximately 150 to 200 base pair fragments with the Covaris (Woburn, MA, USA) E210 and sequence capture performed with Agilent’s SureSelect Human All Exon kit. Samples were hybridized for 24 hours. The initial 19 samples were prepared with the 38 Mb kit and run on Illumina’s Genome Analyzer IIx with each individual being run in two lanes. The remaining 145 samples were captured using the 50 Mb kit, indexed, and multiplexed to run three per lane on the Illumina Hiseq 2000. Paired end 2 × 100 sequencing was performed. Sequencing data was processed using the Illumina Real Time Analysis (RTA) base calling pipeline, initially with version 1.7 and with a subset being run on version 1.8. Alignment to the hg19 human reference genome was executed with the Burrows-Wheeler Aligner (BWA) and variant calling performed with the Genome Analysis Toolkit (GATK) [22, 23]. GATK parameters included base quality score recalibration and duplicate removal [24]. Samples captured by the 38 Mb kit had an average depth of 64.4x, while the remaining samples processed with the 50 Mb kit had an average depth of 55.78x (Table 1). Variants were called at positions with a VQSLOD score greater than zero and minimum read depth of 4x. Alterations were annotated utilizing the SeattleSeq [25], PolyPhen-2 [26], and Sorting Intolerant From Tolerant (SIFT) programs [27]. The SeattleSeq program categorizes the two nucleotides flanking each exon as positions which, when altered, could potentially result in splicing alterations. In addition to samples from the autism extended families, 308 unrelated individuals of European ancestry negative for autism were internally processed at the HIHG. These HIHG control samples were captured with the Agilent SureSelect Human All Exon 50 Mb kit and processed according to the pipeline described above. Figure 1 outlines the steps used to generate and filter the data to the variants of interest. Genome wide SNP genotyping data on 159 samples was utilized to perform a quality check and confirm sample identity [28, 29]. All but two samples passed quality control metrics. Therefore, we compared the single nucleotide variant (SNV) calls between the whole exome sequencing and SNP genotyping in 157 samples to confirm sample identity and found an average concordance of 98.3%.

Flowchart of sequencing and filtering methods to identify and prioritize IBD variants. The data from whole exome sequencing as well as two genotyping platforms, whole genome SNP array and exome chip array, were each independently generated and processed. SNP array genotyping calls were compared to variants identified by exome sequencing as a quality check. Independent confirmation of calls from exome sequencing were made either by genotyping on the HumanExome BeadChip or by traditional Sanger sequencing. IBD, identical by descent.
Genotyping and identity by descent filtering
Of the 164 samples selected for exome sequencing, 159 were also evaluated on one of four Illumina whole genome genotyping arrays: the Human 1Mv1 BeadChip (n = 129), the 1 M-DuoV3 BeadChip (n = 24), the HumanOmniExpress-12 v1.0 BeadChip (n = 4), or the HumanOmni2.5-4v1 BeadChip (n = 2). The 1 M and 1 M-Duo BeadChips were analyzed as previously described [28, 29]. Samples processed on the OmniExpress BeadChip were prepared following Illumina’s Infinium HD Assay Ultra protocol, while those processed on the HumanOmni2.5 BeadChip followed Illumina’s Infinium HD Assay Super protocol. All chips were processed with automation on the Tecan (Männedorf, Switzerland) EVO-1 and BeadChips were scanned by either the Illumina BeadArray Reader or iScan. Data was extracted by the Genome Studio software from data files created by the iScan (Illumina) and a GenCall cutoff score of 0.15 was used. Samples on each of the four types of BeadChips were required to have a genotyping call rate of 98% or higher to pass quality control. Concordance between the genotypes of the variants identified through exome sequencing and genotyping was evaluated using the PLINK program [30]. All but two samples passed quality control metrics. Therefore, we compared the SNV calls between the whole exome sequencing and SNP genotyping in 157 samples to confirm sample identity and found an average concordance of 98.3%.
Genotyping information was further used to delineate IBD regions within each extended family. Only the 100 individuals with a confirmed ASD diagnosis were used to determine each family’s IBD regions. PLINK was employed for linkage disequilibrium (LD) pruning using the CEPH (Centre d'Etude du Polymorphisme Humain) CEU HapMap data for all families except 7606, for which the Yoruban (YRI) HapMap dataset was used [30]. The indep-pairwise option was utilized with a window size of 50, a step of 5, and an r2 threshold of 0.5. Next, these locations and their HapMap allele frequencies were analyzed in our dataset using the extended option in the MERLIN program using the 164 samples that were exome sequenced and 222 additional relatives [31]. To determine the start and stop positions of IBD sharing regions within each family, the MERLIN output was evaluated in a sliding window of ten SNVs, defining IBD as sharing at each location with a threshold >50%. Only regions shared across all available ASD individuals within a family were used to determine the IBD sharing segments.
To identify alterations inherited by all ASD individuals in a family from a single ancestor, whole exome sequencing data was restricted to IBD regions. Priority was also given to novel variants and rare SNVs. We define rare SNVs as those with a minor allele frequency (MAF), less than 5% in each of the three HapMap populations (African, Asian, or European), as well as in the 5,379 samples from the NHLBI Exome Sequencing Project, Exome Variant Server (EVS, version 5400) [32], and 308 HIHG control exomes. Novel variants were defined as those absent from the EVS, the 1000 Genomes Project [33], and dbSNP 134 [34]. Variants were evaluated for conservation with the Genomic Evolutionary Rate Profiling (GERP) score [35] and alterations measured for likelihood of having a damaging consequence on protein function through the PolyPhen-2 [27] and SIFT programs [28]. We also examined our results for overlap in genes previously reported in the literature and publically available databases (that is, SFARI Gene) to be associated with ASDs and other neurological disorders including ADHD, bipolar disorder, developmental delay, epilepsy, intellectual disability, major depression, obsessive compulsive disorder, schizophrenia, speech disorders, and Tourette syndrome [2, 36, 37].
Enrichment of ASD genes
To determine whether there was enrichment of ASD genes in the IBD, damaging variants that were identified, results were compared to a list of 1,075 genes that included known and suspected ASD candidate genes [Additional file 1: Table S3]. This list was compiled from the review by Betancur [2], the ASD and candidate genes lists generated by Pinto and colleagues [37], three autism exome de novo papers [15–17], and the SFARI Gene database [36]. The P value was calculated using a hypergeometric distribution.
Validation of variants
Of the 164 individuals from ASD extended families included in this study, 100 samples were also run on the Infinium HumanExome 12v1 BeadChip (Illumina). Exome chips were prepared following the manufacturer’s Infinium HD Assay Ultra protocol and automated using the Tecan EVO-1, as described above. Samples were required to have a genotyping call rate of 98% or higher to pass quality control. Variant calls were compared between the exome sequencing and genotyping with the PLINK program [30]. Between the exome sequencing and exome genotyping platforms, 446 changes were concordant, while only one variant was found to be discordant. A subset of variants was also validated by Sanger sequencing. Variants present in multiple families were prioritized, as were those occurring in genes with previous evidence implicating them in neurodevelopmental and neuropsychiatric disorders. Primers were created using the Primer3 v0.4.0 program (http://fokker.wi.mit.edu/primer3/input.htm) and the UCSC reference genome (GRCh37/hg19). Sequencing reactions were performed with the Big Dye Terminator v3.1, run on an Applied Biosystems 3730xl DNA Analyzer (Life Technologies, Carlsbad, CA, USA), and evaluated in the Sequencher v4.10.1 program (Gene Codes Corporation, Ann Arbor, MI, USA). Fifty-seven IBD changes were validated via Sanger sequencing, while one position failed to validate. With a total of 503/505 SNVs (99.6%) being concordant in two independent platforms, we determined that there is a relatively low false positive rate of variant calling in the filtered exome sequencing data.
Results and discussion
Identification and validation of rare and potentially damaging variants
Whole exome sequencing was performed in 164 individuals from 40 families to detect potentially causative variants [Additional file 1: Table S1 and S2]. SNP genotyping data on these 164 individuals and 222 relatives was used to isolate genomic areas inherited from a common ancestor, or IBD, and shared between ASD relatives (Figure 1). Following variant calling of the exome data and rigorous quality control (see Methods section), each family had SNVs at approximately 90,000 unique locations. We tested heterozygous, homozygous, and X-linked inheritance models using the same scheme (Figure 1). To investigate the first model, variants were filtered to include only IBD, heterozygous alterations present in all affected individuals within a family and predicted to be detrimental by either altering amino acids or splicing patterns. Variants were further parsed to include only those that were novel or relatively rare (MAF, <5%) in HapMap populations, EVS exomes, and 308 internally processed HIHG control exomes. A total of 742 IBD, heterozygous alterations in 690 genes were identified across the 40 families. This method was repeated with a homozygous model but no alterations survived the filtering process. Sixteen of the 40 families conformed to a possible X-linked pattern of inheritance and variants in three additional genes were found [Additional file 1: Table S1]. Therefore, a total of 745 rare, predicted damaging, alterations in 693 genes were identified. Three families did not demonstrate IBD co-segregation of any SNVs, while the remaining 37 families had at least two segregating SNVs. We then validated 502 of the 745 IBD variants of interest by one of two methods: Sanger sequencing or SNP genotyping [Additional file 1: Table S4].
Genes identified with more than one variant
We identified 36 genes that had segregating SNVs in at least two families (Figure 2, Additional file 1: Table S5). To put these new findings in context, we examined the ASD pedigrees for the presence of additional neurobehavioral features in both affected individuals and obligate carriers of the mutations of interest. While 32 of these genes have not been previously linked to ASDs, one of them, SLIT3, has been associated with another neuropsychiatric disorder (Table 2). Duplications overlapping the SLIT3 gene were previously found to be overrepresented in individuals with major depressive disorder [38]. In our study, the two families carrying an alteration in SLIT3, 17342 and 18074, also presented with a history of depression in three of the four obligate carriers (all female); the fourth obligate carrier was male and had no reported neuropsychiatric traits. In addition, four ASD candidate genes were found with alterations in more than one extended family: CEP290, CSMD1, FAT1, and STXPB5. Alterations in CEP290 have been connected to ASDs and intellectual disability and were previously identified in patients diagnosed with a wide variety of ciliopathies including Bardet-Biedl, Joubert, Meckel-Gruber, and Senior-Løken syndromes (http://medgen.ugent.be/cep290base/) [39]. Along with being linked to ASDs, CSMD1 has been associated with schizophrenia and is a known target of mir-137, a microRNA that regulates neuronal maturation and adult neurogenesis [40–43]. FAT1 is a candidate for both ASD and bipolar disorder and is a member of the cadherin gene family [15, 44]. While we did not identify individuals with mutations in FAT1 who presented with bipolar disorder, the two obligate carriers (female) in family 17545 reported a history of major depression. Lastly, a deletion across STXBP5 was previously reported in a patient with autism, intellectual disabilities, and seizures [45]. STXBP5 functions in neuronal guidance and synaptic transmission [46]. In our first family identified with a STXBP5 variant, 37994, we observed intellectual disability in both of the ASD individuals as well as seizures in the proband (1). In addition, the mother (1001) of the proband was reported to have bipolar disorder. In the second family carrying a mutation in STXBP5, 7623, we observed seizures in one of the affected individuals (101) as well as migraines in his mother (1007), an obligate carrier. These results augment the growing evidence supporting a genetic overlap between a wide variety of neurodevelopmental and neuropsychiatric disorders [7–11].

Diagram of genes of high interest. The area of each circle corresponds to the number of genes identified in one of four categories: genes previously implicated as ASD candidates, non-ASD genes that have been implicated in other neurodevelopmental and neuropsychiatric disorders, genes found to have damaging variants in more than one family, and genes that carry two damaging alterations in the same family. ASD, autism spectrum disorder.
Fourteen genes segregated two heterozygous and damaging SNVs in cis within a single family; 12 of these genes have no prior evidence of a connection to ASDs (Figure 2, Additional file 1: Table S6). Two novel genes have been identified in other neurodevelopmental and neuropsychiatric disorders: PRICKLE1 and OFCC1 (Table 3). Family 37232 carries two changes in PRICKLE1, a gene involved in neurite outgrowth which has been linked to epilepsy and neural tube defects [47–50]. Interestingly, the proband of this family (1) was reported to have a history of seizures. Independently, Paemka and colleagues implicated PRICKLE1 as an ASD gene through extensive in vivo and in vitro functional analysis [51], thereby demonstrating the power of the extended family approach to identify novel ASD candidates. Moreover, OFCC1, a gene linked with Tourette syndrome [52], was found to have both a missense and a nonsense alteration in family 7606, the one family of African ancestry in this study. In this family, we observed self-injurious behaviors in all three affected individuals as well as seizures in one affected (2061). In addition, two ASD genes were found to carry variants in cis: ABHD14A and FAT1. Family 17351 carries two changes in ABHD14A, a gene involved in cerebellar development [53, 54]. In this family, one affected individual (105) was described as having intellectual disability and a male obligate carrier (1000) was reported to have bipolar disorder. Another ASD candidate gene, FAT1, has two alterations in family 17545, as well as a distinct variant detected in family 37037 [15, 44]. As mentioned above, family 17545 reported that both obligate carriers in the family have a history of major depression.
Single alterations found in genes related to neurological disorders
Three additional genes related to other neuropsychiatric and neurodevelopmental disorders were each recognized to carry a single IBD alteration: AP4M1 (intellectual disability [55]), CLCN2 (epilepsy [56]), and WDR60 (intellectual disability and schizophrenia [57], Additional file 1: Table S7). Moreover, an additional 38 known or suspected ASD genes were identified with one variant, including AGAP1[58], CDH9[28, 59], DLGAP2[60], FBXO40[61], GRIN3B[13], NRXN2[62], and SYNE2[14]. Following an X-linked pattern of inheritance, hemizygous alterations were identified and validated in SYN1, a gene which encodes a synaptic vesicle phosphoprotein and has been previously connected to both autism and epilepsy [63–65]. Both ASD individuals carrying the SYN1 alteration in family 37674 have a history of epilepsy. Interestingly, Paemka and colleagues independently show that SYN1 co-immunoprecipitated with PRICKLE1 in mouse brain and that the proteins co-localized in Drosophila neurons [51], demonstrating a conserved physical interaction between a newly identified candidate gene, PRICKLE1, and an established ASD gene, SYN1, and supporting the validity of our exome filtering method. This provides additional evidence that investigating genes and pathways that interact with known ASD candidates will likely prove a fruitful area for the identification of ASD related genes.
Further potentially novel ASD candidates identified by exome sequencing include genes with previous clinical and molecular evidence supporting a neuronal function such as CIC, GLUD2, NTSR2, RODG1, and SEZ6. CIC is a genetic modifier of the neurodegenerative disorder spinocerebellar ataxia [66]. GLUD2 plays a role in postsynaptic density formation in the cerebellum and modifies the age of onset of Parkinson’s disease patients [67, 68]. NTSR2 is a G protein-coupled receptor for neurotensin that is widely expressed throughout the brain [69]. RODG1 mutations result in Kohlschütter-Tönz syndrome, a disorder that presents with epilepsy and developmental delay, both features found in children with autism [70]. Lastly, SEZ6 acts in dendritic arborization and has been associated with seizures in a Chinese cohort [71, 72]. This category of genes, along with genes identified to carry alterations in more than one family, may be the most fruitful groups of novel ASD candidates for future investigations.
IBD filtering enriches for ASD candidate genes
In order to determine whether our filtering method enriched for ASD specific variants, we generated a list of 1,075 known and suspected ASD candidates from the literature and the SFARI Gene database [2, 15–17, 36, 37]. When this ASD candidate list was compared to the 693 genes carrying variants meeting the above filter criteria, we found that 5.7% of the genes in the ASD list met our criteria compared to only 3.4% of all genes captured by the exome sequencing; thus, we were 1.65 times more likely to find a variant in an ASD candidate gene than a random gene captured by exome sequencing (P = 8.55 × 10-5). We therefore concluded that there was a significant enrichment of ASD candidate genes carrying damaging, segregating variants, supporting our hypothesis that whole exome sequencing of extended autism families is a reliable approach to identify new autism genes.
Conclusions
Our results have identified several new genes that likely play a role in ASD and provide additional support for the role of other proposed ASD genes. These data reinforce two emerging observations about ASDs. The first is the extreme level of genetic heterogeneity, with no locus contributing to more than 1% of ASD cases [2, 73]. The second is that there is significant overlap between the genetic etiology of ASD and other neuropsychiatric and neurodevelopmental disorders [7–11]. These data, taken together with the increasing amount of functional data available for many of these genes such as PRICKLE1[51], highlight the enormous complexity of ASD and the difficulties in resolving this enigma.
Abbreviations
- ADHD:
-
Attention deficit hyperactivity disorder
- ADI-R:
-
Autism diagnostic interview-revised
- ASD:
-
Autism spectrum disorder
- BWA:
-
Burrows-Wheeler Aligner
- CEU:
-
The CEPH (Centre d'Etude du Polymorphisme Humain) HapMap cohort
- DSM-IV:
-
Diagnostic and statistical manual of mental disorders fourth edition
- EVS:
-
Exome variant server
- GATK:
-
Genome analysis toolkit
- GERP:
-
Genomic evolutionary rate profiling
- HIHG:
-
Hussman institute for human genomics
- IBD:
-
Identical by descent
- IQ:
-
Intelligence quotient
- IRB:
-
Institutional review board
- LD:
-
Linkage disequilibrium
- MAF:
-
Minor allele frequency
- MSEL:
-
Mullen scales of early learning
- RTA:
-
Real time analysis
- SIFT:
-
Sorting intolerant from tolerant
- SNP:
-
Single nucleotide polymorphism
- SNV:
-
Single nucleotide variant
- UCSC:
-
University of California Santa Cruz
- UM:
-
University of Miami
- VABS:
-
Vineland adaptive behavior scale
- YRI:
-
Yoruban HapMap cohort.
References
American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders (DSM-IV). 1994, Washington, DC: American Psychiatric Press, Inc, Fourth
Betancur C: Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res. 2011, 1380: 42-77.
Autism and Developmental Disabilities Monitoring Network Surveillance Year: Prevalence of autism spectrum disorders - autism and developmental disabilities monitoring network, 14 sites, United States, 2008. MMWR Surveill Summ. 2008, 2012 (61): 1-19.
McClellan J, King MC: Genetic heterogeneity in human disease. Cell. 2010, 141: 210-217. 10.1016/j.cell.2010.03.032.
State MW, Levitt P: The conundrums of understanding genetic risks for autism spectrum disorders. Nat Neurosci. 2011, 14: 1499-1506. 10.1038/nn.2924.
Malhotra D, Sebat J: CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell. 2012, 148: 1223-1241. 10.1016/j.cell.2012.02.039.
Carroll LS, Owen MJ: Genetic overlap between autism, schizophrenia and bipolar disorder. Genome Med. 2009, 1: 102-10.1186/gm102.
Guilmatre A, Dubourg C, Mosca AL, Legallic S, Goldenberg A, Drouin-Garraud V, Layet V, Rosier A, Briault S, Bonnet-Brilhault F, Laumonnier F, Odent S, Le Vacon G, Joly-Helas G, David V, Bendavid C, Pinoit JM, Henry C, Impallomeni C, Germano E, Tortorella G, Di Rosa G, Barthelemy C, Andres C, Faivre L, Frebourg T, Saugier Veber P, Campion D: Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch Gen Psychiatry. 2009, 66: 947-956. 10.1001/archgenpsychiatry.2009.80.
Saus E, Brunet A, Armengol L, Alonso P, Crespo JM, Fernandez-Aranda F, Guitart M, Martin-Santos R, Menchon JM, Navines R, Soria V, Torrens M, Urretavizcaya M, Valles V, Gratacos M, Estivill X: Comprehensive copy number variant (CNV) analysis of neuronal pathways genes in psychiatric disorders identifies rare variants within patients. J Psychiatr Res. 2010, 44: 971-978. 10.1016/j.jpsychires.2010.03.007.
Lionel AC, Crosbie J, Barbosa N, Goodale T, Thiruvahindrapuram B, Rickaby J, Gazzellone M, Carson AR, Howe JL, Wang Z, Wei J, Stewart AF, Roberts R, McPherson R, Fiebig A, Franke A, Schreiber S, Zwaigenbaum L, Fernandez BA, Roberts W, Arnold PD, Szatmari P, Marshall CR, Schachar R, Scherer SW: Rare copy number variation discovery and cross-disorder comparisons identify risk genes for ADHD. Sci Transl Med. 2011, 3: 95ra75-
Fernandez TV, Sanders SJ, Yurkiewicz IR, Ercan-Sencicek AG, Kim YS, Fishman DO, Raubeson MJ, Song Y, Yasuno K, Ho WS, Bilguvar K, Glessner J, Chu SH, Leckman JF, King RA, Gilbert DL, Heiman GA, Tischfield JA, Hoekstra PJ, Devlin B, Hakonarson H, Mane SM, Gunel M, State MW: Rare copy number variants in tourette syndrome disrupt genes in histaminergic pathways and overlap with autism. Biol Psychiatry. 2012, 71: 392-402. 10.1016/j.biopsych.2011.09.034.
Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, Shendure J, Bamshad MJ: Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010, 42: 30-35. 10.1038/ng.499.
O'Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, Rieder MJ, Nickerson DA, Bernier R, Fisher SE, Shendure J, Eichler EE: Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011, 43: 585-589. 10.1038/ng.835.
Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, Kendall J, Grabowska E, Ma B, Marks S, Rodgers L, Stepansky A, Troge J, Andrews P, Bekritsky M, Pradhan K, Ghiban E, Kramer M, Parla J, Demeter R, Fulton LL, Fulton RS, Magrini VJ, Ye K, Darnell JC, Darnell RB: De novo gene disruptions in children on the autistic spectrum. Neuron. 2012, 74: 285-299. 10.1016/j.neuron.2012.04.009.
Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, Polak P, Yoon S, Maguire J, Crawford EL, Campbell NG, Geller ET, Valladares O, Schafer C, Liu H, Zhao T, Cai G, Lihm J, Dannenfelser R, Jabado O, Peralta Z, Nagaswamy U, Muzny D, Reid JG, Newsham I, Wu Y: Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012, 485: 242-245. 10.1038/nature11011.
O'Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Rieder MJ, Nickerson DA, Bernier R, Shendure J, Eichler EE: Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012, 485: 246-250. 10.1038/nature10989.
Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, Dilullo NM, Parikshak NN, Stein JL, Walker MF, Ober GT, Teran NA, Song Y, El-Fishawy P, Murtha RC, Choi M, Overton JD, Bjornson RD, Carriero NJ, Meyer KA, Bilguvar K, Mane SM, Sestan N, Lifton RP, Gunel M, Roeder K, Geschwind DH, Devlin B, State MW: De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012, 485: 237-241. 10.1038/nature10945.
Chahrour MH, Yu TW, Lim ET, Ataman B, Coulter ME, Hill RS, Stevens CR, Schubert CR, Greenberg ME, Gabriel SB, Walsh CA, ARRA Autism Sequencing Collaboration: Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet. 2012, 8: e1002635-10.1371/journal.pgen.1002635.
Puffenberger EG, Jinks RN, Wang H, Xin B, Fiorentini C, Sherman EA, Degrazio D, Shaw C, Sougnez C, Cibulskis K, Gabriel S, Kelley RI, Morton DH, Strauss KA: A homozygous missense mutation in HERC2 associated with global developmental delay and autism spectrum disorder. Hum Mutat. 2012, 33: 1639-1646. 10.1002/humu.22237.
Rutter M, LeCouteur A, Lord C: Autism Diagnostic Interview, Revised (ADI-R). 2003, Los Angeles, CA: Western Psychological Services
Sparrow SS, Cicchetti DV, Balla D: Vineland Adaptive Behavior Scales. 2005, Circle Pines, MN: American Guidance Service, 2
Li H, Durbin R: Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009, 25: 1754-1760. 10.1093/bioinformatics/btp324.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA: The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20: 1297-1303. 10.1101/gr.107524.110.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ: A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011, 43: 491-498. 10.1038/ng.806.
SeattleSeq. [http://snp.gs.washington.edu/SeattleSeqAnnotation137/]
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR: A method and server for predicting damaging missense mutations. Nat Methods. 2010, 7: 248-249. 10.1038/nmeth0410-248.
Kumar P, Henikoff S, Ng PC: Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009, 4: 1073-1081. 10.1038/nprot.2009.86.
Ma DQ, Salyakina D, Jaworski JM, Konidari I, Whitehead PL, Andersen A, Hoffman JD, Slifer SH, Hedges DJ, Cukier H, Beecham GW, Wright HH, Abramson RK, Martin ER, Hussman JP, Gilbert JR, Cuccaro ML, Haines JL, Pericak-Vance MA: A genome-wide association study of autism reveals a common novel risk locus at 5p14.1. Ann Hum Genet. 2009, 73: 263-273. 10.1111/j.1469-1809.2009.00523.x.
Salyakina D, Cukier HN, Lee JM, Sacharow S, Nations LD, Ma D, Jaworski JM, Konidari I, Whitehead PL, Wright HH, Abramson RK, Williams SM, Menon R, Haines JL, Gilbert JR, Cuccaro ML, Pericak-Vance MA: Copy number variants in extended autism spectrum disorder families reveal candidates potentially involved in autism risk. PLoS One. 2011, 6: e26049-10.1371/journal.pone.0026049.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007, 81: 559-575. 10.1086/519795.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR: Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002, 30: 97-101. 10.1038/ng786.
The National Heart, Lung, and Blood Institute Exome Sequencing Project Exome Variant Server. [http://evs.gs.washington.edu/EVS/]
Durbin RM, Abecasis GR, Altshuler DL, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME McVean GA, Durbin RM, Abecasis GR, Altshuler DL, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME McVean GA, 1000 Genomes Project Consortium: A map of human genome variation from population-scale sequencing. Nature. 2010, 467: 1061-1073. 10.1038/nature09534.
The National Center for Biotechnology Information’s Database of Single Nucleotide Polymorphisms. [http://www.ncbi.nlm.nih.gov/snp]
Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A, NISC Comparative Sequencing Program: Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15: 901-913. 10.1101/gr.3577405.
Banerjee-Basu S, Packer A: SFARI Gene: an evolving database for the autism research community. Dis Model Mech. 2010, 3: 133-135. 10.1242/dmm.005439.
Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Almeida J, Bacchelli E, Bader GD, Bailey AJ, Baird G, Battaglia A, Berney T, Bolshakova N, Bolte S, Bolton PF, Bourgeron T, Brennan S, Brian J, Bryson SE, Carson AR, Casallo G, Casey J, Chung BH, Cochrane L, Corsello C: Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010, 466: 368-372. 10.1038/nature09146.
Glessner JT, Wang K, Sleiman PM, Zhang H, Kim CE, Flory JH, Bradfield JP, Imielinski M, Frackelton EC, Qiu H, Mentch F, Grant SF, Hakonarson H: Duplication of the SLIT3 locus on 5q35.1 predisposes to major depressive disorder. PLoS One. 2010, 5: e15463-10.1371/journal.pone.0015463.
Coppieters F, Lefever S, Leroy BP, De Baere E: CEP290, a gene with many faces: mutation overview and presentation of CEP290base. Hum Mutat. 2010, 31: 1097-1108. 10.1002/humu.21337.
Glancy M, Barnicoat A, Vijeratnam R, de Souza S, Gilmore J, Huang S, Maloney VK, Thomas NS, Bunyan DJ, Jackson A, Barber JC: Transmitted duplication of 8p23.1-8p23.2 associated with speech delay, autism and learning difficulties. Eur J Hum Genet. 2009, 17: 37-43. 10.1038/ejhg.2008.133.
Havik B, Le Hellard S, Rietschel M, Lybaek H, Djurovic S, Mattheisen M, Muhleisen TW, Degenhardt F, Priebe L, Maier W, Breuer R, Schulze TG, Agartz I, Melle I, Hansen T, Bramham CR, Nothen MM, Stevens B, Werge T, Andreassen OA, Cichon S, Steen VM: The complement control-related genes CSMD1 and CSMD2 associate to schizophrenia. Biol Psychiatry. 2011, 70: 35-42. 10.1016/j.biopsych.2011.01.030.
Kwon E, Wang W, Tsai LH: Validation of schizophrenia-associated genes CSMD1, C10orf26, CACNA1C and TCF4 as miR-137 targets. Mol Psychiatry. 2011, 18: 11-12.
Ripke S, Sanders AR, Kendler KS, Levinson DF, Sklar P, Holmans PA, Lin DY, Duan J, Ophoff RA, Andreassen OA, Scolnick E, Cichon S, St Clair D, Corvin A, Gurling H, Werge T, Rujescu D, Blackwood DH, Pato CN, Malhotra AK, Purcell S, Dudbridge F, Neale BM, Rossin L, Visscher PM, Posthuma D, Ruderfer DM, Fanous A, Stefansson H, Steinberg S: Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011, 43: 969-976. 10.1038/ng.940.
Blair IP, Chetcuti AF, Badenhop RF, Scimone A, Moses MJ, Adams LJ, Craddock N, Green E, Kirov G, Owen MJ, Kwok JB, Donald JA, Mitchell PB, Schofield PR: Positional cloning, association analysis and expression studies provide convergent evidence that the cadherin gene FAT contains a bipolar disorder susceptibility allele. Mol Psychiatry. 2006, 11: 372-383. 10.1038/sj.mp.4001784.
Davis LK, Meyer KJ, Rudd DS, Librant AL, Epping EA, Sheffield VC, Wassink TH: Novel copy number variants in children with autism and additional developmental anomalies. J Neurodev Disord. 2009, 1: 292-301. 10.1007/s11689-009-9013-z.
Sakisaka T, Baba T, Tanaka S, Izumi G, Yasumi M, Takai Y: Regulation of SNAREs by tomosyn and ROCK: implication in extension and retraction of neurites. J Cell Biol. 2004, 166: 17-25. 10.1083/jcb.200405002.
Okuda H, Miyata S, Mori Y, Tohyama M: Mouse Prickle1 and Prickle2 are expressed in postmitotic neurons and promote neurite outgrowth. FEBS Lett. 2007, 581: 4754-4760. 10.1016/j.febslet.2007.08.075.
Bassuk AG, Wallace RH, Buhr A, Buller AR, Afawi Z, Shimojo M, Miyata S, Chen S, Gonzalez-Alegre P, Griesbach HL, Wu S, Nashelsky M, Vladar EK, Antic D, Ferguson PJ, Cirak S, Voit T, Scott MP, Axelrod JD, Gurnett C, Daoud AS, Kivity S, Neufeld MY, Mazarib A, Straussberg R, Walid S, Korczyn AD, Slusarski DC, Berkovic SF, El-Shanti HI: A homozygous mutation in human PRICKLE1 causes an autosomal-recessive progressive myoclonus epilepsy-ataxia syndrome. Am J Hum Genet. 2008, 83: 572-581. 10.1016/j.ajhg.2008.10.003.
Bosoi CM, Capra V, Allache R, Trinh VQ, De Marco P, Merello E, Drapeau P, Bassuk AG, Kibar Z: Identification and characterization of novel rare mutations in the planar cell polarity gene PRICKLE1 in human neural tube defects. Hum Mutat. 2011, 32: 1371-1375. 10.1002/humu.21589.
Tao H, Manak JR, Sowers L, Mei X, Kiyonari H, Abe T, Dahdaleh NS, Yang T, Wu S, Chen S, Fox MH, Gurnett C, Montine T, Bird T, Shaffer LG, Rosenfeld JA, McConnell J, Madan-Khetarpal S, Berry-Kravis E, Griesbach H, Saneto RP, Scott MP, Antic D, Reed J, Boland R, Ehaideb SN, El-Shanti H, Mahajan VB, Ferguson PJ, Axelrod JD: Mutations in prickle orthologs cause seizures in flies, mice, and humans. Am J Hum Genet. 2011, 88: 138-149. 10.1016/j.ajhg.2010.12.012.
Paemka L, Mahajan VB, Skeie JM, Sowers LP, Ehaideb SN, Gonzalez-Alegre P, Sasaoka T, Tao H, Miyagi A, Ueno N, Wu S, Darbro BW, Ferguson PJ, Pieper AA, Britt JK, Wemmie JA, Rudd DS, Wassink T, El-Shanti H, Mefford HC, Carvill GL, Manak JR, Bassuk AG: PRICKLE1 Interaction with SYNAPSIN I Reveals a Role in Autism Spectrum Disorders. PLoS One. 2013, 8: e80737-10.1371/journal.pone.0080737.
Sundaram SK, Huq AM, Sun Z, Yu W, Bennett L, Wilson BJ, Behen ME, Chugani HT: Exome sequencing of a pedigree with Tourette syndrome or chronic tic disorder. Ann Neurol. 2011, 69: 901-904. 10.1002/ana.22398.
Hoshino J, Aruga J, Ishiguro A, Mikoshiba K: Dorz1, a novel gene expressed in differentiating cerebellar granule neurons, is down-regulated in Zic1-deficient mouse. Brain Res Mol Brain Res. 2003, 120: 57-64. 10.1016/j.molbrainres.2003.10.004.
Casey JP, Magalhaes T, Conroy JM, Regan R, Shah N, Anney R, Shields DC, Abrahams BS, Almeida J, Bacchelli E, Bailey AJ, Baird G, Battaglia A, Berney T, Bolshakova N, Bolton PF, Bourgeron T, Brennan S, Cali P, Correia C, Corsello C, Coutanche M, Dawson G, de Jonge M, Delorme R, Duketis E, Duque F, Estes A, Farrar P, Fernandez BA: A novel approach of homozygous haplotype sharing identifies candidate genes in autism spectrum disorder. Hum Genet. 2012, 131: 565-579. 10.1007/s00439-011-1094-6.
AbouJamra R, Philippe O, Raas-Rothschild A, Eck SH, Graf E, Buchert R, Borck G, Ekici A, Brockschmidt FF, Nothen MM, Munnich A, Strom TM, Reis A, Colleaux L: Adaptor protein complex 4 deficiency causes severe autosomal-recessive intellectual disability, progressive spastic paraplegia, shy character, and short stature. Am J Hum Genet. 2011, 88: 788-795. 10.1016/j.ajhg.2011.04.019.
Everett K, Chioza B, Aicardi J, Aschauer H, Brouwer O, Callenbach P, Covanis A, Dooley J, Dulac O, Durner M, Eeg-Olofsson O, Feucht M, Friis M, Guerrini R, Heils A, Kjeldsen M, Nabbout R, Sander T, Wirrell E, McKeigue P, Robinson R, Taske N, Gardiner M: Linkage and mutational analysis of CLCN2 in childhood absence epilepsy. Epilepsy Res. 2007, 75: 145-153. 10.1016/j.eplepsyres.2007.05.004.
Kirov G, Grozeva D, Norton N, Ivanov D, Mantripragada KK, Holmans P, Craddock N, Owen MJ, O'Donovan MC, International Schizophrenia Consortium; Wellcome Trust Case Control Consortium: Support for the involvement of large copy number variants in the pathogenesis of schizophrenia. Hum Mol Genet. 2009, 18: 1497-1503. 10.1093/hmg/ddp043.
Wassink TH, Piven J, Vieland VJ, Jenkins L, Frantz R, Bartlett CW, Goedken R, Childress D, Spence MA, Smith M, Sheffield VC: Evaluation of the chromosome 2q37.3 gene CENTG2 as an autism susceptibility gene. Am J Med Genet B Neuropsychiatr Genet. 2005, 136B: 36-44. 10.1002/ajmg.b.30180.
Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS, Salyakina D, Imielinski M, Bradfield JP, Sleiman PM, Kim CE, Hou C, Frackelton E, Chiavacci R, Takahashi N, Sakurai T, Rappaport E, Lajonchere CM, Munson J, Estes A, Korvatska O, Piven J, Sonnenblick LI, Alvarez-Retuerto AI, Herman EI, Dong H, Hutman T, Sigman M, Ozonoff S, Klin A: Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009, 459: 528-533. 10.1038/nature07999.
Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, Thiruvahindrapduram B, Fiebig A, Schreiber S, Friedman J, Ketelaars CE, Vos YJ, Ficicioglu C, Kirkpatrick S, Nicolson R, Sloman L, Summers A, Gibbons CA, Teebi A, Chitayat D, Weksberg R, Thompson A, Vardy C, Crosbie V, Luscombe S, Baatjes R: Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008, 82: 477-488. 10.1016/j.ajhg.2007.12.009.
Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, Imielinski M, Frackelton EC, Reichert J, Crawford EL, Munson J, Sleiman PM, Chiavacci R, Annaiah K, Thomas K, Hou C, Glaberson W, Flory J, Otieno F, Garris M, Soorya L, Klei L, Piven J, Meyer KJ, Anagnostou E, Sakurai T: Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009, 459: 569-573. 10.1038/nature07953.
Gauthier J, Siddiqui TJ, Huashan P, Yokomaku D, Hamdan FF, Champagne N, Lapointe M, Spiegelman D, Noreau A, Lafreniere RG, Fathalli F, Joober R, Krebs MO, DeLisi LE, Mottron L, Fombonne E, Michaud JL, Drapeau P, Carbonetto S, Craig AM, Rouleau GA: Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia. Hum Genet. 2011, 130: 563-573. 10.1007/s00439-011-0975-z.
Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Sudhof TC: Essential functions of synapsins I and II in synaptic vesicle regulation. Nature. 1995, 375: 488-493. 10.1038/375488a0.
Garcia CC, Blair HJ, Seager M, Coulthard A, Tennant S, Buddles M, Curtis A, Goodship JA: Identification of a mutation in synapsin I, a synaptic vesicle protein, in a family with epilepsy. J Med Genet. 2004, 41: 183-186. 10.1136/jmg.2003.013680.
Fassio A, Patry L, Congia S, Onofri F, Piton A, Gauthier J, Pozzi D, Messa M, Defranchi E, Fadda M, Corradi A, Baldelli P, Lapointe L, St-Onge J, Meloche C, Mottron L, Valtorta F, Khoa Nguyen D, Rouleau GA, Benfenati F, Cossette P: SYN1 loss-of-function mutations in autism and partial epilepsy cause impaired synaptic function. Hum Mol Genet. 2011, 20: 2297-2307. 10.1093/hmg/ddr122.
Fryer JD, Yu P, Kang H, Mandel-Brehm C, Carter AN, Crespo-Barreto J, Gao Y, Flora A, Shaw C, Orr HT, Zoghbi HY: Exercise and genetic rescue of SCA1 via the transcriptional repressor Capicua. Science. 2011, 334: 690-693. 10.1126/science.1212673.
Plaitakis A, Latsoudis H, Kanavouras K, Ritz B, Bronstein JM, Skoula I, Mastorodemos V, Papapetropoulos S, Borompokas N, Zaganas I, Xiromerisiou G, Hadjigeorgiou GM, Spanaki C: Gain-of-function variant in GLUD2 glutamate dehydrogenase modifies Parkinson's disease onset. Eur J Hum Genet. 2010, 18: 336-341. 10.1038/ejhg.2009.179.
Yuzaki M: The ins and outs of GluD2–why and how Purkinje cells use the special glutamate receptor. Cerebellum. 2012, 11: 438-439. 10.1007/s12311-011-0328-4.
Vita N, Oury-Donat F, Chalon P, Guillemot M, Kaghad M, Bachy A, Thurneyssen O, Garcia S, Poinot-Chazel C, Casellas P, Keane P, Le Fur G, Maffrand JP, Soubrie P, Caput D, Ferrara P: Neurotensin is an antagonist of the human neurotensin NT2 receptor expressed in Chinese hamster ovary cells. Eur J Pharmacol. 1998, 360: 265-272. 10.1016/S0014-2999(98)00678-5.
Schossig A, Wolf NI, Fischer C, Fischer M, Stocker G, Pabinger S, Dander A, Steiner B, Tonz O, Kotzot D, Haberlandt E, Amberger A, Burwinkel B, Wimmer K, Fauth C, Grond-Ginsbach C, Koch MJ, Deichmann A, von Kalle C, Bartram CR, Kohlschutter A, Trajanoski Z, Zschocke J: Mutations in ROGDI Cause Kohlschütter-Tönz Syndrome. Am J Hum Genet. 2012, 90: 701-707. 10.1016/j.ajhg.2012.02.012.
Gunnersen JM, Kim MH, Fuller SJ, De Silva M, Britto JM, Hammond VE, Davies PJ, Petrou S, Faber ES, Sah P, Tan SS: Sez-6 proteins affect dendritic arborization patterns and excitability of cortical pyramidal neurons. Neuron. 2007, 56: 621-639. 10.1016/j.neuron.2007.09.018.
Yu ZL, Jiang JM, Wu DH, Xie HJ, Jiang JJ, Zhou L, Peng L, Bao GS: Febrile seizures are associated with mutation of seizure-related (SEZ) 6, a brain-specific gene. J Neurosci Res. 2007, 85: 166-172. 10.1002/jnr.21103.
Kumar RA, Christian SL: Genetics of autism spectrum disorders. Curr Neurol Neurosci Rep. 2009, 9: 188-197. 10.1007/s11910-009-0029-2.
Acknowledgements
The authors gratefully acknowledge the participation of the individuals with ASD and their families, without whom this research would not be possible. The authors also wish to acknowledge Dr Dale Hedges and James Jaworski’s contributions in the exome sequencing analysis and Sandhya Ramsook and Bianca Barrionuevo’s efforts in the validation of ASD specific variants.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HNC, JLH, MLC, JRG, and MAPV conceived and designed the experiments. HNC prepared samples for exome sequencing, analyzed exome sequencing data, and wrote the manuscript. JML and MLC collected the samples, made diagnoses, and interpreted the clinical data from the ASD families. PLW, EL, NL, IK, RCG, and WFH performed the exome sequencing and exome chip genotyping. SHS, DVB, MAS, ERM, and MAPV analyzed the exome sequencing data. NDD analyzed the exome chip data. VM and NKH performed the Sanger sequencing validation. NDD, ERM, JLH, MLC, JRG, and MAPV edited the manuscript. The authors jointly discussed the experimental results throughout the duration of the study. All authors read and approved the final manuscript.
Electronic supplementary material
13229_2013_99_MOESM1_ESM.doc
Additional file 1: This file contains the seven tables listed below: Table S1 - Extended families structure. Table S2 - Clinical information on individuals with ASDs. Table S3 - ASD candidate genes. Table S4 - Variants identified in exome sequencing and validated by a second platform. Table S5 - Genes with damaging, validated variants in more than one family. Table S6 - Families with multiple damaging, validated variants in the same gene. Table S7 - Damaging, validated variants in genes previously implicated in ASD or other disorders. (DOC 2 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Cukier, H.N., Dueker, N.D., Slifer, S.H. et al. Exome sequencing of extended families with autism reveals genes shared across neurodevelopmental and neuropsychiatric disorders. Molecular Autism 5, 1 (2014). https://doi.org/10.1186/2040-2392-5-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2040-2392-5-1