
Abstract
The method used to cultivate the popular Japanese mushroom Grifola frondosa (maitake), called 'wood log cultivation', comprised two steps: (1) the mycelium is grown around a wood log in a plastic bag, and (2) the mycelium that has spread on the wood log is transferred into casing substrates in a forest. This method is still popular in Japan due to its low cost and high-quality crop production. The importance of the microbiome that inhabits mushroom-cultivation surroundings has recently attracted attention, but no study of the microbial communities in maitake cultivation has been published. We investigated how the bacterial communities changed in wood logs in comparison with a control group (without inoculation) and their interaction with maitake during the first to fourth years of maitake wood log cultivation. A maitake biomass was detected by quantitative PCR in wood logs but not in the casings, and we thus decided to investigate the bacterial communities in wood log samples for control and first- to fourth-year cultivation. The results indicate that the phyla Proteobacteria, Firmicutes, and Gemmatimonadota play key roles in changes of the microbiome composition for maitake wood log cultivation. In a functional profile, bacteria communities in the wood logs during maitake cultivation showed higher relative abundance in cellulolysis, glycolysis, TCA cycle, and many biosynthesis pathways, whereas the control group showed higher relative abundance in fermentation. These results suggested that (i) the bacterial communities which inhabited maitake cultivated wood logs may help the maitake degrade wood cellulose, and (ii) part of the glucose from the cellulose degraded by both maitake and bacteria was used for the bacterial TCA cycle instead of fermentation. Bacteria also produce some chemicals that maitake mycelium may need. It is also likely that some potential intracellular parasites dwell with maitake. The different cultivation stages showed different network structures. A network analysis indicated that Class Gammaproteobacteria is a potential keystone taxon for the microbiome network stability of maitake cultivated wood logs. These results contribute to the understanding of the microbiome in maitake-cultivation surroundings and will improve maitake wood log cultivation.
Similar content being viewed by others

Introduction
Grifola frondosa (maitake) mushroom is a basidiomycete that grows on decaying wood, and it is an economically important edible mushroom [1]. Due to advances in cultivation and breeding technology, maitake mushroom is available in grocery markets [2]. In Japan, two methods are used for the cultivation of G. frondosa mushrooms: (i) bottle or plastic bag cultivation, and (ii) wood log cultivation. An artificial environment and aseptic processing are necessary for the bottle or plastic bag cultivation method, but a natural forest environment is used for the wood log cultivation method. Although the bottle or plastic bag cultivation method provides a short fruiting period, the wood log cultivation method can produce tastier and more natural-tasting fruiting bodies at a lower cost compared to the bottle/plastic bag cultivation method [3]. Shortening the long fruiting period of the wood log cultivation method is desired, and the recent attention given to microbiomes may provide useful information for this purpose. However, there are no published studies of the wood log cultivation method related to mushroom microbiomes.
Generally, there are two steps involved in wood log cultivation method for G. frondosa: in the first step, mycelium grows around a wood log in a plastic bag, and the mycelium that has spread on the wood log is then transferred into casing substrates in a forest. Previous studies have shown that the microbiome of the casing substrate is an important factor for the cultivation of several types of mushroom. In Agaricus mushrooms, the addition of a casing substrate is applied to achieve high productivity and induce the transition from vegetative to reproductive growth [4]. Casing microbiomes can also suppress the development of diseases during cultivation [5, 6]. A new method for cultivating morels (Morchella spp.) in non-axenic soils under greenhouse environments was developed in China [7]. With further research, soil bacterial communities are considered an important factor for primordia differentiation and ascocarp growth [8].
The two above-described mushroom cultivation methods are based on a compost or sawdust substrate for growing mycelium. We speculated that not only the casing microbiome but also the surrounding bacterial communities might also be important factors for the fungal growth and life cycle. Positive effects of these factors on the co-culturing of fungi with bacteria have been reported; for example, coexisting bacteria promote mycelium growth [9] and the basidiospore germination rate [10]. Even some truffle aromas, which are thiophene volatiles, might be synthesized by surrounding bacteria [11, 12]. These studies suggested that bacterial communities may play a key role in the fungal life cycle, and we thus suspected that (i) the bacteria communities in wood logs during maitake cultivation or in the casing substrate may potentially have some effects that are beneficial to maitake, and (ii) the determination of the composition of the surrounding microbiome may provide a new and effective method for the development of an artificial surrounding bacterial community that can be used to improve the production of maitake mushrooms.
We thus conducted the present study to understand the dynamics of the bacterial microbiome and to elucidate the interaction between the bacterial community and maitake in wood logs as the first step for improving maitake mushroom production. To achieve this, we investigated: (1) the bacterial community, diversity, and functionality during mushroom cultivation; (2) the co-occurrence networks of microbial communities under different cultivation stages; and (3) the coexisting keystone taxa in wood logs during maitake cultivation.
Materials and methods
Cultivation and sampling
Logs of the deciduous tree Quercus serrata (konara), which are typically used in maitake cultivation, were used as the test logs in this study. The test wood logs had a diameter of 10–15 cm and a length of 12–16 cm. Before being inoculated with G. frondosa mycelium, the wood logs were soaked in water for 24 h and then sterilized by autoclaving. The inoculation was performed in January (in 2019, 2020, 2021 and 2022), and maitake wood logs were incubated for 5–6 months in plastic bags. In July, the maitake wood logs were buried in sand in a forest (Misato, Miyazaki). The production of maitake mushrooms in the wood logs was observed from September to October.
Every year in November, fruiting bodies were harvested. In November 2022, after fruiting bodies harvesting, we collected the following as wood log samples for this study: control wood logs (buried in sand from July to November without inoculation), first-year wood logs (at 1 year of cultivation), second-year wood logs (at 2 years' cultivation), third-year wood logs (3 years' cultivation), and fourth-year wood logs (4 years' cultivation). Four logs were examined for each group's sample. We thus used 20 samples (5 wood log sample types × 4 replicates). We also collected 20 sand casings surrounding each wood log in November 2022 as sand samples for this study (5 sand sample types × 4 replicates).
Wood log and casing sand DNA extraction, and PCR amplification and sequencing
DNA from the 20 wood logs and the 20 casing sand samples was extracted using the NucleoSpin® soil kit (Macherey–Nagel, Düren, Germany) in accord with the manufacturer's instructions. The NanoDrop quantification method was used to measure the DNA concentrations.
To expand our understanding of the biomass of bacteria and maitake, we conducted a real-time PCR, which revealed 16S rRNA and cytC genes. Thermal cycling and fluorescence detection were conducted using the CFX connect real-time polymerase chain reaction (RT-PCR) system (Bio-Rad, Hercules, CA, USA) for determine the number of copies of rRNA gene (16S rRNA gene) from bacteria (968R: 5’-AACGCGAAGAACCTTAC-3’; 1401F: 5’-CGGTGTGTACAAGACCC-3’) [13] and cytochrome C gene (cytC) [14] from G. frondosa (CYTc-F1: 5’-GGTGCCGGTCTTTTCAAGACTC-3’; CYTc-R1: 5’-CTGCATGGTATCGTGTTGTCCT-3’) in total DNA. DNA was diluted to 0.5 ng/μL, and 1-μL samples were used for the real-time PCR analysis. The real-time PCR reactions were performed in a final volume of 10 μL with a final concentration of 0.4 μM for each primer set, using TB Green Premix Ex Taq II (Takara Bio, Otsu, Japan) according to the manufacturer's instructions.
The PCR amplification was performed as follows: initial denaturation at 95 °C for 1 min, followed by 40 cycles of denaturation at 95 °C for 10 s, and annealing 30 s (56 °C for 16S rRNA gene amplification; 60 °C for cytC gene amplification), and extension at 72 °C (30 s for 16S rRNA gene amplification; 60 s for cytC gene amplification). At the end of the real-time PCR, a melting curve analysis was performed as a final step that consists of the measurement of fluorescence signal intensities from 65 °C to 95 °C.
Due to maitake biomass was detected only in the wood logs, only twenty wood log samples were used for sequencing (for details, see the discussion section). The PCR amplification of the 16S rRNA gene was performed using the V3V4 sequencing protocol (Part #15044223 Rev. B). TaKaRa Ex Taq HS (Takara Bio) in Ex Taq buffer was used for the PCR with the following program: 2 min at 94 °C, followed by 25 cycles of 30 s at 94 °C, 30 s at 55 °C and 30 s at 72 °C, and a final extension of 5 min at 72 °C. A high-throughput sequencing of PCR amplicons was performed on an Illumina MiSeq sequencer (Illumina, San Diego, CA) using the MiSeq Reagent Kit v3 in accord with the manufacturer's protocol. However, there was a problem in the PCR amplification in one fourth-year sample; for the subsequent analysis we thus used only three samples as the fourth-year samples. The raw reads were submitted to the DDBJ Sequence Read Archive under the accession number DRA016788. A QIIME2 (ver. 2021.11) (https://qiime2.org/) plugged DADA2 (dada2 denoise-paired) pipeline was used to filter chimeric sequences and merge the filtered forward and reverse sequences.
The taxonomic assignment, diversity indices, and functional profile annotation of the bacterial communities
The taxonomic assignment was performed for the amplicon sequence variants (ASVs) using the qiime2-feature-classifier [15] by the SILVA bacterial 16S database (ver. 138). A database of representative sequences from operational taxonomic units was generated via the clustering of SILVA sequences at 99% identity. Species-level feature tables were created by QIIME2.
The similarity of the microbial communities in each sample was evaluated by a principal-coordinate analysis (PCoA). Sample variation at the species level was represented by an ordination using an Aitchison distance matrix in the R package vegan [16]. The R package ggplot2 [17] was used for visualization. We examined the differences among the communities of all groups by conducting a permutational multivariate analysis of variances (PERMANOVA) on the Aitchison distance using the adonis2 function in vegan with 9,999 permutations. To determine whether there were differences in the variance (dispersion) between groups, we used the multivariate homogeneity of group dispersions test (betadisper) in vegan.
Alpha diversities were assessed on the ASV read counts. ASV read counts were rarefied to a sequence depth of 22,991 reads per sample in Qiime2. Alpha diversity indices, including Chao1, Simpson evenness, Faith's phylogenetic diversity (Faith's PD), Simpson, and Shannon indices were calculated using Qiime2. The Kruskal–Wallis test was used for analyzing significant difference among groups.
For the functional profiling of bacterial communities, PICRUSt2 [18] and FAPROTAX [19] were used separately. In PICRUSt2, representative sequences that were outputted by Qiime2 were used to predict the metagenomic functions of the bacterial communities via marker gene profiles. In FAPROTAX, to obtain the best optimized results, species-level feature tables were produced by the SILVA 132 database. Based on the existing literature regarding cultured representatives, metabolic or ecologically relevant functions were predicted for the feature tables' members. A statistical analysis of taxonomic and functional profiles (STAMP) [20] was used for the visualization and the statistical analysis.
Co-occurrence networks of bacterial communities
To understand the interactions of microbiomes, we calculated the correlation between the relative abundance of bacteria at the species level by using the Spearman's rank correlation coefficient test in the R package psych [21]. For further analysis, co-occurrence networks were constructed using the R packages psych, Hmisc, and igraph [22] based on the Spearman's correlation matrixes. The Benjamini–Hochberg false discovery rate was used to adjust the p-values in the correlation [23]. In this analysis, 15 samples were separated to two stages (early stage, four first-year samples and four second-year samples; late stage, four third-year samples and three fourth-year samples). Only species that occurred with ≥ 0.05% relative abundance in all samples were included in the analysis. Centered log-ratio (clr) translation was used for the species abundance treatment, and the co-occurrence networks for bacteria were constructed based on the Spearman's correlation using only significant correlations (adjusted p < 0.05) of r > 0.6 or r < − 0.6 in the R packages NetCoMi [24] and psych.
The Louvain method was used for a modularity analysis; the eight highest proportions of modularity classes were colored in the plot, and the results of the correlation matrix were visualized using Gephi software (https://gephi.org) [25]. The degree and betweenness centrality were used to statistically identify microbial keystone species. We selected species with a degree above the 80th percentile and betweenness centrality below the 20th percentile as potential keystone taxa.
The network comparison and differential network analysis were performed with the NetCoMi package. Only species with the 100 highest variances were selected for the network construction. The figure of network comparison required to subset to species, which presented in both bacterial communities (early stage and late stage) under investigation. We used Spearman's rank correlation coefficient test to calculate the correlation relationships between the relative abundance of bacteria at the species level. Hubs of the network comparison were identified as nodes above the 70% quantile of the distribution of the two networks' metrics degree and eigenvector centrality. The nodes of the differential network were determined using the Fisher test, requiring p < 0.05. Values were adjusted to multiple testing using the Benjamini–Hochberg false discovery rate for multiple testing.
Results
The biomass, composition, and diversity of the microbial communities in the wood logs and casing sand samples
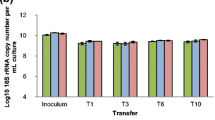
To prove whether maitake existed and know how many bacteria existed in wood logs, real-time quantitative PCR was used in the first step of our study. As shown in Fig. 1A, the real-time PCR data demonstrated that cytC gene from maitake was not detected in any of the casing sand samples or wood log control samples. In contrast to cytC gene, 16S rRNA gene was detected in all samples (Fig. 1B). The 16S rRNA gene copy numbers in the casing sand used for maitake cultivation (1 to 4 years of cultivation) were significantly higher than those in the casing sand used for the control wood logs. In the wood logs used for maitake cultivation, the 16S rRNA gene copy numbers showed an upward trend with the increase in the cultivation time (Fig. 1B). In contrast, the ratio of cytC gene to 16S rRNA gene exhibited a downward trend with the increase in cultivation time (Fig. 1C).

Copy numbers of the 16S rRNA gene and cytC gene. Copy numbers were detected in genomic DNA isolated from the wood logs and casing sand used for maitake cultivation for different durations (years) as determined by quantitative (q)PCR). A Log copy number of cytC gene per ng DNA in wood log and casing sand used for maitake cultivation during different years. B Log copy number of 16S rRNA gene per ng DNA in the wood logs and casing sand used for maitake cultivation during different durations. C Log ratio of cytC gene to 16S rRNA gene in the wood logs and casing sand used for maitake cultivation of different durations. Error bars: standard error of the mean. Means with the same letters were not significantly different at p < 0.05 among the different samples
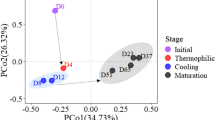
To understand bacterial community structures, principal-coordinate analysis (PCoA) and relative abundance analysis were conducted. The principal-coordinate analysis ordination plots and PERMANOVA analysis results demonstrated that the bacterial communities were significantly different among the five groups (R2 = 0.28, p < 0.001) and was not due to sample group dispersion (p = 0.691) (Fig. 2, Table 1). Regarding alpha diversity, we observed a significant difference in Simpson evenness; however, the data revealed no significant difference in Chao1, Faith's pd, Shannon, or Simpson diversities (Table 2). The bacterial composition results revealed that Proteobacteria had the highest relative abundance in all five groups (Fig. 3A).

The principal-coordinate analysis (PCoA) ordination plots of the bacterial communities in wood logs during maitake cultivation

The bacterial composition at the different years of G. frondosa cultivated on wood logs. A The relative abundance of the dominant bacterial phyla. B The relative abundance of secondary dominant bacterial phyla
Significant differences in relative abundance among all groups were revealed in three phyla: Proteobacteria, Firmicutes and Gemmatimonadota. Compared to the control group, the third- and fourth-year groups showed significantly lower relative Proteobacteria abundance (Fig. 3A). The control group had significantly higher relative Firmicutes abundance (Fig. 3B). The second-year group showed significantly higher relative Gemmatimonadota abundance than the control, first-year and fourth-year groups (Fig. 3B).
Functional profiles of the bacterial communities in the wood logs during maitake cultivation
To understand bacterial functional profile, FAPROTAX and PICRUSt2 were used. The PCA plot of functional prediction retrieved from the bacterial communities showed a similar pattern in the FAPROTAX and PICRUSt2 results. The control group was clearly separated from the other groups (Fig. 4). In detail, based on the FAPROTAX database, the relative abundance of bacterial functions, ligninolysis, and xylanolysis regarding the wood degradation and chitinolysis regarding fungal cell-wall degradation was not significantly different among the five groups (Fig. 5). In contrast, the relative abundance of cellulolysis regarding wood degradation, fermentation, and intracellular parasites was significantly different among all groups. The control group showed the highest relative abundance of fermentation but the lowest cellulolysis and intracellular parasites functions. In the PICRUSt2 results, significant differences among all groups were identified in 73 pathways (50 biosynthesis, 14 degradation, and nine other pathways) (Fig. 6). Aerobic respiration I (cytochrome c) in particular showed the highest relative abundance in 73 pathways, and that of control group was the lowest relative abundance among all groups (Fig. 6C).

The principal components analysis (PCA) plot of functional prediction. A The plot of FAPROTAX functional prediction. B The plot of PICRUSt2 functional prediction

The relative abundance of bacterial functions in the FAPROTAX database. Different colors indicate the relative abundance of functions in the individual samples. The samples are grouped according to their similarity to each other. Brown: the function with higher relative abundance in the corresponding sample. White: the function with lower relative abundance in the corresponding sample. P-values are from the ANOVA. *p < 0.10, **p < 0.05, ***p < 0.01

The heatmap and boxplot of significantly different functions inferred by PICRUSt2. A The plot of higher relative abundance functions. B The plot of lower relative abundance functions. C The plot of highest relative abundance functions. Red colors represent higher abundance, and blue colors represent lower abundance. Significant differences (adjusted p < 0.05) were detected among different wood log samples
Co-occurrence networks and differential networks
Generally, maitake wood log cultivation was conducted for four years. We identified first half of the cultivation time as early-stage (first- and second-year) and the last half of the cultivation time as late-stage (third- and fourth-year). To increase our understanding of interactions within bacterial communities, we constructed co-occurrence networks using early-stage and late-stage samples. The network of bacterial communities demonstrated distinct co-occurrence patterns with the different cultivation stages. The early stage showed 0.853 modularity, and the late stage showed 0.913 modularity (data not shown). The results revealed that the number of edges of the early-stage network was twice that of the late-stage network based on a similar number of nodes (Fig. 7). The network showed a high number of edges, which means that the interaction between bacteria in a community is high.

The bacterial co-occurrence networks at the two stages of G. frondosa cultivated wood logs. Nodes represent individual species; edges represent significant positive or negative Spearman's correlations (r > 0.6 or < − 0.6 and adjusted p < 0.05). Different colors, including purple, green, blue, black, orange, red, dark green, and tan, represent the eight largest modules. The early stage includes the first-year and second-year samples. The late stage includes the third-year and fourth-year samples
Twenty-two potential keystone taxa (Suppl. Table S1) were detected in the early-stage network, and 38 potential keystone taxa (Suppl. Table S2) were observed in the late-stage network. At the early stage, Gammaproteobacteria and Acidimicrobiia showed the highest proportion at > 20% each, followed by Alphaproteobacteria (13.64%) (Table 3). At the late stage, Gammaproteobacteria also showed the highest proportion at > 20%, followed by Thermoleophilia and Alphaproteobacteria.
To understand how the early stage and late stage differ, we constructed a network comparison plot and a differential network (Fig. 8, Table 4, Suppl. Table S3). Figure 8A summarizes the hub bacteria that are implicated in the reorganization or transition from the early to late stage. We selected 38 taxa to construct the plot for the comparison of the networks. Seven hub taxa were detected in the early stage, and five hub taxa were detected in the late stage. Only hub taxon S126 (which is an unclassified bacterial taxon from the Acidothermus genus, Actinobacteria class) was observed in both the early and late networks.

Comparison of the networks and differential networks. A The plot of the network comparison. Node sizes are according to degree. A red edge indicates a negative association, and a green edge indicates a positive association. Node colors represent clusters. B The plot of differential networks. Two nodes are connected if they are differentially associated between the two groups. Edge colors represent the direction of associations in the two groups. C Association networks generated form the early and late stages of the bacterial community in G. frondosa cultivated wood logs, including only the differentially associated species
The modularity of global network properties was significantly higher (adjusted p < 0.05) in the early-stage network (0.593) than in the late-stage network (0.392) (data not shown). A significant difference in the average path length (adjusted p < 0.05) was detected between the early- and late-stage networks (2.375 and 1.495, respectively). Based on the differences between the two networks, we selected 14 taxa for the construction of a differential network (Fig. 8B). Sorted in terms of the proportion of the selected taxa numbers at the class level, the selected microorganisms were Alphaproteobacteria (42.86%), Acidobacteria (28.57%), Gammaproteobacteria (14.29%), Actinobacteria (7.14%), and Vicinamibacteria (7.14%) (Table 4).
Discussion
A study of morel cultivation showed that non-sterilized soils were highly colonized by Morchella mycelium [7]. The morel mycelium appears to potentially coexist with the bacterial microbiome, leading to primordia differentiation and ascocarp growth [7, 8]. Those findings suggested that the microbiome is an important factor in morel cultivation. For the production of maitake mushrooms, wood log cultivation is carried out in natural forest conditions in non-sterilized sand. In the present study, we detected the bacterial biomass in the wood logs and the surrounding casing sand; however, maitake biomass was detected only in the wood logs. Based on the morel cultivation experience and our results, we decided to first investigate the microbiome in the wood logs.
The first-year to fourth-year wood logs showed significant differences in the pattern of bacterial communities compared to the control group; however, when wood logs were colonized by maitake, a conservative pattern was observed during the cultivation, and no significant differences were detected among the wood logs of the different groups. Moreover, a significant difference was observed only in the diversity of Simpson evenness. Although no difference was detected in the pattern of bacterial communities, differences in bacterial compositions including Proteobacteria, Firmicutes, and Gemmatimonadota were observed among the wood log samples. Proteobacteria was dominant in the bacterial communities of the wood logs. The dynamic trend of the relative abundance of Proteobacteria detected in the wood logs was similar to that observed in the Agaricus bisporus compost process (phase III), with a dominant Proteobacteria population in the beginning that decreased by the end of the cultivation in compost [5].
In another edible mushroom, Actinobacteria, Chloroflexi, and Proteobacteria were found at high relative abundance from the soils used in morel cultivation [7]. Proteobacteria, Acidobacteria, and Actinobacteria were dominant in the soils from the forestland where the mushroom Stropharia rugosoannulata [26] was cultivated. In the cultivation of the mushroom Pleurotus eryngii, Proteobacteria remained in highest relative abundance from the growth of the P. eryngii hyphae to the filling of the cultivation bags [27]. Actinobacteria, Proteobacteria, and Firmicutes are also considered important phyla in forest decayed wood [28, 29]. Based on these results, it is apparent that the relative abundances of Proteobacteria, Firmicutes, Gemmatimonadota play key roles in the microbiome composition in wood logs during the cultivation of maitake.
The present study's functional profiles showed that the bacterial communities from the first-year to fourth-year groups presented higher relative abundance in cellulolysis, glycolysis III (from glucose), aerobic respiration I (cytochrome c), and the TCA cycle, but lower relative abundance in fermentation compared to the control group. Cellulose can be converted to glucose by many bacterial strains in both aerobic and anaerobic conditions [30], and the glucose is then converted to pyruvate by glycolysis. Under anaerobic conditions, NADH2 is usually used as a reductant for the production of ethanol from pyruvate. In contrast, under aerobic conditions, pyruvate is converted to acetyl-CoA and enters the TCA cycle (aerobic respiration) [31]. The erosion and penetration of hyphae were observed in fungal decaying wood [32], and the wood components cellulose, hemicellulose, and lignin were degraded by white rot fungi [33]. Based on these results, with the progression of wood logs' degradation by fungi, which makes aerobic conditions, the coexisting bacteria may help maitake degrade wood cellulose and prefer entrance into the TCA cycle instead of a fermentation pathway with the degradation of glucose. It is possible that the metabolism of glucose into the TCA cycle by coexisting bacteria contributes to a suppression of the decrease in cellulolytic activity (negative feedback) by consuming the glucose that has accumulated in decaying wood under aerobic conditions.
Our analyses also revealed that 50 biosynthesis pathways showed significant differences among the wood logs of the different groups, and most of these pathways showed higher relative abundance in the maitake cultivated wood logs compared to the control group. It has been reported that the fungus Aspergillus nidulans and the bacterium Bacillus subtilis establish a mutualistic growth mechanism that involves bacterial thiamine [34], and that the bacterium Klebsiella aerogenes produced dopamine as a possible substrate that was used by the fungus Cryptococcus neoformans [35]. These findings suggested that interactions between bacteria and fungi include an exchange of chemicals. Our present findings indicate that some chemicals produced during biosynthesis may contribute to the growth of the maitake mycelium, but further studies are necessary before any conclusions can be made.
We also observed a high relative abundance of intracellular parasites in the first-year to fourth-year groups. Endobacteria (bacterial endosymbionts) that dwell inside fungal cells were recently described. Mycoplasma-related endobacteria (MRE) (class Betaproteobacteria) and Burkholderia-related endobacteria (BRE) (class Mollicutes) were detected in the division Glomeromycotina of the arbuscular mycorrhizal fungi Mortierellomycotina and Mucoromycota [36, 37]. The endobacteria of fungi exhibit mutualism or antagonism behavior with a fungal host. For example, the sexual reproduction of Mortierella sp. was inhibited by endobacteria [38]. However, there is no report or endobacteria being detected in mushrooms. The intracellular parasites identified in the present study belonged to the classes Chlamydiae, Alphaproteobacteria, and Gammaproteobacteria. Based on these results, it appears that there are some potential intracellular parasites dwelling in maitake; however, these intracellular parasites are obligate or facultative endosymbiotic bacteria or free-living bacteria, and further investigation is necessary.
Our network results showed that the early stage and late stage of maitake cultivation involve different interactions among bacterial communities, which suggests that the two cultivation stages present different microbiome network stability. Potential keystone taxa were also detected in both stages, and class Gammaproteobacteria showed the highest proportion of keystone taxa. Other research has indicated that keystone taxa have important roles in maintaining the network structure relative to the other taxa in the network [39,40,41]. Keystone taxa may therefore play a role in maintaining ecosystem stability. Alphaproteobacteria and Gammaproteobacteria were considered potential keystone taxa in the rhizosphere [40], and Gammaproteobacteria play an important role in several ecosystems as the keystone taxa of the bacterial community [42]. Pseudomonas (one genus of class Gammaproteobacteria) interacts with morels to induce primordium formation [43], and it promotes mycelium growth in A. bisporus [44] and Pleurotus eryngii [45]. Gammaproteobacteria may thus be a keystone or biomarker in the cultivation of many species of mushroom. Our analysis of bacterial differential networks also identified Gammaproteobacteria. Together these results suggest that Gammaproteobacteria is an important potential keystone taxon for microbiome network stability in maitake cultivation.
A limitation of our study is that it was conducted only in wood log samples. Further studies are necessary to reveal the interaction between maitake and bacterial communities. As a next step in our research, we will try to isolate coexisting bacteria and examine their interaction with maitake in vitro and investigate the presence of intracellular parasites and chemical exchanges. We will then attempt to determine the precise bacterial and fungal communities in casing sand.
Conclusions
Our results demonstrated the presence of maitake biomass in wood logs but not in casing sand, and we thus investigated the bacterial communities in wood log samples during maitake cultivation. Differences in microbial diversity, bacterial community, and the network were observed among the control samples and the different years of maitake cultivated wood logs. The relative abundance of Proteobacteria, Firmicutes, and Gemmatimonadota play key roles in the microbiome composition. Bacterial communities and maitake may create a mutualistic mechanism. During the degradation of wood logs, the bacterial communities may help maitake degrade wood cellulose, and part of the glucose from the cellulose degraded by both maitake and bacteria may be used for the bacterial TCA cycle. Bacteria also produce some chemicals that maitake mycelium may need. Some potential intracellular parasites probably dwell within maitake. Regarding the microbiome network, the different cultivation stages showed different network structures, and Gammaproteobacteria was an important potential keystone taxon for the microbiome network stability of maitake cultivated on wood logs. These insights contribute to our understanding of the microbiome in wood logs during maitake cultivation and will help improve the wood log cultivation of maitake mushrooms.
Availability of data and materials
All the raw sequence data of the bacterial 16S rDNA genes were submitted in the Sequence Read Archive of DDBJ database under the accession number DRA016788.
Abbreviations
- RT-PCR:
-
Real-time polymerase chain reaction
- ASVs:
-
Amplicon sequence variants
- PCoA:
-
Principal-coordinate analysis
- PERMANOVA:
-
Permutational multivariate analysis of variances
- STAMP:
-
Statistical analysis of taxonomic and functional profiles
- Clr:
-
Centered log-ratio
- MRE:
-
Mycoplasma-related endobacteria
- BRE:
-
Burkholderia-related endobacteria
References
Montoya S, Orrego CE, Levin L (2012) Growth, fruiting and lignocellulolytic enzyme production by the edible mushroom Grifola frondosa (maitake). World J Microbiol Biotechnol 28:1533–1541
Kawaguchi N, Hayashi M, Chen FC, Shimomura N, Yamaguchi T, Aimi T (2019) Genetic analyses of causal genes of albinism (white fruiting body) in Grifola frondosa. J Wood Sci 65:32
Tabata T, Yamasaki Y, Ogura T (2004) Comparison of chemical compositions of Maitake (Grifola frondosa (Fr.) Gray) cultivated on logs and sawdust substrate. Food Sci Technol Res 10:21–24
Dias ES, Zied DC, Pardo-Giménez A (2021) Revisiting the casing layer: casing materials and management In Agaricus Mushroom cultivation. Ciênc Agrotec 45:e0001R21
Carrasco J, Tello ML, de Toro M, Tkacz A, Poole P, Pérez-Clavijo M, Preston G (2019) Casing microbiome dynamics during button mushroom cultivation: implications for dry and wet bubble diseases. Microbiol 165:611–624
Taparia T, Hendriks E, Nijhuis E, Hendriks M, de Boer W, van der Wolf J (2021) Casing soil microbiome mediates suppression of bacterial blotch of mushrooms during consecutive cultivation cycles. Soil Biol Biochem 155:108161
Benucci GMN, Longley R, Zhang P, Zhao Q, Bonito G, Yu F (2019) Microbial communities associated with the black morel Morchella sextelata cultivated in greenhouses. PeerJ 7:e7744
Liu Q, Liu H, Chen C, Wang J, Han Y, Long Z (2017) Effects of element complexes containing Fe, Zn and Mn on artificial morel’s biological characteristics and soil bacterial community structures. PLoS ONE 12:e0174618
Kamei I, Yoshida T, Enami D, Meguro S (2012) Coexisting Curtobacterium bacterium promotes growth of white-rot fungus Stereum sp. Curr Microbiol 64:173–178
Mizutani K (2013) Stimulation of basidiospore germination of the ectomycorrhizal fungus Tricholoma matsutake by bacteria of the genus Pseudomonas. Jpn J Mycol 54:60e65 (In Japanese)
Antony-Babu S, Deveau A, Van Nostrand JD, Zhou J, Le Tacon F, Robin C, Frey-Klett P, Uroz S (2014) Black truffle-associated bacterial communities during the development and maturation of Tuber melanosporum ascocarps and putative functional roles. Environ Microbiol 16:2831–2847
Splivallo R, Deveau A, Valdez N, Kirchhoff N, Frey-Klett P, Karlovsky P (2015) Bacteria associated with truffle-fruiting bodies contribute to truffle aroma. Environ Microbiol 17:2647–2660
Cébron A, Norini MP, Beguiristain T, Leyval C (2008) Real-Time PCR quantification of PAH-ring hydroxylating dioxygenase (PAH-RHDalpha) genes from Gram positive and Gram negative bacteria in soil and sediment samples. J Microbiol Methods 73:148–159
Numata F, Kawaguchi N, Yamada C, Ota Y, Chen FC, Hayashi M, Shimomura N, Yamaguchi T, Aimi T (2019) Mitochondrial DNA dynamics during fruiting body formation in Grifola frondosa. Mycoscience 60:147–150
Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, Huttley GA, Gregory Caporaso J (2018) Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6:90
Oksanen J, Simpson GL, Blanchet FG, Kindt R, Legendre P, Minchin PR, O'Hara RB, Solymos P, Stevens MHH, Szoecs E, et al (2022) vegan: community ecology package. R package version 2.6–2 https://CRAN.R-project.org/package=vegan
Wickham H (2016) ggplot2: elegant graphics for data analysis. https://ggplot2.tidyverse.org
Douglas GM, Maffei VJ, Zaneveld JR, Yurgel SN, Brown JR, Taylor CM, Huttenhower C, Langille M (2020) PICRUSt2 for prediction of metagenome functions. Nat Biotechnol 38:685–688
Louca S, Parfrey LW, Doebeli M (2016) Decoupling function and taxonomy in the global ocean microbiome. Science 353:1272–1277
Parks DH, Tyson GW, Hugenholtz P, Beiko RG (2014) STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30:3123–3124
Revelle WR (2017) psych: Procedures for personality and psychological research. R package version 2.1.9. https://CRAN.R-project.org/package=psych
Csardi G, Nepusz T (2006) The igraph software package for complex network research. InterJ Complex Syst 1695:1–9
Benjamini Y, Krieger AM, Yekutieli D (2006) Adaptive linear step-up procedures that control the false discovery rate. Biometrika 93:491–507
Peschel S, Müller CL, von Mutius E, Boulesteix AL, Depner M (2021) NetCoMi: network construction and comparison for microbiome data in R. Brief Bioinform 22:bbaa290
Bastian M, Heymann S, Jacomy M (2009) Gephi: an open source software for exploring and manipulating networks. Proc Int AAAI Conf Web Social Media 3:361–362
Gong S, Chen C, Zhu J, Qi G, Jiang S (2018) Effects of wine-cap Stropharia cultivation on soil nutrients and bacterial communities in forestlands of northern China. PeerJ 6:e5741
Chen L, Yan M, Qian X, Yang Z, Xu Y, Wang T, Cao J, Sun S (2022) Bacterial community composition in the growth process of Pleurotus eryngii and growth-promoting abilities of isolated bacteria. Front Microbiol 13:787628
Velasco-Rodríguez Ó, Fil M, Heggeset TMB, Degnes KF, Becerro-Recio D, Kolsaková K, Haugen T, Jønsson M, Toral-Martínez M, García-Estrada C, Sola-Landa A, Josefsen KD, Sletta H, Barreiro C (2022) Characterization of microbial diversity in decayed wood from a Spanish forest: an environmental source of industrially relevant microorganisms. Microorganisms 10:1249
Kuramae EE, Leite MFA, Suleiman AKA, Gough CM, Castillo BT, Faller L, Franklin RB, Syring J (2019) Wood decay characteristics and interspecific interactions control bacterial community succession in Populus grandidentata (Bigtooth Aspen). Front Microbiol 10:979
Gupta P, Samant K, Sahu A (2012) Isolation of cellulose-degrading bacteria and determination of their cellulolytic potential. Int J Microbiol 2012:578925
Chen X, Sun Y, Xiu Z, Li X, Zhang D (2006) Stoichiometric analysis of biological hydrogen production by fermentative bacteria. Int J Hydrogen Energy 31:539–549
Rudakiya DM, Gupte A (2017) Degradation of hardwoods by treatment of white rot fungi and its pyrolysis kinetics studies. Int Biodeterior Biodegrad 120:21–35
Kuuskeri J, Häkkinen M, Laine P, Smolander OP, Tamene F, Miettinen S, Nousiainen P, Kemell M, Auvinen P, Lundell T (2016) Time-scale dynamics of proteome and transcriptome of the white-rot fungus Phlebia radiata: growth on spruce wood and decay effect on lignocellulose. Biotechnol Biofuels 9:192
Abeysinghe G, Kuchira M, Kudo G, Masuo S, Ninomiya A, Takahashi K, Utada AS, Hagiwara D, Nomura N, Takaya N, Obana N, Takeshita N (2020) Fungal mycelia and bacterial thiamine establish a mutualistic growth mechanism. Life Sci Alliance 3:e202000878
Peleg AY, Hogan DA, Mylonakis E (2010) Medically important bacterial-fungal interactions. Nat Rev Microbiol 8:340–349
Bonfante P, Desirò A (2017) Who lives in a fungus? The diversity, origins and functions of fungal endobacteria living in Mucoromycota. ISME J 11:1727–1735
Naumann M, Schüssler A, Bonfante P (2010) The obligate endobacteria of arbuscular mycorrhizal fungi are ancient heritable components related to the Mollicutes. ISME J 4:862–871
Takashima Y, Degawa Y, Nishizawa T, Ohta H, Narisawa K (2020) Aposymbiosis of a Burkholderiaceae-Related endobacterium impacts on sexual reproduction of its fungal host. Microb Environ 35:ME19167
Olesen JM, Bascompte J, Dupont YL, Jordano P (2007) The modularity of pollination networks. Proc Natl Acad Sci USA 104:19891–19896
Shi S, Nuccio EE, Shi ZJ, He Z, Zhou J, Firestone MK (2016) The interconnected rhizosphere: high network complexity dominates rhizosphere assemblages. Ecol Lett 19:926–936
Faust K, Raes J (2012) Microbial interactions: from networks to models. Nat Rev Microbiol 10:538–550
Liao H, Lin X, Li Y, Qu M, Tian Y (2020) Reclassification of the taxonomic framework of orders Cellvibrionales, Oceanospirillales, Pseudomonadales, and Alteromonadales in Class Gammaproteobacteria through phylogenomic tree analysis. MSystems 5:e00543-e620
Zhang C, Shi X, Zhang J, Zhang Y, Wang W (2023) Dynamics of soil microbiome throughout the cultivation life cycle of morel (Morchella sextelata). Front Microbiol 14:979835
Chen S, Qiu C, Huang T, Zhou W, Qi Y, Gao Y, Shen J, Qiu L (2013) Effect of 1-aminocyclopropane-1-carboxylic acid deaminase producing bacteria on the hyphal growth and primordium initiation of Agaricus bisporus. Fungal Ecol 6:110–118
Kim MK, Math RK, Cho KM, Shin KJ, Kim JO, Ryu JS, Lee YH, Yun HD (2008) Effect of Pseudomonas sp. P7014 on the growth of edible mushroom Pleurotus eryngii in bottle culture for commercial production. Biores Technol 8:3306–3308
Acknowledgements
The authors would like to thank Mr. Takesi Imanisi, Dr. Taku Tsuyama and Dr. Takeshi Nitta for their contributions to the samples collection.
Funding
This work was supported by University of Miyazaki.
Author information
Authors and Affiliations
Contributions
FC designed this study and analyzed the study data and was a major contributor in writing the manuscript. MT, KI and KY contributed to the discussion of the obtained results. All of the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
The keystone taxa in early stage bacterial networks. Table S2. The keystone taxa in late stage bacterial networks. Table S3. The selected taxa in bacterial differential networks.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Chen, FC., Motoda, T., Kamei, I. et al. Characterization of microbial communities during Grifola frondosa (maitake) wood log cultivation. J Wood Sci 69, 39 (2023). https://doi.org/10.1186/s10086-023-02111-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10086-023-02111-3