
Abstract
Introduction
The receptor for advanced glycation end products (RAGE) has been considered as one of the major pattern recognition receptors and plays an important role in the development of sepsis and multiple organ dysfunction in critical illnesses. Although genetic variants of the RAGE gene have been shown to be well associated with susceptibility to some inflammatory diseases, little is known about their clinical relevance in the development of sepsis in critical ill patients.
Methods
Four genetic variants were selected from the entire RAGE gene and genotyped using pyrosequencing and polymerase chain reaction-length polymorphism methods. Association studies were performed in two independent Chinese Han populations.
Results
Among the four genetic variants, only the rs1800625 polymorphism was significantly associated with sepsis morbidity rate and multiple organ dysfunction (MOD) scores in patients with major trauma both in Chongqing (n = 496) and Zhejiang (n = 232) districts, respectively. Results from ex vivo responsiveness of peripheral blood leukocytes indicated that the rs1800625 polymorphism was well associated with decreased production of TNFα. In addition, the rs1800625 polymorphism could significantly inhibit the promoter activities of the RAGE gene.
Conclusions
The rs1800625 polymorphism is a functional variant, which might be used as a relevant risk estimate for the development of sepsis and multiple organ dysfunction syndrome in patients with major trauma.
Similar content being viewed by others

Introduction
The receptor for advanced glycation end products (RAGE), not only limiting to mediation of advanced glycation end products (AGEs), has been recognized as a multiligand receptor, especially playing pivotal roles in innate immune responses as a pattern-recognition receptor (PRR) in sensing both pathogen-associated molecular patterns (PAMPs) and endogenous damage-associated molecular patterns (DAMPs) [1–6]. RAGE could up-regulate the capacity of leukocytes to kill bacteria [2] and facilitates host defense during bacterial infection [3, 4]. Besides its anti-microbial effects, RAGE also acts as an endothelial adhesion receptor for leukocyte integrins and promotes leukocyte recruitment [5]. Engagement of RAGE by its diverse ligands, such as lipopolysaccharides, high-mobility group box 1 protein, AGEs, results in receptor-dependent signaling and amplification of pro-inflammatory responses [1]. RAGE is up-regulated at levels of both mRNA and protein in sepsis [6]. Administration of AGE-modified protein has the potential to activate RAGE/NF-kB-mediated inflammatory reactions, causing increased mortality in experimental peritonitis [7]. In addition, the genomic deletion of RAGE[8, 9], or inhibition of RAGE signaling [8, 10] results in markedly decreased septic response and significantly increased survival rates in a mouse model of sepsis. Therefore, RAGE has been considered to be one of the major PRRs responsible for the development of sepsis [11] and its subsequent organ dysfunction [12–14]. It might be used as a potential molecular marker and therapeutic target for sepsis [15–17].
The RAGE gene is located on chromosome 6p21.3 in the major histocompatibility complex (MHC) locus in the class III region [18]. Growing evidence indicates that allelic variation within the key domains of RAGE gene may influence the magnitude of proinflammatory response, thereby affecting susceptibility to acute and chronic inflammatory diseases [19]. A total of 97 genetic variants have been identified so far in the entire RAGE gene. However, only three of them (rs1800625, rs1800624 and rs2070600) have been well studied in relation to their clinical relevance. The rs1800625 is revealed in association with increased risk for diabetes [20], diabetic retinopathy [21] and colorectal cancer [22]. The rs1800624 polymorphism was shown to be associated with increased susceptibility to Crohn's disease [23], coronary artery disease [24], in-stent restenosis after coronary stent implantation [25], ischemic heart disease [26], and myocardial infarction [27]. The rs2070600 polymorphism revealed increased risk of rheumatoid arthritis [28], multiple sclerosis [29] and coronary artery disease in non-diabetics [30]. In spite of the above findings, there also have been plenty of controversial reports with respect to the clinical relevance of the above three gene polymorphisms [26, 27, 29, 31–36]. In addition, other polymorphisms, such as 2184A/G, 2245G/A, rs17493811 and rs9469089, also have been reported in relation to diabetes [37–39]. However, little is known about the clinical relevance of the genetic variants of RAGE gene in relation to the development of sepsis and multiple organ dysfunction syndrome (MODS).
To comprehensively assess the clinical relevance of the common genetic variants within the entire RAGE gene, haplotype bins were constructed to identify haplotype tagging single nucleotide polymorphisms (SNPs) (htSNPs) within the entire RAGE gene and its surrounding regions. In view of that, the majority of trauma patients are young people with little history of pre-existing diseases [40]. The trauma patient cohort is appropriate to investigate the association of genetic factors with susceptibility or resistance to sepsis in critically ill patients. Four genetic variants were selected, and two independent Chinese Han populations were recruited in this study cohort. The rs1800625 polymorphism (-429T/C) was demonstrated to be clinically relevant and have functional activity.
Materials and methods
Study population and clinical evaluation
A total of 728 unrelated patients with major trauma recruited in this study were Han Chinese and from Chongqing in south-western China (n = 496) and Zhejiang in eastern China (n = 232), respectively. The trauma patients were admitted to the Department of Trauma Surgery in the Daping Hospital and the Chongqing Emergency Medical Center between 1 January 2005 and 1 May 2011, and to the Department of Trauma and Emergency in the Second Affiliated Hospital, Zhejiang University between 1 January 2008 and 1 May 2011. They were enrolled in the study if they met the following inclusion criteria: 1) were between 18 and 65 years of age, 2) expected an Injury Severity Score (ISS) greater than 16 combined with the presence of at least one life threatening injury and at least one additional severe injury in another part of the body, and 3) had a probability of survival greater than 48 h. Patients were not eligible if they had penetrating injuries or preexisting cardiovascular, respiratory, renal, hepatic, hematologic or immunologic diseases. ISS was performed according to the abbreviated injury scale by independent evaluators [41]. Standard demographic, laboratory and clinical data were extracted from a prospectively collected database. The protocol was approved by the Ethical and Protocol Review Committee of the Third Military Medical University, and informed consent was obtained from the patients and the patients' next of kin. Patient confidentiality was preserved according to the guidelines for studies of human subjects.
The patients with major trauma were prospectively monitored after admission by physicians who did not know the genotypes. The definitions of sepsis and infection were shown in Additional file 1. Daily physiologic and laboratory data were collected during the ICU stay and clinical events were recorded thereafter, until death or hospital discharge. Multiple organ dysfunction scores were calculated as the sum of the simultaneously obtained individual organ scores on each hospital day [42]. Neurological scoring was not performed because every patient was sedated. MODS is defined as a Marshall score of 4 or more for at least two consecutive days based on the comparative studies reported by A Sauaia [43].
Tag SNP selection
The human RAGE gene (Accession Number: NC_000006) was pinpointed to chromosome 6, position 32148745-32152022 (data retrieved from Genbank in the website of NCBI). The RAGE 5' flanking region from -505 in a 5'direction has been shown to overlap with PBX2, a gene that has a pseudogene copy on chromosome 3, which might make any studies of polymorphisms in this duplicated region potentially fraught with error [44]. Therefore, besides all exons and introns of the RAGE gene, 505-bp upstream of the transcription start site and 5,000-bp downstream of the stop codon were included (8.872 kb total).
Genetic variation data for the entire RAGE gene and its surrounding selected regions were obtained from the healthy Chinese Han Beijing (CHB) population of HapMap. From this database, a total of 11 SNPs have been identified in the CHB population (Table 1). Ten of them were common SNPs with a minor allele frequency (MAF) more than or equal to 0.05, which were then selected for the analysis of htSNPs (Figure 1). Haplotype blocks (bins) were constructed using LDselect (Christopher S. Carlson and Deborah A. Nickerson, Seattle, Washington, USA), a software package that provides computation of linkage disequilibrium (LD) statistics and population haplotype patterns from genotype data [45]. This algorithm selects a subset of htSNPs that efficiently describe all common patterns of variants in a gene, based on two primary criteria: 1) the MAF is more than or equal to 0.05 and 2) the minimum level of association between assayed and unassayed SNPs, measured by the linkage disequilibrium statistic r2 is more than or equal to 0.8. Given these parameters, LDSelect selects a subset of htSNPs that captures all known common genetic variants within the entire gene.

Overview of selected tSNPs within the entire RAGE gene. (A) The pair-wise analysis of linkage disequilibrium (LD), based on r2, among the 10 SNPs with a minor allele frequency ³5% within the RAGE gene and 505 bp up- and 5,000 bp down-stream regions. The selected tSNPs are indicated by trigones. A LD-plot of the 10 SNPs is displayed by using r2-black and white color scheme. Black represents very high LD (r2 = 0.8-1), and white indicates the absence of correlation (r2 = 0-0.2) between SNPs. (B) The two tSNPs and SNPs that are indirectly measured by them are listed with corresponding r2-values. Major and minor alleles of the selected tSNPs are given with their frequencies, based on the HapMap data for the CHB population.
To determine the possible functionality of the htSNPs selected from the 5'-flanking region of the RAGE gene, on-line software was used to analyze the effect of these SNPs on potential transcription factor binding sites.
Genotyping
Blood specimens were collected in tripotassium ethylenediamine tetraacetic acid sterile tubes from trauma patients immediately after admission to avoid the effect of blood transfusion. The genomic DNA was isolated from whole blood using the Wizard genomic DNA purification kit (Promega, Madison, WI, USA) according to the manufacturer's protocol. Pyrosequencing was used for genotyping of rs1800625, rs1800624 and rs2070600 polymorphisms according to our previous reports [46]. Polymerase chain reaction (PCR) - length polymorphism (LP) approach was used for genotyping of the 63 bp ins/del variant. The PCR primers and the annealing temperature were shown in Additional file 2, Table S1. Genotyping was performed in a blinded fashion without knowledge of the patients' clinical data, and approximately 10% of the samples were genotyped in duplicate to monitor genotyping quality.
Ex vivo tumor necrosis factorα production
A human whole-blood assay was used as in our previous method. In brief, aliquots of whole blood collected from the trauma patients immediately after admission were mixed 1:1 with Roswell Park Memorial Institute (RPMI) 1640 culture medium (Thermo Scientific, Beijing, China), and incubated with 100 ng/ml LPS (Escherichia coli O26:B6, Difco Laboratories, Detroit, MI, USA) in a sample mixer at 37°C for 4 h. After centrifugation, the supernatants were aspirated and aliquoted for stoRAGE at -80°C. Tumor necrosis factor-alpha (TNF-α) in the supernatants was assayed with a sandwich enzyme linked immunosorbent assay (ELISA), according to the manufacturer's instructions (Endogen, Woburn, MA, USA). The detection limits of the assay were 4 pg/ml.
Functionality of the rs1800625 polymorphism
The possible effect of rs1800625, which is located in the 5'-flanking region (-429T/C), on the promoter activity was investigated using a reporter gene assay system [47]. A 531-bp sequence (-504 approximately +26) of the RAGE gene containing T or C allele at position -429 were obtained according to the method described in Additional file 3 and were directly inserted into a promoterless pGL3-Basic vector (Promega, Madison, WI, USA) containing the firefly luciferase gene as a reporter. Human U937 were cultured in RPMI 1640 medium (Hyclone, Logan, UT, USA) containing 10% fetal calf serum, 3 mM glutamine, penicillin-streptomycin (100 U/mL for each), and 23 mM sodium bicarbonate at 37°C in a humidified 5% CO2 air atmosphere. After incubation for 24 hours, the cultured cells were co-transfected with 0.8 μg of the constructed vectors or pGL3-Basic original plasmid and 20 ng control Renilla luciferase reporter plasmid pRL-CMV using Lipofectmine 2000 system (Invitrogen, Carlsbad, CA, USA). At 24 hours post-transfection, the cells were treated with LPS (100 ng/mL) for 24 hours. Then luciferase activity of the transfected cells was measured using the Luciferase Assay System (Promega) following the supplier's protocol on a Luminoskan Ascent luminometer (Thermo Labsystems, Helsinki, Finland). Transfection efficiency was normalized by measuring the luciferase activity of control plasmid pRL-CMV. Luminescence experiments were performed in triplicate with each transfection. Three independent transfections were performed for each constructed vector. Results are expressed as fold increase in relative luciferase activity of the RAGE promoter construct vectors compared with the relative luciferase activity of pGL3-Basic.
Statistical analysis
Sample size was calculated using online Power and Sample Size Program software (William D Dupont and Walton D Plummer, Nashville, Tennessee, USA) [48]. The desired power of our study was set at 80% with a significance level of 0.05 in a two-sided test. We chose the log-additive inheritance model, which is the most suitable for polygenic diseases.
Allele frequencies for each SNP were determined by gene counting. Genotype distribution of each SNP was tested for departure from Hardy-Weinberg equilibrium (HWE) using χ2 analyses. The extent of pair-wise linkage disequilibrium between polymorphisms was determined by the Haploview (version 4.0) software (Jeffrey C Barrett and Mark J Daly, Cambridge, MA, USA). The association between polymorphisms and supernatant TNFα was determined using one-way analysis of variance. The association between polymorphisms and MOD scores was performed using analysis of one-way ANOVA testing with age, sex ratio and injury severity to adjust for possible confounding effects. Three genetic models (dominant, recessive and allele-dose effects) were used. The association of genotypes with sepsis morbidity rate was determined by χ2 analysis. Odds ratios with 95% confidence intervals were calculated by multiple logistic regression analyses to estimate the relative risk of sepsis. Age, sex ratio and injury severity were used as covariances of multiple logistic regression. P- values smaller than 0.05 after Bonferroni correction for multiple testing were considered significant. All statistic analysis was carried out using SPSS Version 13.0.
Results
Construction of haplotype bins and selection of htSNPs
There are a total of 10 SNPs with a minor allele frequency of more than or equal to 5% in the CHB population, which constructed two haplotype bins (Figure 1). Based on the analysis of tagging threshold of r2 of SNPs in each bin, one htSNP was selected from each bin for genotyping. In combination with previous association studies [21], the rs1800625 and rs1800624 polymorphisms were selected as htSNP of the respective bins (Figure 1). The rs2070600 polymorphism, though not forming any bin with other SNPs, was still selected due to its nonsynonymous variation, showing a substitution of the conserved glycine to serine at amino acid 82 (Gly82Ser) [19]. In addition, a deletion variant (63 bp ins/del) was identified in the region of 5'-flanking region (-407 to -345). This variant, although not being included in the current public gene polymorphism database, was shown to affect the promoter activities of the RAGE gene [21]. As a result, this variant was also selected in this study cohort.
Overall clinical characteristics of patients with major trauma
There were two independent patient cohorts, consisting of 496 and 232 individuals, respectively. All patients survived at least 48 hours after admission and completed genotyping. Baseline data of the patients are shown in Table 2. Patients were severely injured and mostly young. Sepsis morbidity rate was 40.7% and 37.9%, respectively in the Chongqing and Zhejiang cohorts. No pathogens were identified in blood cultures in 53.0% and 50.0% of the patients in both cohorts as the causative microorganism for sepsis, although they had an identified site of infection. The common pathogens identified in this study cohort were Staphylococcus aureus, coagulase-negative staphylococci, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, Escherichia coli, Enterococcus spp. and Enterobacter cloacae. Gram negative infection accounted for about 21% and 24.6%, Gram positive infection for about 18% and 15.9%, and mixed infection for about 5.8% and 7.3%, respectively, in the Chongqing and Zhejiang cohorts. Median time point for sepsis occurrence in the whole study cohort was 6.0 days (interquartile range 5.0 to 9.0 days. Chongqing: median 7.0 days, interquartile range 5.0 to 8.0 days; Zhejiang: median 6.0 days, interquartile range 5.0 to 7.0 days). Organ dysfunction occurred in 72.1% and 73.2% of the patients in both cohorts, among whom 213 (42.9%) and 96 (41.4%), respectively, had two or more organ dysfunctions. Among the patients with MODS, those with sepsis accounted for 40.7% and 37.9% in Chongqing and Zhejiang populations, respectively. Among the patients with sepsis, the median time point for MODS occurrence was shown to be 8.0 days (interquartile range 6.5 to 10.5 days) in Chongqing, and 9.0 days (interquartile range 7.0 to 11.0 days) in Zhejiang patients, respectively. With respect to the patients without sepsis, the median time point for MODS occurrence was 5.0 days (interquartile range 4.0 to 8.0 days) in Chongqing, and 5.5 days (interquartile range 3.5 to 7.5 days) in Zhejiang patients, respectively.
Allele frequencies and genotype distribution
The MAF of the rs2070600, rs1800624 and rs1800625 polymorphisms among the trauma patients were similar to those among the CHB population in the HapMap database (Table 3). The MAF of the 63 bp ins/del variant was 7.7% and 6.3% in the Chongqing and Zhejiang districts, respectively. This seems to be higher in the Chinese Han population when compared with that in Western populations, among whom the MAF of the 63 bp ins/del variant is less than 1% [21]. The genotype distribution of the four genetic variants was in agreement with the Hardy-Weinberg equilibrium (P >0.05, Table 3), indicating that both allele and genotype frequencies of these variants in the population remain constant; that is, they are in equilibrium from generation to generation.
Clinical relevance of the four genetic variants in trauma patients in the Chongqing district
We first selected 496 Chinese Han patients with major trauma in the Chongqing district to investigate the clinical relevance of the four variants (63 bp ins/del, rs2070600, rs1800624 and rs1800625) of the RAGE gene. As shown in Table 4, there were no significant differences in age, gender ratio and ISS among patients stratified according to the different genotypes of each variant. Among the four genetic variants selected in this study, only the rs1800625 was shown to be significantly associated with the risk for development of sepsis and MODS in major trauma patients. The patients carrying the variant C allele revealed a significantly lower sepsis morbidity rate and MOD scores, when compared with those carrying the T allele (P = 0.003 for sepsis morbidity rate and P = 0.015 for MOD scores in case of dominant effect, P = 0.032 for MOD scores in case of recessive effect). Data from regression analyses further indicated that the association of this polymorphism was in significant allele-dose effect with sepsis morbidity rate (OR = 0.428, 95% CI: 0.276 to 0.886, P = 0.026), and MOD scores (P = 0.045), respectively. There were no significant differences in the sepsis morbidity rate and MOD scores among different groups when patients were stratified according to the genotypes of the other three genetic variants.
The clinical relevance of the four variants in trauma patients in the Zhejiang district
In order to confirm the biological significance of the four genetic variants selected in this study, we further investigated their clinical relevance in another Chinese Han population of trauma patients, who lived in the Zhejiang province of eastern China. As expected, only rs1800625 was shown to be significantly associated with risk of sepsis and MODS in trauma patients in the Zhejiang district. The sepsis morbidity rates and MOD scores were significantly lower in the patients carrying the variant C allele compared with those carrying the T allele (P = 0.016 and P = 0.042 for sepsis morbidity rates and MOD scores in case of dominant effect, Table 4). Data from linear regression analysis revealed that the association of this polymorphism was in borderline significant allele-dose effect with MOD scores (P = 0.048).
Effect of rs1800625 on LPS-induced TNF-α production
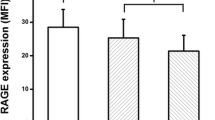
As shown in Figure 2, the rs1800625 polymorphism was well associated with the LPS responsiveness of peripheral blood leukocytes. LPS-induced TNF-α production was significantly lower in patients with the variant C allele than that in those with wild T allele (CC genotype: 5423.4 ± 2059.4 pg/ml, TC genotype: 5763.1 ± 2771.8 pg/ml and TT genotype: 6146.7 ± 3978.4 pg/ml, P = 0.017 for dominant effect).

Effect of the rs1800625 (-429T/C) on LPS-induced TNF-α production. One-way ANOVA was used to assess statistical significance. *P = 0.017 for dominant association (TT + TC vs. TT).
Effect of the rs1800625 on promoter activity
In view of the location of the rs1800625 in the 5'-flanking region of the RAGE gene, we further hypothesized that the T→C variation of this polymorphism might affect the promoter activities of the RAGE gene. Figure 3 showed that the fold increase of relative luciferase activity (RLA) was significantly lower in cells transfected with variant C allele than those transfected with wild T allele (1.36 ± 0.11 vs. 1.57 ± 0.27, P = 0.032).

Effect of the -429T/C polymorphism on the transcription activity of the RAGE promoter. Relative luciferase activity (RLA) was measured in U937 cells transfected with -429T or -429C plasmid constructs as described in methods. The fold increase of RLA was significantly lower in cells transfected with variant C allele than those transfected with wild T allele (P = 0.032).
Discussion
It has been demonstrated that inappropriate immune inflammatory response contributes to the development of sepsis and MODS in critically ill patients [40]. Increasing evidence suggests that genetic variants, particularly single nucleotide polymorphisms (SNPs), are critical determinants for inter-individual differences in both inflammatory responses and clinical outcome [48, 49]. Delineating the variation in genes and associated differences in immune inflammatory response might contribute to the development of new genetically tailored diagnostic and therapeutic interventions that will improve outcome in critically ill patients. RAGE, a member of the immunoglobulin superfamily of cell surface molecules, was initially identified and characterized as a cellular interaction site for AGEs [6]. Growing evidence has indicated that RAGE is an important PRR, involved in amplification of pro-inflammatory response and antimicrobial host defense [7]. The present study, to our knowledge for the first time, investigated the potential clinical relevance of the genetic variants identified within the entire RAGE gene in the Han Chinese population. Three SNPs (rs1800625, rs1800624 and rs2070600) are selected from 10 SNPs with a minor allele frequency of more than or equal to 5% based on LD threshold and their possible functionalities. The 63 bp ins/del variant, although being rare in Western populations with an MAF of less than 1% [21] and not being included in the current public database of the Chinese Han population, is shown to be a common polymorphism in the Chinese Han population, showing 7.7% in Chongqing and 6.3% in Zhejiang populations, respectively, in our study cohorts. Although the rs2070600 polymorphism occurs in less than 5% in many populations, such as Brazilian and Finnish populations, the MAF was approximately 20% in Asia populations [50]. This is further confirmed in our study cohorts, showing an MAF of 29.4% and 27.4% in the Chongqing and Zhejiang districts. Therefore, the four genetic variants selected in this study cohort are all common gene polymorphisms in the Chinese Han population.
Case-control study is a common and convenient association study design for finding a genetic basis of disease. However, a major limitation in this approach is the potential for population stratification when inappropriate patient-control matching occurs, such as using healthy blood donors as the control group in the study. To avoid confounding association, we only selected trauma patients and prospectively followed them to determine whether those who had genetic variants had a lesser or higher risk of post-traumatic MODS and sepsis. In addition, the patients recruited in this study cohort were all Han Chinese. In the context of a biologically relevant phenotype and a racially uniform population, this might maximize the likelihood of finding a meaningful genetic association. Furthermore, two independent patient populations from different geographic regions were used in this study cohort. Our results indicate that among the four genetic variants, only the rs1800625 polymorphism reveals a strong clinical relevance, showing lower sepsis morbidity rate and MOD scores in the patients with the variant C allele both in Chongqing and Zhejiang cohorts. This is in contrast to its association with other diseases, showing increased risk of diabetes [18], diabetic retinopathy [19] and colorectal cancer [20]. This might be because RAGE plays different roles in the pathogenesis of different diseases. The other three genetic variants (rs1800624, rs2070600 and 63bp ins/del), being associated with an increased risk of some inflammatory diseases [18, 26–28], are not shown to be associated with risk of sepsis and MODS in patients with major trauma in the Chongqing and Zhejiang cohorts. In combination with previous reports showing that the rs1800625 polymorphism is a common allele in other ethnic populations with the minor allele frequency of more than 10% [51–53], it might be used as a relevant risk estimate for sepsis and MODS in critical ill patients.
Given the functional significance of the rs1800625 polymorphism, we further investigated the association of this SNP with LPS-induced responsiveness of peripheral leukocytes obtained from trauma patients. The whole blood samples were taken immediately after admission in an attempt to avoid the potential effects of blood transfusion and fluid resuscitation. As we expected, the rs1800625 polymorphism is significantly associated with lower responsiveness of peripheral blood leukocytes in response to LPS stimulation, showing much lower levels of TNFα in patients carrying the variant C allele. This is in accordance with the clinical association of the rs1800625 with risk of sepsis and MODS in patients with major trauma. The rs1800625 polymorphism is located in the promoter region of the RAGE gene and induces T→C substitution at position -429 [54]. Bioinformatics analysis reveals that binding sites for some transcription factors (GATA-1, GATA-2 and sp1) (motif.genome.jp/) are present in close proximity to nucleotide -429 in the promoter. In order to further determine the functionality of the rs1800625 polymorphism, we investigated the effect of the rs1800625 polymorphism on the RAGE promoter activity using reporter gene assay system. Using the U937 cell line, we show that the fold increase of RLA is significantly lower in the cells transfected with vectors containing -429 C allele. It suggests that T→C variation at position -429 could significantly reduce the transcriptional activity of the RAGE promoter. Taken together, The T→ C variation at position -429 might reduce DNA-protein interaction, and then inhibit RAGE gene transcription, which contributes to decreased TNF production by peripheral blood leukocytes in response to ex vivo LPS stimulation, leading to decreased risk of sepsis and MODS in patients with major trauma.
There are some limitations to our current study. First, the sample size of both patient groups recruited in this study was relatively small, especially in the Zhejiang cohort. The values of power for rs1800625 in the Zhejiang cohort are shown to be 51.7% and 36.2% for sepsis morbidity rate and MOD scores, respectively, at a significance of 0.05. The too small number size of patients with the rs1800625 TC genotype (n = 5) and their higher incidence of sepsis (40%) in the Zhejiang cohort might explain the discrepancy in sepsis morbidity rate between the Chongqing and Zhejiang cohorts. The clinical relevance of the rs1800625 polymorphism needs to be validated in a larger population. Second, the population we studied was only Han Chinese who live in the Chongqing and Zhejiang districts. Our results may not be generalized to other populations. Third, difficulties in obtaining additional blood samples did not allow us to determine plasma sRAGE levels, or RAGE expression on peripheral blood leukocytes; therefore, the in vivo association between the rs1800625 polymorphism and RAGE activities needs to be confirmed. Despite the limited power of the clinical association study, our results demonstrate that the rs1800625 polymorphism might be a causal risk allele for sepsis and MODS in patients with major trauma
Conclusions
The present study investigated the clinical relevance of the genetic variations within the entire RAGE gene by means of construction of haplotype bins in patients with major trauma. We demonstrate that rs1800625 polymorphism is shown to affect TNFα production, and might be used as a relevant risk estimate for sepsis and MODS in trauma patients. In addition, the rs1800625 polymorphism could significantly enhance the promoter activities of the RAGE gene.
Key messages
-
Among the four tagSNPs of RAGE, only rs1800625 reveals a strong clinical relevance, showing lower sepsis morbidity rate and MOD scores in the patients with the variant C allele in both the Chongqing and Zhejiang cohorts.
-
The rs1800625 polymorphism is significantly associated with lower responsiveness of peripheral blood leukocytes in response to LPS stimulation, showing much lower levels of TNFα in patients carrying the variant C allele.
-
The rs1800625 polymorphism could significantly enhance the promoter activities of the RAGE gene.
Author's contributions
LZ and A-QZ were the main researchers for this study and co-contributed to the writing of this manuscript. WG, JZ, L-YZ, D-YD, MZ, JY and CY were involved in the collecting of blood samples and other clinical data. H-YW did the technical work. J-XJ planned the study, wrote the protocol and was involved in the genetic and clinical aspects of data analyses and revised the manuscript. All authors read and approved the final manuscript.
Abbreviations
- AGEs:
-
advanced glycation end products
- CHB:
-
Chinese Han Beijing
- DAMPs:
-
damage-associated molecular patterns
- DNA:
-
deoxyribonucleic acid
- ELISA:
-
enzyme-linked immunoabsorbent assay
- htSNPs:
-
haplotype tagging single nucleotide polymorphisms
- HWE:
-
Hardy-Weinberg equilibrium
- ISS:
-
Injury Severity Score
- LD:
-
linkage disequilibrium
- LP:
-
length polymorphism
- LPS:
-
lipopolysaccharide
- MAF:
-
minor allele frequency
- MHC:
-
major histocompatibility complex
- MODS:
-
multiple organ dysfunction syndrome
- OR:
-
odds ratios
- PAMPs:
-
pathogen-associated molecular patterns
- PCR:
-
polymerase chain reaction
- PRR:
-
pattern-recognition receptor
- RAGE:
-
receptor for advanced glycation end products
- RLA:
-
relative luciferase activity
- RPMI:
-
Roswell Park Memorial Institute
- SNPs:
-
single nucleotide polymorphisms
- TNFα:
-
tumor necrosis factor alpha
References
van Zoelen MA, Achouiti A, van der Poll T: RAGE during infectious diseases. Front Biosci 2011, 3: 1119-1132.
Tadie JM, Bae HB, Banerjee S, Zmijewski JW, Abraham E: Differential activation of RAGE by HMGB1 modulates neutrophil associated NADPH oxidase activity and bacterial killing. Am J Physiol Cell Physiol 2011, 302: 249-256.
van Zoelen MA, Schmidt AM, Florquin S, Meijers JC, de Beer R, de Vos AF, Nawroth PP, Bierhaus A, van der Poll T: Receptor for advanced glycation end products facilitates host defense during Escherichia coli -induced abdominal sepsis in mice. J Infect Dis 2009, 200: 765-773. 10.1086/604730
Clynes R, Herold K, Schmidt AM: RAGE : exacting a toll on the host in response to polymicrobial sepsis and Listeria monocytogenes. Crit Care 2007, 11: 183. 10.1186/cc6193
Frommhold D, Kamphues A, Hepper I, Pruenster M, Lukic IK, Socher I, Zablotskaya V, Buschmann K, Lange-Sperandio B, Schymeinsky J, Ryschich E, Poeschl J, Kupatt C, Nawroth PP, Moser M, Walzog B, Bierhaus A, Sperandio M: RAGE and ICAM-1 cooperate in mediating leukocyte recruitment during acute inflammation in vivo . Blood 2010, 116: 841-849. 10.1182/blood-2009-09-244293
Hu YM, Pai MH, Yeh CL, Hou YC, Yeh SL: Glutamine administration ameliorates sepsis-induced kidney injury by downregulating the high-mobility group box protein-1-mediated pathway in mice. Am J Physiol Renal Physiol 2012, 302: F150-F158. 10.1152/ajprenal.00246.2011
Humpert PM, Lukic IK, Thorpe SR, Hofer S, Awad EM, Andrassy M, Deemer EK, Kasper M, Schleicher E, Schwaninger M, Weigand MA, Nawroth PP, Bierhaus A: AGE-modified albumin containing infusion solutions boosts septicaemia and inflammation in experimental peritonitis. J Leukoc Biol 2009, 86: 589-597. 10.1189/jlb.1008646
Lutterloh EC, Opal SM, Pittman DD, Keith JC Jr, Tan XY, Clancy BM, Palmer H, Milarski K, Sun Y, Palardy JE, Parejo NA, Kessimian N: Inhibition of RAGE products increases survival in exprimental models of severe sepsis and systemic infection. Crit Care 2007, 11: R112. 10.1186/cc6161
Liliensiek B, Weigand MA, Beirhaus A, Nicklas W, Kasper M, Hofer S, Plachky J, Gröne HJ, Kurschus FC, Schmidt AM, Yan SD, Martin E, Schleicher E, Stern DM, Hämmerling GJ, Nawroth PP, Arnold B: Receptor for advanced glycation end products ( RAGE ) regulates sepsis but not the adaptive immune response. J Clin Invest 2004, 113: 1641-1650.
Christaki E, Opal SM, Keith JC Jr, Kessimian N, Palardy JE, Parejo NA, Tan XY, Piche-Nicholas N, Tchistiakova L, Vlasuk GP, Shields KM, Feldman JL, Lavallie ER, Arai M, Mounts W, Pittman DD: A monoclonal antibody against RAGE alters gene expression and is protective in experimental models of sepsis and pneumococcal pneumonia. Shock 2011, 35: 492-498. 10.1097/SHK.0b013e31820b2e1c
Yamamoto Y, Harashima A, Saito H, Tsuneyama K, Munesue S, Motoyoshi S, Han D, Watanabe T, Asano M, Takasawa S, Okamoto H, Shimura S, Karasawa T, Yonekura H, Yamamoto H: Septic shock is associated with receptor for advanced glycation end products ligation of LPS. J Immunol 2011, 186: 3248-3257. 10.4049/jimmunol.1002253
Calfee CS, Ware LB, Eisner MD, Parsons PE, Thompson BT, Wickersham N, Matthay MA, NHLBI ARDS Network: Plasma receptor for advanced glycation end products and clinical outcomes in acute lung injury. Thorax 2008, 63: 1083-1089. 10.1136/thx.2008.095588
Boyd JH, Kan B, Roberts H, Wang Y, Walley KR: S100A8 and S100A9 mediate endotoxin-induced cardiomyocyte dysfunction via the receptor for advanced glycation end products. Circ Res 2008, 102: 1239-1346. 10.1161/CIRCRESAHA.107.167544
Sadik NA, Mohamed WA, Ahmed MI: The association of receptor of advanced glycated end products and inflammatory mediators contributes to endothelial dysfunction in a prospective study of acute kidney injury patients with sepsis. Mol Cell Biochem 2012, 359: 73-81. 10.1007/s11010-011-1001-4
Bopp C, Hofer S, Weitz J: s RAGE is elevated in septic patients and associated with patients outcome. J Surg Res 2008, 147: 79-83. 10.1016/j.jss.2007.07.014
Nakamura T, Sato E, Fujiwara N, Kawagoe Y, Suzuki T, Ueda Y, Yamada S, Shoji H, Takeuchi M, Ueda S, Matsui T, Adachi H, Okuda S, Yamagishi S: Circulating levels of advanced glycation end products (AGE) and interleukin-6 (IL-6) are independent determinants of serum asymmetric dimethylarginine (ADMA) levels in patients with septic shock. Pharmacol Res 2009, 60: 515-518. 10.1016/j.phrs.2009.05.002
Creagh-Brown BC, Quinlan GJ, Evans TW, Burke-Gaffney A: The RAGE axis in systemic inflammation, acute lung injury and myocardial dysfunction: an important therapeutic target? Intens Care Med 2010, 36: 1644-1656. 10.1007/s00134-010-1952-z
Hudson BI, Stickland MH, Grant PJ, Futers TS: Characterization of allelic and nucleotide variation between the RAGE gene on chromosome 6 and a homologous pseudogene sequence to its 5'-regulatory region on chromosome 3. Diabetes 2001, 50: 2646-2651. 10.2337/diabetes.50.12.2646
Kim OY, Jo SH, Jang Y, Chae IS, Kim JY, Hyun YJ, Lee JH: G allele at RAGE SNP82 is associated with proinflammatory markers in obese subjects. Nutr Res 2009, 29: 106-113. 10.1016/j.nutres.2009.01.006
Laki J, Kiszel P, Vatay A: The HLA 8.1 ancestral haplotype is strongly linked to the C allele of -429T>C promoter polymorphism of receptor of the advanced glycation endproduct ( RAGE ) gene. Haplotype-independent association of the -429C allele with high hemoglobinA1C levels in diabetic patients. Mol Immunol 2007, 44: 648-655. 10.1016/j.molimm.2006.01.011
Hudson BI, Stickland MH, Futers TS, Grant PJ: Effects of novel polymorphisms in the RAGE gene on transcriptional regulation and their association with diabetic retinopathy. Diabetes 2001, 50: 1505-1511. 10.2337/diabetes.50.6.1505
Tóth EK, Kocsis J, Madaras B, Bíró A, Pocsai Z, Fust G, Blaskó B, Karádi I, Adány R, Laki J: The 8.1 ancestral MHC haplotype is strongly associated with colorectal cancer risk. Int J Cancer 2007, 121: 1744-1748. 10.1002/ijc.22922
Dabritz J, Friedrichs F, Weinhage T, Hampe J, Kucharzik T, Lügering A, Broeckel U, Schreiber S, Spieker T, Stoll M, Foell D: The functional -374T/A polymorphism of the receptor for advanced glycation end products may modulate Crohn's disease. Am J Physiol Gastrointest Liver Physiol 2011, 300: G823-G832. 10.1152/ajpgi.00115.2010
Falcone C, Geroldi D, Buzzi MP, Emanuele E, Yilmaz Y, Fontana JM, Vignali L, Boiocchi C, Sbarsi I, Cuccia M: The -374T/A RAGE polymorphism protects against future cardiac events in nondiabetic patients with coronary artery disease. Arch Med Res 2008, 39: 320-325. 10.1016/j.arcmed.2007.11.003
Falcone C, Emanuele E, Buzzi MP, Ballerini L, Repetto A, Canosi U, Mazzucchelli I, Schirinzi S, Sbarsi I, Boiocchi C, Cuccia M: The -374T/A variant of the RAGE gene promoter is associated with clinical restenosis after coronary stent placement. Int J Immunopathol Pharmacol 2007, 20: 771-777.
dos Santos KG, Canani LH, Gross JL, Tschiedel B, Pires Souto KE, Roisenberg I: The -374A allele of the receptor for advanced glycation end products gene is associated with a decreased risk of ischemic heart disease in African-Brazilians with type 2 diabetes. Mol Genet Metab 2005, 85: 149-156. 10.1016/j.ymgme.2005.02.010
Pettersson-Fernholm K, Forsblom C, Hudson BI, Perola M, Grant PJ, Groop PH: The functional -374 T/A RAGE gene polymorphism is associated with proteinuria and cardiovascular disease in type 1 diabetic patients. Diabetes 2003, 52: 891-894. 10.2337/diabetes.52.3.891
Hofmann MA, Drury S, Hudson BI, Gleason MR, Qu W, Lu Y, Lalla E, Chitnis S, Monteiro J, Stickland MH, Bucciarelli LG, Moser B, Moxley G, Itescu S, Grant PJ, Gregersen PK, Stern DM, Schmidt AM: RAGE and arthritis: the G82S polymorphism amplifies the inflammatory response. Genes Immun 2002, 3: 123-135. 10.1038/sj.gene.6363861
Li K, Zhao B, Dai D, Yao S, Liang W, Yao L, Yang Z: A functional p.82G>S polymorphism in the RAGE gene is associated with multiple sclerosis in the Chinese population. Mult Scler 2011, 17: 914-921. 10.1177/1352458511403529
Kucukhuseyin O, Yilmaz-Aydogan H, Isbir CS, Isbir T: Is there any association between GLY82 ser polymorphism of RAGE gene and Turkish diabetic and non diabetic patients with coronary artery disease? Mol Biol Rep 2012, 39: 4423-4428. 10.1007/s11033-011-1230-3
Ng ZX, Kuppusamy UR, Tajunisah I, Fong KC, Chua KH: Association analysis of -429T/C and -374T/A polymorphisms of receptor of advanced glycation end products ( RAGE ) gene in Malaysian with type 2 diabetic retinopathy. Diabetes Res Clin Pract 2012, 95: 372-377. 10.1016/j.diabres.2011.11.005
Kalousová M, Brabcová I, Germanová A, Jáchymová M, Matl I, Mestek O, Bandúr S, Zima T, Viklický O: RAGE polymorphisms, renal function and histological finding at 12 months after renal transplantation. Clin Biochem 2009, 42: 347-352. 10.1016/j.clinbiochem.2008.12.006
Globocnik PM, Steblovnik K, Peterlin B, Pitrovic D: The - 429 T/C and - 374 T/A gene polymorphisms of the receptor of advanced glycation end products gene are not risk factors for diabetic retinopathy in Caucasians with type 2 diabetes. Klin Monbl Augenheilkd 2003, 220: 873-876.
Ramprasad S, Radha V, Mathias RA, Majumder PP, Rao MR, Rema M: RAGE gene promoter polymorphisms and diabetic retinopathy in a clinic-based population from South India. Eye (Lond) 2007, 21: 395-401. 10.1038/sj.eye.6702239
Lu L, Jin Pu L, Chen QJ, Wang L, Peng W, Yan X, Zhang Q, Yan Zhang R, Gong PH, Qiu JP, Shen WF: Increased glycated albumin and decreased es RAGE concentrations are associated with in-stent restenosis in Chinese diabetic patients. Clin Chim Acta 2008, 396: 33-37. 10.1016/j.cca.2008.06.019
Prevost G, Fajardy I, Fontaine P, Danze PM, Besmond C: Human RAGE GLY82SER dimorphism and HLA class II DRB1-DQA1-DQB1 haplotypes in type 1 diabetes. Eur J Immunogenet 1999, 26: 343-348. 10.1046/j.1365-2370.1999.00168.x
Ng ZX, Kuppusamy UR, Tajunisah I, Fong KC, Koay AC, Chua KH: 2245G/A polymorphism of the receptor for advanced glycation end-products ( RAGE ) gene is associated with diabetic retinopathy in the Malaysian population. Br J Ophthalmol 2012, 96: 289-292. 10.1136/bjophthalmol-2011-300658
Forbes JM, Soderlund J, Yap FY, Knip M, Andrikopoulos S, Ilonen J, Simell O, Veijola R, Sourris KC, Coughlan MT, Forsblom C, Slattery R, Grey ST, Wessman M, Yamamoto H, Bierhaus A, Cooper ME, Groop PH: Receptor for advanced glycation end-products ( RAGE ) provides a link between genetic susceptibility and environmental factors in type 1 diabetes. Diabetologia 2011, 54: 1032-1042. 10.1007/s00125-011-2058-z
Kanková K, Stejskalová A, Pácal L, Tschoplová S, Hertlová M, Krusová D, Izakovicová-Hollá L, Beránek M, Vasků A, Barral S, Ott J: Genetic risk factors for diabetic nephropathy on chromosomes 6p and 7q identified by the set-association approach. Diabetologia 2007, 50: 990-999. 10.1007/s00125-007-0606-3
Jiang JX, Li L: The development and perspective of wound and trauma in wound. Med J Chin PLA 2010, 35: 781-784.
The Abbreviated Injury Scale: 2005 revision. Association for the Advancement of Automotive Medicine2005. [http://www.AAAM.org]
Marshall JC, Cook DJ, Christou NV, Bernard GR, Sprung CL, Sibbald WJ: Multiple organ dysfunction score: a reliable descriptor of a complex clinical outcome. Crit Care Med 1995, 23: 1638-1652. 10.1097/00003246-199510000-00007
Sauaia A, Moore EE, Johnson JL, Ciesla DJ, Biffl WL, Baneriee A: Validation of Postinjury Multiple Organ Failure scores. Shock 2009, 31: 438-447. 10.1097/SHK.0b013e31818ba4c6
Hudson BI, Stickland MH, Grant PJ, Futers TS: Characterization of allelic and nucleotide variation between the RAGE gene on chromosome 6 and a homologous pseudogene sequence to its 5' regulatory region on chromosome 3: implications for polymorphic studies in diabetes. Diabetes 2001, 50: 2646-2651. 10.2337/diabetes.50.12.2646
Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA: Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet 2004, 74: 106-120. 10.1086/381000
Zeng L, Gu W, Zhang AQ, Zhang M, Zhang LY, Du DY, Huang SN, Jiang JX: A functional variant of lipopolysaccharide binding protein predisposes to sepsis and organ dysfunction in patients with major trauma. Ann Surg 2012, 255: 147-157. 10.1097/SLA.0b013e3182389515
Gu W, Shan YA, Zhou J, Jiang DP, Zhang LY, Du DY, Wang ZG, Jiang JX: Functional significance of gene polymorphisms in the promoter of myeloid differentiation-2. Ann Surg 2007, 46: 151-158.
Dupont WD, Plummer WD Jr: PS: Power and Sample Size Calculation version 3.0.2009. [http://biostat.mc.vanderbilt.edu/twiki/bin/view/Main/PowerSampleSize]
Zeng L, Gu W, Chen KH, Jiang DP, Zhang LY, Du DY, Hu P, Liu Q, Huang SN, Jiang JX: Clinical relevance of the interleukin 10 promoter polymorphisms in Chinese Han patients with major trauma: genetic association studies. Crit Care 2009, 13: 1-8.
Liu LM, Xiang KS: RAGE Gly82Ser polymorphism in diabetic microangiopathy. Diabetes Care 1999, 22: 645-654. 10.2337/diacare.22.4.645
Prasad P, Tiwari AK, Kumar KM, Ammini AC, Gupta A, Gupta R, Thelma BK: Association analysis of ADPRT1, AKR1B1, RAGE , GFPT2 and PAI-1 gene polymorphisms with chronic renal insufficiency among Asian Indians with type-2 diabetes. BMC Medical Genetics 2010, 11: 52.
Santos IC, Daga DR, Frigeri HR, Réa RR, Almeida AC, Souza EM, Pedrosa FO, Fadel-Picheth CM, Picheth G: The functional polymorphisms -429T>C and -374T>A of the RAGE gene promoter are not associated with gestational diabetes in Euro-Brazilians. Genet Mol Res 2010, 9: 1130-1135. 10.4238/vol9-2gmr817
Kirbis J, Milutinović A, Steblovnik K, Teran N, Terzić R, Zorc M: The -429 T/C and -374 T/A gene polymorphisms of the receptor of advanced glycation end products gene ( RAGE ) are not risk factors for coronary artery disease in Slovene population with type 2 diabetes. Coll Antropol 2004, 28: 611-616.
Li J, Schmidt AM: Characterization and functional analysis of the promoter of RAGE , the receptor for advanced glycation end products. J Biol Chem 1997, 272: 16498-16506. 10.1074/jbc.272.26.16498
Acknowledgements
This work is supported by National Key Technology R&D Programme (2012BAI11B00).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Ling Zeng, An-qiang Zhang contributed equally to this work.
Electronic supplementary material
13054_2012_693_MOESM1_ESM.DOC
Additional file 1:The definition of sepsis and infection. We evaluated sepsis and infection of major trauma patients' according to these criteria. (DOC 28 KB)
13054_2012_693_MOESM2_ESM.DOC
Additional file 2:Table S1. Primers of the four variants of the RAGE gene and their PCR conditions. The PCR primers, sequencing primers and the annealing temperatures of the four variants of the RAGE gene were shown in Table S1. (DOC 32 KB)
13054_2012_693_MOESM3_ESM.DOC
Additional file 3:The methods of plasmid construction. The possible effect of -429T/C on the promoter activity was investigated using a reporter gene assay system. Two plasmids which contained the -429T promoter and -429C promoter were constructed. (DOC 24 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Zeng, L., Zhang, Aq., Gu, W. et al. Identification of haplotype tag single nucleotide polymorphisms within the receptor for advanced glycation end products gene and their clinical relevance in patients with major trauma. Crit Care 16, R131 (2012). https://doi.org/10.1186/cc11436
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/cc11436