
Abstract
Introduction
The PACS01 trial has demonstrated that a docetaxel addition to adjuvant anthracycline-based chemotherapy improves disease-free survival (DFS) and overall survival of node-positive early breast cancer (EBC). We searched for prognostic and predictive markers for docetaxel's benefit.
Methods
Tumor samples from 1,099 recruited women were analyzed for the expression of 34 selected proteins using immunohistochemistry. The prognostic and predictive values of each marker and four molecular subtypes (luminal A, luminal B, HER2-overexpressing, and triple-negative) were tested.
Results
Progesterone receptor-negativity (HR = 0.66; 95% CI 0.47 to 0.92, P = 0.013), and Ki67-positivity (HR = 1.53; 95% CI 1.12 to 2.08, P = 0.007) were independent adverse prognostic factors. Out of the 34 proteins, only Ki67-positivity was associated with DFS improvement with docetaxel addition (adjusted HR = 0.51, 95% CI 0.33 to 0.79 for Ki67-positive versus HR = 1.10, 95% CI 0.75 to 1.61 for Ki67-negative tumors, P for interaction = 0.012). Molecular subtyping predicted the docetaxel benefit, but without providing additional information to Ki67 status. The luminal A subtype did not benefit from docetaxel (HR = 1.16, 95% CI 0.73 to 1.84); the reduction in the relapse risk was 53% (HR = 0.47, 95% CI 0.22 to 1.01), 34% (HR = 0.66, 95% CI 0.37 to 1.19), and 12% (HR = 0.88, 95% CI 0.49 to 1.57) in the luminal B, HER2-overexpressing, and triple-negative subtypes, respectively.
Conclusions
In patients with node-positive EBC receiving adjuvant anthracycline-based chemotherapy, the most powerful predictor of docetaxel benefit is Ki67-positivity.
Similar content being viewed by others

Introduction
The use of adjuvant chemotherapy has improved prognosis of early breast cancer [1]. Benefits in terms of survival, first demonstrated in the 1970s with the CMF (cyclophosphamide, methotrexate, fluorouracil) regimen, were improved with the addition of anthracycline in the 1980s [2]. Recently, third-generation regimens, based on the addition of taxane, were shown as even more efficient [3].
However, patients do not benefit equally from the same drugs and regimens. Current histo-clinical prognostic factors and the factors predictive for response to a given therapy are not sufficient to solve this heterogeneity, basing the choice of adjuvant chemotherapy regimen upon the risks of relapse and toxicity, comorbidities and physician's experience, rather than upon the probability of efficiency. Today, no factor predictive for efficiency of third-generation regimens has been validated, and all node-positive patients empirically receive adjuvant regimens based on anthracycline and taxane [3], although the optimal role for taxanes in this setting remains controversial. The 20 first-generation taxane trials reported to date that compared taxane-based versus taxane-free adjuvant regimens [4–22] and three meta-analyses [23–25] have shown that the taxane-associated absolute benefit is modest, that is, a mere 5% for disease-free survival (DFS) and 3% for overall survival (OS). Taxanes are associated with many side effects and with greater deterioration of quality of life [26]. They are expensive and data are scarce regarding their long-term toxicity. Meta-analyses of first-generation taxane trials have shown that the DFS benefit associated with taxane addition is independent of age and menopausal status, degree of node involvement, estrogen receptor (ER) expression, type of taxane and schedule of administration [24]. In the absence of clear guidance on which patients may benefit from taxanes, their inclusion in adjuvant regimens has also been advocated as a means to reduce exposure to anthracyclines and the risk of associated late toxicity. Furthermore, the type of taxane (docetaxel or paclitaxel), dose and schedule are still debated. Today, an important question is whether subgroups of patients benefit more or less from taxanes in the adjuvant setting. This benefit likely depends upon molecular determinants that remain to be defined.
Randomized clinical trials provide an opportunity for identifying such predictive biomarkers in the adjuvant setting. To date, only immunohistochemistry (IHC)-based studies have been retrospectively reported. Five of them analyzed one to four markers (including ER, progesterone receptor (PR), HER2, and/or Ki67) in a series ranging from 798 to 3,329 tumor samples [9, 18, 27–31]. A few data suggest that taxanes might benefit ER-negative and/or HER2-positive and/or luminal B breast cancer patients [9, 28, 29], but results are inconsistent, notably with a recent study [32], negative for 15 proteins analyzed in 1,350 samples.
The PACS01 trial was a multicenter, prospective, randomized, phase III, open-label trial comparing six cycles of fluorouracil, epirubicin (100 mg/m2), and cyclophosphamide (FEC) with three cycles of FEC followed by three cycles of docetaxel (100 mg/m2; FEC-D), as adjuvant chemotherapy in node-positive operable breast cancer [8]. A total of 1,999 patients were enrolled between 1997 and 2000. With a median follow-up of 60 months, the five-year DFS was 73% with FEC and 78% with FEC-D (18% reduction in the relative risk of relapse). In an analysis restricted to ER-positive tumors, Ki67 expression identified a subgroup of patients who could benefit from docetaxel [30]. Here, we have analyzed the expression of 34 selected IHC markers in a subset of 1,099 patients included in the trial, regardless of their ER status. Our objective was to assess the prognostic and/or predictive value of these markers and the molecular subtypes for the benefit of docetaxel in terms of DFS.
Materials and methods
Patients
This biomarker study is ancillary to the PACS01 trial. A tumor block representative of the primary tumor was collected for 1,190 out of the 1,999 enrolled patients. All samples were obtained from operated tumors before any systemic therapy. Patients provided written informed consent for research use, and the study was approved by the ethics committee/institutional review board. Post-menopausal women with hormone receptor (HR)-positive tumors (ER and/or PR-positive) received tamoxifen after completion of chemotherapy. In December 1998, the protocol was amended to require tamoxifen for pre-menopausal women with HR-positive disease. Radiotherapy was mandatory for all patients who had undergone breast-conservative surgery, and was recommended after mastectomy. No patient with an HER2-positive tumor received trastuzumab in the adjuvant setting. The primary end-point was DFS, defined as the time from randomization until the first event: relapse (local, regional, or metastatic), contralateral breast cancer, or death from any cause.
For translational studies, the 1,190 tumors had been centrally immunostained with ER, PR, Ki67 and HER2 specific antibodies on standard slides [30]. Here, we considered ER and PR staining as positive when at least 1% of tumor cells were stained. For Ki67, the positivity cut-off value was 20%. The HER2 status was evaluated with the Dako scale (HercepTest kit scoring guidelines, DakoCytomation, Copenhagen, Denmark): positivity corresponded to 3+ IHC score, or 2+ score with Fluorescent In Situ Hybridisation (FISH) amplification. FISH results were obtained from the previously reported centralized reading on standard slides [30]; a HER2/CEP17 ratio higher than 2.2 defined amplification. From the 1,190 cases, a tissue microarray (TMA) was prepared for 1,099 cases. Histo-clinical characteristics and magnitude of docetaxel efficacy in the study group were similar to those of the whole population of the trial (Additional file 1, Table S1), suggesting its representativity. Histo-clinical features of patients whose tumor samples were and were not centrally tested were similar (data not shown).
Tissue microarrays and immunohistochemistry
Tissue microarrays (TMAs) were prepared as previously described from formalin-fixed and paraffin-embedded tissues [33]. For each tumor, three representative areas were selected from a hematoxylin-eosin-safran stained section of a donor block. Core cylinders with a diameter of 0.6 mm each were punched from each of these areas and deposited into three separate recipient paraffin blocks using a specific arraying device (Alphelys, Plaisir, France). Five-μm sections of the resulting microarray blocks were made and used for IHC after transfer to glass slides.
The selection of the 30 additional proteins to be tested was based on known or putative importance in breast cancer as prognostic/predictive markers or in resistance to taxane, and availability and suitability of a corresponding antibody for paraffin-embedded tissues. They explored different pathways: cell differentiation and adhesion (Cytokeratins CK5/6, CK8/18, CK14, P-Cadherin, E-Cadherin, α-Catenin, β-Catenin, Afadin/AF6, Mucin MUC1, Caveolin CAV1, Moesin, CD10, CD44), proliferation and cell cycle (Aurora A, TACC2, TACC3, Cyclin D1, P21, P27), ER-associated (GATA3), tyrosine kinase signaling (EGFR, FGFR1, MET), apoptosis (BCL2), checkpoints and tumor suppression (P53, PTEN, FHIT), and others (Angiogenin, Topoisomerase II α TOPO2A, microtubule-associated protein TAU).
Immunohistochemical analysis on TMA sections was done as previously described [33] using Dako LSABR2 Kit in the autoimmunostainer (Dako Autostainer, Glostrup, Denmark). Sections were deparaffinized in Histolemon (Carlo Erba Reagenti, Rodano, Italy) and rehydrated in graded ethanol solutions. Details of antibodies are given in Additional file 2 (Table S2). The dilution of each antibody was established on the basis of negative and positive controls and staining with a range of dilutions. For each antibody, the selected titer was in the linear range and allowed the extinction of the negative control and the persistence of the positive control. Results were evaluated by two pathologists (JJ, ECJ) under a light microscope with the Spot Browser system (Alphelys, Plaisir, France). Only invasive tumor components were scored, using the quick score (QS, range from 0 to 300). For each tumor, the mean of the score of a minimum of two core biopsies was calculated. For each antibody, a tumor was considered as positive when the QS was superior to 0.
Statistical analysis
Distributions of molecular markers and other categorical histo-clinical variables were compared between groups using the Chi2 test. The primary end-point was that of the PACS01 trial, DFS, as defined above. Data concerning patients without any event at last follow-up were censored. The follow-up was calculated from the date of randomization to the time of the first event or time of last follow-up for censored patients. Survival curves were derived from Kaplan-Meier estimates and compared by log-rank test. Uni- and multivariate analyses were done using Cox regression analysis. The variables tested in univariate analyses included patients' age, pathological tumor size, number of pathologically involved axillary lymph nodes, Scarff-Bloom Richardson (SBR) grade, and IHC status of the 34 tested markers. The prognostic influence of markers and IHC-defined molecular subtypes was assessed in multivariate analysis by the Cox proportional hazard models, using the adjustment variables preplanned for the PACS01 trial: age, number of pathologically involved axillary lymph nodes, pathological tumor size, SBR grade, and HR status, and the therapeutic arm. The predictive value of each marker and molecular subtype for the docetaxel benefit was assessed using a Cox regression model with terms for treatment by marker interaction, with and without adjustment with the variables quoted above. Survival rates and hazard ratios (HR) are presented with their 95% confidence intervals (95% CI). Statistical tests were two-sided at the 5% level of significance without adjustment for multiple comparisons. All analyses were done using SAS Version 9.1 (Evry-Grégy-sur-Yerres, France). The paper is written in accordance with reporting recommendations for tumor marker prognostic studies (REMARK) criteria.
Results
Patients' characteristics and survival
Out of the 1,099 cases included in our analysis, 546 had been treated in the FEC arm and 553 in the FEC-D arm. Patients' characteristics were balanced between the two treatment arms, except for the pathological tumor size, more frequently inferior to 20 mm in the FEC-D arm as observed in the PACS01 trial (Table 1). Almost all treated patients received radiotherapy. Tamoxifen use was not different between arms, in both pre- and post-menopausal women. The median follow-up was 60 months. The five-year DFS was 76% (95% CI 73.7 to 78.9). A total of 268 events were reported during the study period, 150 in the FEC arm (27%) and 118 in the FEC-D arm (21%). Respective five-year DFS was 72% (95% CI 68.1 to 76.0) and 79% (95% CI 76.1 to 83.0; P = 0.0125, log-rank test; Figure 1). In multivariate analysis (Additional file 3, Table S3), the features associated with shorter DFS (Wald test) were age inferior to 50 years (HR = 1.31), more than three involved axillary lymph nodes (HR = 1.94): pathological tumor size superior or equal to 20 mm (HR = 1.78), SBR grade superior to 1 (HR = 1.77 for grade 2, HR = 2.46 for grade 3), and negative hormone receptor status (HR = 1.76), validating the pre-planned choice of these variables as adjustment variables for multivariate analyses. The adjusted HR for an event associated with docetaxel was 0.78 (95% CI 0.60 to 1.02, P = 0.066, Wald test), similar to that observed in the whole PACS01 trial.

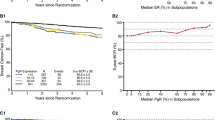
Disease-free survival in patients included in this sub-study. Kaplan-Meier DFS curves in patients treated without (FEC: black curve) and with (FEC-D: dashed curve) docetaxel.
In most of the subgroups defined according to these histo-clinical prognostic features (age, lymph nodes, size, grade, HR status), addition of docetaxel reduced the risk of relapse, with significantly greater benefit in patients aged 50 years or older, pathological tumor size inferior to 20 mm, four or more positive lymph nodes, and negative HR status (data not shown). However, in multivariate analysis, none of these features showed significant interaction with the chemotherapy arm (Additional file 4, Figure S1).
Protein markers, survival and docetaxel benefit
The expression of 34 proteins in tumor samples was centrally analyzed by IHC: ER, PR, Ki67 and HER2 on standard slides, and the 30 additional proteins on TMAs. Results are detailed in Table 2. Staining was heterogeneous between tumors. The percent of positive tumors varied from 12.5% for Moesin to 98% for CK8/18.
Table 2 reports the correlation between each single marker and DFS. The 12 proteins associated with shorter DFS in univariate analysis (negativity of BCL2, CK8/18, ER, GATA3, MUC1, PR, and TAU, and positivity of Aurora A, HER2, Ki67, P-Cadherin, and P53) were included in multivariate analysis, together with pre-planned histo-clinical variables. The status of two proteins remained associated with shorter DFS (Wald test): PR-negativity (HR = 0.66; 95% CI 0.47 to 0.92, P = 0.013), and Ki67-positivity (HR = 1.53; 95% CI 1.12 to 2.08, P = 0.007).
We then searched for an association between each protein and the benefit of docetaxel in terms of DFS. Results of univariate and multivariate analyses are detailed in Additional file 5, Table S4. On multivariate analysis (Table 3 and Figure 2), the addition of docetaxel reduced the risk of an event (HR for relapse inferior to 1) in most of the subgroups defined by the positive status and the negative status of most of the 34 proteins, significantly (P <0.05) for 11 proteins (Angiogenin, β-Catenin, CAV1, CD44, E-Cadherin, CK8/18, Ki67, MET, MUC1, P27, and PTEN). However, an interaction between the protein expression and the addition of docetaxel was significant only for Ki67 (P = 0.012). Docetaxel was associated with a 49% reduction in the risk of an event (HR = 0.51, 95% CI 0.33 to 0.79; P = 0.003 Wald test) in Ki67-positive patients (Figure 3A), but with no reduction (HR = 1.10 95% CI 0.75 to 1.61; P = 0.612) in Ki67-negative patients (Figure 3B). Interaction of borderline significance (P ≤0.15) was observed for CK14, Angiogenin, and β-Catenin, with a trend towards docetaxel benefit for patients with a marker-positive tumor.

Adjusted hazard ratios associated with docetaxel addition. Forest plots showing the adjusted hazard ratios associated with docetaxel addition according to expression of 34 proteins.

Disease-free survival according to Ki67 status and docetaxel. A. Kaplan-Meier DFS curves in patients with Ki67-positive status according to docetaxel addition. B. Similar to B, but in patients with Ki67-negative status.
Molecular subtypes, survival and docetaxel benefit
Genomics has revealed at least four subtypes of breast cancer: luminal A, luminal B, basal, and HER2/ERBB2-overexpressing [34]. These subtypes may be approximately, but more easily for clinical routine, defined by four IHC markers (ER, PR, HER2, and Ki67). Based on these markers, we classified the 1,099 samples into four subtypes: luminal A (HR-positive, HER2-negative, Ki67-negative: N = 525, 54%), luminal B (HR-positive, HER2-negative, Ki67-positive; N = 125, 13%), HER2-overexpressing (HER2-positive, whatever HR; N = 175, 18%), and triple-negative (HR-negative, HER2-negative; N = 148, 15%).
As expected, these subtypes correlated with histo-clinical variables (Additional file 6, Table S5). Triple-negative tumors and HER2-overexpressing tumors were more frequently grade 3 than luminal tumors (P <0.0001), as were luminal B tumors when compared with luminal A tumors. HER2-overexpressing tumors presented more frequently more than three involved axillary lymph nodes, followed by luminal B tumors, then luminal A, then triple-negative tumors (P = 0.015). Classical basal markers (CK14, EGFR, Moesin, P-Cadherin, P53), were more frequently positive in triple-negative tumors (P ≤0.001). Five-year DFS was different among the subtypes (P <0.0001, log-rank test): 83% for luminal A (95% CI 79.4to 6.2), 73% for luminal B (95% CI 63.8 to 79.8), 66% for HER2-overexpressing (95% CI 58.3 to 72.7), and 65% for triple-negative (95% CI 56.9 to 72.4).
The benefit of docetaxel was analyzed per subtype (Additional file 7, Figure S2). Results of uni- and multivariate analyses are shown in Additional file 8, Table S6. In multivariate analysis, docetaxel was associated with a 53% reduction in the risk of relapse in the luminal B subtype (HR = 0.47, 95% CI 0.22 to 1.01; P = 0.05, Wald test), a 34% reduction in the HER2-overexpressing subtype (HR = 0.66, 95% CI 0.37 to 1.19; P = 0.14), and a 12% reduction (HR = 0.88, 95% CI 0.49 to 1.57, P = 0.67) in the triple-negative subtype. By contrast, the risk of an event was 16% higher with vs without docetaxel in luminal A tumors (HR = 1.16, 95% CI 0.73 to 1.84, P = 0.52). The interaction between benefit of docetaxel and each subtype was significant for luminal B (P = 0.047), borderline for HER2-overexpressing (P = 0.14), and not significant for triple-negative (P = 0.46).
We explored the added predictive value of molecular subtyping compared with Ki67 status alone by testing the interaction between the molecular subtype factor and the treatment arm in a Cox model adjusted for the pre-planned histo-clinical variables and Ki67 by treatment interaction. Analysis using the likelihood ratio test did not show any significant added predictive value to that provided by the Ki67 status alone for DFS (P = 0.88).
Discussion
Identifying the patients who would and those who would not benefit from taxanes is crucial to moving away from the "one shoe fits all" strategy. Here, we have analyzed the expression of 34 selected proteins in a subset of 1,099 patients included in the PACS01 trial.
We show that a Ki67-positive status is not only independently associated with shorter DFS, but also with the benefit of a docetaxel addition in women treated with adjuvant anthracycline-based chemotherapy. Ki67, expressed during the cell cycle, is a well-established cell proliferation marker. Its expression in breast cancer correlates with poor prognosis [35, 36] and higher response to chemotherapy. In the neo-adjuvant setting, correlation between Ki67 positivity and response to taxanes, either as monotherapy [37] or in association with anthracyclines [38], has been reported, although the relationship was not observed in other small series [39, 40]. We found a prognostic correlation much more important in docetaxel-free patients (P <0.001, log-rank test) than in docetaxel-treated patients (P = 0.048), in relation to the interaction observed with docetaxel benefit. Such interaction has been previously studied in randomized trials of adjuvant chemotherapy. Higher efficiency of the CMF regimen vs. no chemotherapy (P = 0.16 for interaction) was reported in Ki67-positive ER-positive patients treated in the NSABP-20 trial [41], whereas Ki67 labeling index was not predictive of better response to adjuvant chemotherapy in endocrine-responsive tumors [42]. Bartlett et al. analyzed data of the UK NEAT/BR9601 trial, which showed benefit for the addition of anthracyclines to CMF regimen, and did not detect any interaction with anthracycline benefit for Ki67 status [43]. The benefit of docetaxel (P = 0.11 for interaction) in Ki67-positive ER-positive patients enrolled in the PACS01 trial has been reported [30]. Our present analysis, applied to all available PACS01 samples regardless of the ER status, showed a significant interaction, with an adjusted HR for the risk of relapse in Ki67-positive patients (HR = 0.51) equal to that previously reported for ER-positive patients only, and superior to that observed in the unselected whole population (HR = 0.81). By contrast, in an analysis of the BCIRG001 trial [32], Ki67 did not present any interaction with docetaxel benefit. In this study, the positivity threshold was the median value of the tested population, whereas we used the more classical 20% cut-off [36]. We did not find in the literature any pre-clinical or clinical data in the neo-adjuvant or the metastatic setting regarding Ki67 status and the response to or the benefit associated with docetaxel specifically. Of course, our observations will require validation before application in routine. But already, they clearly suggest that patients with Ki67-positive breast cancer potentially derive a high benefit from adjuvant docetaxel (adjusted HR = 0.51), and it might be a candidate for intensifying taxane delivery (six cycles and/or a dose-dense scheme). By contrast, we did not observe any additional benefit for docetaxel in patients with Ki67-negative tumors (adjusted HR = 1.10), who represent more than two-thirds of the tested patients. A potential application might be to omit docetaxel for these patients and go back to six cycles of FEC. However, because that would increase the risk of cardiotoxicity and leukemia, it is reasonable to think that the delivery of three FEC three docetaxel cycles would remain "as good" as six FEC cycles and worthwhile giving. Whether Ki67 is mechanistically involved in the response to docetaxel remains to be demonstrated, but even if it is not, it may be a marker of a phenotype more sensitive to docetaxel. One may suppose that docetaxel, which exerts its cytotoxic effects in the G2/M phase of the cycle by inhibiting the microtubule disassembly (antimitotic effect), is more active on rapidly proliferating cells (high Ki67).
We did not find any interaction among the 33 other markers and docetaxel benefit. In fact, the relapse risk was decreased by docetaxel addition in nearly all subgroups defined according to these markers, but without significant difference for benefit between the marker-positive and marker-negative subgroups. Univariate analysis showed higher reduction of risk in ER-negative patients than in ER-positive ones, but the interaction was not significant. Similar observation was reported in the pooled analysis of two randomized trials [27], and the analyses of BCIRG001 [32], NSABP-B28 [44], CALGB9344 [45], and TACT [9] trials. Today, the predictive value of ER status for response to taxanes is not demonstrated when considering ER alone, and additional markers are required for identifying subgroups of ER-positive and ER-negative who most benefit from these drugs. Our previous [30] and present data suggest that Ki67 is a potential candidate. This predictive value is also debated for HER2. We found a significant interaction with HER2 status in univariate analysis, with more benefit of docetaxel addition in HER2-positive patients (HR for relapse: 0.46 vs 0.84), but the interaction lost significance in multivariate analysis. Analysis of two other adjuvant trials reported a benefit from the addition of taxanes in HER2-positive patients [9, 28], with significant interaction in one [28], but analyses of other trials yielded conflicting results [16, 18, 32, 46]. Regarding the other markers, we observed a trend towards a benefit from docetaxel in patients whose tumor was positive for CK14, Angiogenin, and β-Catenin, with a statistically borderline interaction, which deserves further analysis in larger series.
More complex molecular combinations could prove more informative than single markers for predicting docetaxel benefit [9, 29–31, 47]. Here, we have defined molecular subtypes according to the status of ER, PR, HER2 and Ki67 proteins. These subtypes displayed expected histo-clinical features. Notably, five-year DFS was relatively good in the luminal A subtype, poor in HER2-overexpressing and the triple-negative subtypes, and intermediate in the luminal B subtype, in close agreement with survival rates observed in the BCIRG001 [29] and GEICAM9906 trials [31], even if the definition of luminal B and HER2-overexpressing subtypes was a little different. Clearly, the subtype that did not benefit from docetaxel in our series was luminal A, as reported in the BCIRG001 trial [29], and for ER-positive, HER2-negative tumors (assimilated to luminal A) in the CALGB9344 [28] and TACT [9] trials. By contrast, luminal A tumors unexpectedly benefited from paclitaxel in the GEICAM9906 trial [18]. The reasons for discrepancy between this latter observation and the former ones are unclear, and longer follow-up is required. The benefit we observed in the triple-negative tumors (adjusted HR = 0.88) was not different from the benefit observed in the whole-population, and interaction was not significant. Two subtypes benefited from docetaxel addition: luminal B and HER2-overexpressing. In luminal B patients, the absolute benefit in five-year DFS was 19% (DFS: 64% with FEC vs 83% with FEC-D); docetaxel reduced the risk of relapse by 53% after adjustment for histo-clinical variables, and the interaction was significant. Similarly, luminal B BCIRG001 patients showed a 44% reduction of relapse risk with docetaxel [29]. This was not confirmed in the GEICAM9906 sub-study [31], which, however, used the same definition as the BCIRG001 trial (HR-positive, and either Ki67-positive or HER2-positive). In these two studies, the positivity cut-off was similar for ER and PR (1%), but different for Ki67 (10% in the GEICAM9906, 13% in the BCIRG001 study). From literature data and our comparison of luminal A vs luminal B tumors (Additional file 6, Table S5), some biological features of luminal B vs luminal A tumors might speculatively explain this differential benefit from docetaxel: not only the higher proliferation rate (Ki67), but also the lesser expression of BCL2 and TAU whose expression has been associated with taxane resistance [48, 49], and the higher expression of P53, whose mutations have been associated with resistance to DNA-damaging agents such as anthracyclines [50], and relative sensitivity to taxanes [51]. However, none of these markers, except Ki67, was associated with docetaxel benefit in our univariate analysis (Additional file 5, Table S4). Finally, docetaxel led to a 34% adjusted reduction of the relapse risk (HR = 0.66) in the HER2-overexpressing subtype, with a borderline interaction (P = 0.14). No adjuvant trastuzumab was given in our series, but it is likely that trastuzumab would not decrease the observed docetaxel benefit. Significant interaction between HER2, treatment and outcome was found in the CALGB9344 trial, independently of ER status [28]. Higher benefit of taxanes was also reported in HER2-positive, HR-negative BCIRG001 [29] and TACT [9] patients, but not in the GEICAM9906 trial [31]. Because the current IHC definition of luminal B and HER2-overexpressing subtypes is not consensual, we repeated analyses using definitions used by others in the BCIRG001 [29] and GEICAM9906 trials [31]: luminal B tumors were defined as HR-positive/HER2-negative/Ki67-positive or HR-positive/HER2-positive (N = 206, 21%), and HER2-overexpressing tumors as HR-negative/HER2-positive (N = 86, 9%). Luminal A and triple-negative subtypes were not changed. As shown in Additional file 9, Table S7, unadjusted and adjusted HR for relapse were very similar to previous analysis regarding the benefit of docetaxel per subtype, with more benefit in the luminal B and HER2-overexpressing subtypes, and no benefit in the luminal A subtype..
Our study presents several strengths (randomized prospective trial, high number of samples representative of the whole trial population and of tested proteins, including novel markers), but, like retrospective subset biomarker studies reported in adjuvant trials, suffers also from several limitations: a relatively small number of analyzed markers when compared with high-throughput profiling of frozen samples, a relatively limited proportion of available samples (55%), and limitations intrinsic to unplanned analyses. The relatively low number of events and the relatively small benefit of the experimental arm (HR = 0.81) make the study not powered enough to detect small interactions between markers and docetaxel benefit. The PACS01 trial, like other trials, was not designed to detect the benefit of taxanes in patient subgroups defined by markers. Conversely, unplanned analyses confer a risk of false positive results. For both reasons, meta-analysis of first-generation taxane trials incorporating molecular data, ideally centrally generated, is warranted in the context of international collaborations (planned future EBCTCG meta-analysis), as well as validation in ongoing second-generation taxane trials. For example, a validation study is planned to test the predictive value of Ki67 in the PACS04 trial, which compared three cycles of FEC three of docetaxel vs. six of ED (Epirubicine Docetaxel).
Other limitations are methodological (IHC) and conceptual (breast cancer heterogeneity). Breast cancer is heterogeneous. Given the extent of differences between the four molecular subtypes (luminal A and B, HER2-overexpressing, and triple-negative), and because a signal relevant in a given subtype may be diluted and undetectable in the whole population, and conversely a signal relevant in the whole population may not be detected in a given subtype, another promising approach is to redefine prognostic and predictive markers in each subtype [52]. Results of univariate prognostic analyses are detailed in Additional file 10, Table S8, showing, for example, that the negativity of BCL2, EGFR, and TAU were significant prognosticators in the luminal A subtype, but not in the other subtypes, or that TACC2-positivity had an unfavorable prognostic value in the HER2-overexpressing subtype, but favorable in the triple-negative subtype. However, given the relatively small size of each subtype in our series, this deserves to be reassessed in a larger series and in multivariate analysis.
Regarding IHC, it is known to be non-quantitative, and it is difficult to know the most biologically relevant cut-offs for prognostic and predictive analyses. In the present study, all cut-offs were predefined before statistical analyses. For ER, PR and HER2, we used the Saint-Gallen cut-offs [53]. For the other proteins, the challenge is more substantial. For Ki67, important guidelines are under development [42, 54–57]. A meta-analysis of 46 studies published in 2007 reported many different Ki67 cut-offs, ranging from 3.5% to 34% [35]. The 2011 Saint-Gallen cut-off has been fixed at 14%, based on comparison with gene array data (PAM50) as a prognostic factor [54]. However, as stated in the recommendations [53], the "optimal cut-points in Ki67 labeling index for prediction of efficacy of endocrine or cytotoxic therapy may vary". Here, to remain consistent with our previous PACS01 sub-study [30], we used a 20% cut-off. For the other 30 tested proteins, and in the absence of consensual guidelines, we used a 1% cut-off. We are aware that the chosen cut-offs may not be optimal in terms of prognostic and/or predictive values. We have thus planned the analysis of different cut-offs for each protein, which will require identification and validation in independent series. In the present series, the prognostic value of Ki67 and its value predictive for docetaxel benefit remained significant in multivariate analysis when we applied a 15% cut-off (data not shown), regardless of the cut-off for ER and PR: 1% (P = 0.028 and P for interaction = 0.033 respectively) or 10% (P = 0.022 and P for interaction = 0.031 respectively). By contrast, the favorable impact of the luminal B subtype for the docetaxel benefit lost its significance (P-value for interaction = 0.208 in multivariate analysis) when we used a 15% cut-off for Ki67 (Additional file 11, Table S9). Another analytic approach is to study the prognostic and predictive values of continuous IHC variables. As a preliminary approach, we used quartiles of IHC measurements to categorize our population (percent of stained tumor cells for the data generated on standard slides and quick score QS for the TMA data; Additional file 12, Table S10). We then assessed for each protein the magnitude of the prognostic and treatment effects as a function of such categorization. The results of multivariate analyses (Additional file 13, Table S11) showed a differential DFS according to the value of Ki67 and PR, with a maximum difference for the highest quartile (effect unfavorable for Ki67 and favorable for PR). There was also a trend towards a differential benefit of docetaxel versus no docetaxel according to the value of Ki67 and Aurora A, with a maximum benefit for the highest quartile for both proliferation markers (Additional file 14, Table S12). Complementary analyses, such as STEPP analysis (subpopulation treatment effect pattern plot) [58], are warranted.
Conclusions
We confirm that ER, PR or HER2 status alone does not predict the benefit of docetaxel as adjuvant therapy in node-positive early breast cancer patients treated with anthracycline-based chemotherapy. We show that Ki67 status alone may be informative. We show also that the molecular subclassification may predict which patient may not benefit from docetaxel (luminal A) and may benefit more than the unselected population (luminal B, HER2-overexpressing), but does not provide additional information to Ki67 status alone. To go further in analyses, a supervised analysis [33] will be applied to our dataset, using semi-quantitative and quantitative data, to attempt identifying a multiprotein predictor for docetaxel benefit, overall and per subtype.
Abbreviations
- CMF:
-
cyclophosphamide: methotrexate: fluorouracil
- DFS:
-
disease-free survival
- EBC:
-
early breast cancer
- ER:
-
estrogen receptor
- FEC:
-
fluorouracil: epirubicin: cyclophosphamide
- FEC-D:
-
FEC-docetaxel
- FISH:
-
Fluorescent In Situ Hybridisation
- HR:
-
hormone receptor
- IHC:
-
immunohistochemistry
- OS:
-
overall survival
- PR:
-
progesterone receptor
- QS:
-
quick score
- SBR:
-
Scarff-Bloom Richardson
- STEPP:
-
subpopulation treatment effect pattern plot
- TMA:
-
tissue microarray.
References
Early Breast Cancer Trialists' Collaborative Group: Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomized trials. Lancet. 2005, 365: 1687-1717.
Sparano JA, Hortobagyi GN, Gralow JR, Perez EA, Comis RL: Recommendations for research priorities in breast cancer by the Coalition of Cancer Cooperative Groups Scientific Leadership Council: systemic therapy and therapeutic individualization. Breast Cancer Res Treat. 2010, 119: 511-527. 10.1007/s10549-009-0433-y.
Bedard PL, Di Leo A, Piccart-Gebhart MJ: Taxanes: optimizing adjuvant chemotherapy for early-stage breast cancer. Nat Rev Clin Oncol. 2010, 7: 22-36. 10.1038/nrclinonc.2009.186.
Buzdar AU, Singletary SE, Valero V, Booser DJ, Ibrahim NK, Rahman Z, Theriault RL, Walters R, Rivera E, Smith TL, Holmes FA, Hoy E, Frye DK, Manuel N, Kau SW, McNeese MD, Strom E, Thomas E, Hunt K, Ames F, Berry D, Hortobagyi GN: Evaluation of paclitaxel in adjuvant chemotherapy for patients with operable breast cancer: preliminary data of a prospective randomized trial. Clin Cancer Res. 2002, 8: 1073-1079.
Henderson IC, Berry DA, Demetri GD, Cirrincione CT, Goldstein LJ, Martino S, Ingle JN, Cooper MR, Hayes DF, Tkaczuk KH, Fleming G, Holland JF, Duggan DB, Carpenter JT, Frei E, Schilsky RL, Wood WC, Muss HB, Norton L: Improved outcomes from adding sequential Paclitaxel but not from escalating Doxorubicin dose in an adjuvant chemotherapy regimen for patients with node-positive primary breast cancer. J Clin Oncol. 2003, 21: 976-983. 10.1200/JCO.2003.02.063.
Mamounas EP, Bryant J, Lembersky B, Fehrenbacher L, Sedlacek SM, Fisher B, Wickerham DL, Yothers G, Soran A, Wolmark N: Paclitaxel after doxorubicin plus cyclophosphamide as adjuvant chemotherapy for node-positive breast cancer: results from NSABP B-28. J Clin Oncol. 2005, 23: 3686-3696. 10.1200/JCO.2005.10.517.
Martin M, Pienkowski T, Mackey J, Pawlicki M, Guastalla JP, Weaver C, Tomiak E, Al-Tweigeri T, Chap L, Juhos E, Guevin R, Howell A, Fornander T, Hainsworth J, Coleman R, Vinholes J, Modiano M, Pinter T, Tang SC, Colwell B, Prady C, Provencher L, Walde D, Rodriguez-Lescure A, Hugh J, Loret C, Rupin M, Blitz S, Jacobs P, Murawsky M, et al: Adjuvant docetaxel for node-positive breast cancer. N Engl J Med. 2005, 352: 2302-2313. 10.1056/NEJMoa043681.
Roche H, Fumoleau P, Spielmann M, Canon JL, Delozier T, Serin D, Symann M, Kerbrat P, Soulié P, Eichler F, Viens P, Monnier A, Vindevoghel A, Campone M, Goudier MJ, Bonneterre J, Ferrero JM, Martin AL, Genève J, Asselain B: Sequential adjuvant Epirubicin-based and Docetaxel Chemotherapy for node-positive breast cancer patients: the PACS01 trial. J Clin Oncol. 2006, 24: 5664-5671. 10.1200/JCO.2006.07.3916.
Ellis P, Barrett-Lee P, Johnson L, Cameron D, Wardley A, O'Reilly S, Verrill M, Smith I, Yarnold J, Coleman R, Earl H, Canney P, Twelves C, Poole C, Bloomfield D, Hopwood P, Johnston S, Dowsett M, Bartlett JM, Ellis I, Peckitt C, Hall E, Bliss JM: Sequential docetaxel as adjuvant chemotherapy for early breast cancer (TACT): an open-label, phase III, randomised controlled trial. Lancet. 2009, 373: 1681-1692. 10.1016/S0140-6736(09)60740-6.
Joensuu H, Kellokumpu-Lehtinen PL, Bono P, Alanko T, Kataja V, Asola R, Utriainen T, Kokko R, Hemminki A, Tarkkanen M, Turpeenniemi-Hujanen T, Jyrkkio S, Flander M, Helle L, Ingalsuo S, Johansson K, Jaaskelainen AS, Pajunen M, Rauhala M, Kaleva-Kerola J, Salminen T, Leinonen M, Elomaa I, Isola J: Adjuvant docetaxel or vinorelbine with or without trastuzumab for breast cancer. N Engl J Med. 2006, 354: 809-820. 10.1056/NEJMoa053028.
Burnell M, Levine MN, Chapman JA, Bramwell V, Gelmon K, Walley B, Vandenberg T, Chalchal H, Albain KS, Perez EA, Rugo H, Pritchard K, O'Brien P, Shepherd LE: Cyclophosphamide, epirubicin, and Fluorouracil versus dose-dense epirubicin and cyclophosphamide followed by Paclitaxel versus Doxorubicin and cyclophosphamide followed by Paclitaxel in node-positive or high-risk node-negative breast cancer. J Clin Oncol. 2009, 28: 77-82.
Evans TR, Yellowlees A, Foster E, Earl H, Cameron DA, Hutcheon AW, Coleman RE, Perren T, Gallagher CJ, Quigley M, Crown J, Jones AL, Highley M, Leonard RC, Mansi JL: Phase III randomized trial of doxorubicin and docetaxel versus doxorubicin and cyclophosphamide as primary medical therapy in women with breast cancer: an anglo-celtic cooperative oncology group study. J Clin Oncol. 2005, 23: 2988-2995. 10.1200/JCO.2005.06.156.
Fountzilas G, Skarlos D, Dafni U, Gogas H, Briasoulis E, Pectasides D, Papadimitriou C, Markopoulos C, Polychronis A, Kalofonos HP, Siafaka V, Kosmidis P, Timotheadou E, Tsavdaridis D, Bafaloukos D, Papakostas P, Razis E, Makrantonakis P, Aravantinos G, Christodoulou C, Dimopoulos AM: Postoperative dose-dense sequential chemotherapy with epirubicin, followed by CMF with or without paclitaxel, in patients with high-risk operable breast cancer: a randomized phase III study conducted by the Hellenic Cooperative Oncology Group. Ann Oncol. 2005, 16: 1762-1771. 10.1093/annonc/mdi366.
Francis P, Crown J, Di Leo A, Buyse M, Balil A, Andersson M, Nordenskjold B, Lang I, Jakesz R, Vorobiof D, Gutierrez J, van Hazel G, Dolci S, Jamin S, Bendahmane B, Gelber RD, Goldhirsch A, Castiglione-Gertsch M, Piccart-Gebhart M: Adjuvant chemotherapy with sequential or concurrent anthracycline and docetaxel: Breast International Group 02-98 randomized trial. J Natl Cancer Inst. 2008, 100: 121-133. 10.1093/jnci/djm287.
Gianni L, Baselga J, Eiermann W, Porta VG, Semiglazov V, Lluch A, Zambetti M, Sabadell D, Raab G, Cussac AL, Bozhok A, Martinez-Agullo A, Greco M, Byakhov M, Lopez JJ, Mansutti M, Valagussa P, Bonadonna G: Phase III trial evaluating the addition of paclitaxel to doxorubicin followed by cyclophosphamide, methotrexate, and fluorouracil, as adjuvant or primary systemic therapy: European Cooperative Trial in Operable Breast Cancer. J Clin Oncol. 2009, 27: 2474-2481. 10.1200/JCO.2008.19.2567.
Jones S, Holmes FA, O'Shaughnessy J, Blum JL, Vukelja SJ, McIntyre KJ, Pippen JE, Bordelon JH, Kirby RL, Sandbach J, Hyman WJ, Richards DA, Mennel RG, Boehm KA, Meyer WG, Asmar L, Mackey D, Riedel S, Muss H, Savin MA: Docetaxel With cyclophosphamide is associated with an overall survival benefit compared with doxorubicin and cyclophosphamide: 7-year follow-up of US Oncology Research Trial 9735. J Clin Oncol. 2009, 27: 1177-1183. 10.1200/JCO.2008.18.4028.
Goldstein LJ, O'Neill A, Sparano JA, Perez EA, Shulman LN, Martino S, Davidson NE: Concurrent doxorubicin plus docetaxel is not more effective than concurrent doxorubicin plus cyclophosphamide in operable breast cancer with 0 to 3 positive axillary nodes: North American Breast Cancer Intergroup Trial E 2197. J Clin Oncol. 2008, 26: 4092-4099. 10.1200/JCO.2008.16.7841.
Martin M, Rodriguez-Lescure A, Ruiz A, Alba E, Calvo L, Ruiz-Borrego M, Munarriz B, Rodriguez CA, Crespo C, de Alava E, Lopez Garcia-Asenjo JA, Guitian MD, Almenar S, Gonzalez-Palacios JF, Vera F, Palacios J, Ramos M, Gracia Marco JM, Lluch A, Alvarez I, Segui MA, Mayordomo JI, Anton A, Baena JM, Plazaola A, Modolell A, Pelegri A, Mel JR, Aranda E, Adrover E, et al: Randomized phase 3 trial of fluorouracil, epirubicin, and cyclophosphamide alone or followed by Paclitaxel for early breast cancer. J Natl Cancer Inst. 2008, 100: 805-814. 10.1093/jnci/djn151.
Polyzos A, Malamos N, Boukovinas I, Adamou A, Ziras N, Kalbakis K, Kakolyris S, Syrigos K, Papakotoulas P, Kouroussis C, Karvounis N, Vamvakas L, Christophyllakis C, Athanasiadis A, Varthalitis I, Georgoulias V, Mavroudis D: FEC versus sequential docetaxel followed by epirubicin/cyclophosphamide as adjuvant chemotherapy in women with axillary node-positive early breast cancer: a randomized study of the Hellenic Oncology Research Group (HORG). Breast Cancer Res Treat. 2010, 119: 95-104. 10.1007/s10549-009-0468-0.
Cognetti F, De Laurentiis M, De Matteis A, Manzione L, Boni C, Palazzo S, Di Palma M, Papaldo P, De Placido S, Bianco AR: Sequential epirubicin-docetaxel-CMF as adjuvant therapy for node-positive early breast cancer: updated results of the TAXIt216 randomized trial. Ann Oncol. 2008, 19: a1820-
Del Mastro L, Costantini M, Durando A, Michelotti A, Danese S, Aitini E, Olmeo N, Pronzato P, Venturini M, Intergruppo GONO-M: Cyclophosphamide, epirubicin, and 5-fluorouracil versus epirubicin plus paclitaxel in node-positive early breast cancer patients: A randomized, phase III study of Gruppo Oncologico Nord Ovest-Mammella Intergruppo Group. J Clin Oncol. 2008, 26: a516-
Nitz U, Huober JB, Lisboa B, Harbeck N, Fischer H, Moebus V, Hoffmann G, Augustin D, Weiss E, Kuhn W, West German Study Group/AGO-Mamma: Interim results of Intergroup EC-Doc Trial: a randomized multicenter phase III trial comparing adjuvant CEF/CMF to EC- docetaxel in patients with 1-3 positive lymph nodes [abstract]. J Clin Oncol. 2008, 26: a515-10.1200/JCO.2007.13.8131.
Bria E, Nistico C, Cuppone F, Carlini P, Ciccarese M, Milella M, Natoli G, Terzoli E, Cognetti F, Giannarelli D: Benefit of taxanes as adjuvant chemotherapy for early breast cancer: pooled analysis of 15,500 patients. Cancer. 2006, 106: 2337-2344. 10.1002/cncr.21886.
De Laurentiis M, Cancello G, D'Agostino D, Giuliano M, Giordano A, Montagna E, Lauria R, Forestieri V, Esposito A, Silvestro L, Pennacchio R, Criscitiello C, Montanino A, Limite G, Bianco AR, De Placido S: Taxane-based combinations as adjuvant chemotherapy of early breast cancer: a meta-analysis of randomized trials. J Clin Oncol. 2008, 26: 44-53. 10.1200/JCO.2007.11.3787.
Ferguson T, Wilcken N, Vagg R, Ghersi D, Nowak AK: Taxanes for adjuvant treatment of early breast cancer. Cochrane Database Syst Rev. 2007, CD004421-
Martin M, Lluch A, Segui MA, Ruiz A, Ramos M, Adrover E, Rodriguez-Lescure A, Grosse R, Calvo L, Fernandez-Chacon C, Roset M, Anton A, Isla D, del Prado PM, Iglesias L, Zaluski J, Arcusa A, Lopez-Vega JM, Munoz M, Mel JR: Toxicity and health-related quality of life in breast cancer patients receiving adjuvant docetaxel, doxorubicin, cyclophosphamide (TAC) or 5-fluorouracil, doxorubicin and cyclophosphamide (FAC): impact of adding primary prophylactic granulocyte-colony stimulating factor to the TAC regimen. Ann Oncol. 2006, 17: 1205-1212. 10.1093/annonc/mdl135.
Andre F, Broglio K, Roche H, Martin M, Mackey JR, Penault-Llorca F, Hortobagyi GN, Pusztai L: Estrogen receptor expression and efficacy of docetaxel-containing adjuvant chemotherapy in patients with node-positive breast cancer: results from a pooled analysis. J Clin Oncol. 2008, 26: 2636-2643. 10.1200/JCO.2007.14.9146.
Hayes DF, Thor AD, Dressler LG, Weaver D, Edgerton S, Cowan D, Broadwater G, Goldstein LJ, Martino S, Ingle JN, Henderson IC, Norton L, Winer EP, Hudis CA, Ellis MJ, Berry DA: HER2 and response to paclitaxel in node-positive breast cancer. N Engl J Med. 2007, 357: 1496-1506. 10.1056/NEJMoa071167.
Hugh J, Hanson J, Cheang MC, Nielsen TO, Perou CM, Dumontet C, Reed J, Krajewska M, Treilleux I, Rupin M, Magherini E, Mackey J, Martin M, Vogel C: Breast cancer subtypes and response to docetaxel in node-positive breast cancer: use of an immunohistochemical definition in the BCIRG 001 trial. J Clin Oncol. 2009, 27: 1168-1176. 10.1200/JCO.2008.18.1024.
Penault-Llorca F, Andre F, Sagan C, Lacroix-Triki M, Denoux Y, Verriele V, Jacquemier J, Baranzelli MC, Bibeau F, Antoine M, Lagarde N, Martin AL, Asselain B, Roche H: Ki67 expression and docetaxel efficacy in patients with estrogen receptor-positive breast cancer. J Clin Oncol. 2009, 27: 2809-2815. 10.1200/JCO.2008.18.2808.
Martin M, Rodriguez-Lescure A, Ruiz A, Alba E, Calvo L, Ruiz-Borrego M, Santaballa A, Rodriguez CA, Crespo C, Abad M, Dominguez S, Florian J, Llorca C, Mendez M, Godes M, Cubedo R, Murias A, Batista N, Garcia MJ, Caballero R, de Alava E: Molecular predictors of efficacy of adjuvant weekly paclitaxel in early breast cancer. Breast Cancer Res Treat. 2010, 123: 149-157. 10.1007/s10549-009-0663-z.
Dumontet C, Krajewska M, Treilleux I, Mackey JR, Martin M, Rupin M, Lafanechere L, Reed JC: BCIRG 001 molecular analysis: prognostic factors in node-positive breast cancer patients receiving adjuvant chemotherapy. Clin Cancer Res. 2010, 16: 3988-3997. 10.1158/1078-0432.CCR-10-0079.
Jacquemier J, Ginestier C, Rougemont J, Bardou VJ, Charafe-Jauffret E, Geneix J, Adelaide J, Koki A, Houvenaeghel G, Hassoun J, Maraninchi D, Viens P, Birnbaum D, Bertucci F: Protein expression profiling identifies subclasses of breast cancer and predicts prognosis. Cancer Res. 2005, 65: 767-779.
Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Eystein Lonning P, Borresen-Dale AL: Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001, 98: 10869-10874. 10.1073/pnas.191367098.
de Azambuja E, Cardoso F, de Castro G, Colozza M, Mano MS, Durbecq V, Sotiriou C, Larsimont D, Piccart-Gebhart MJ, Paesmans M: Ki-67 as prognostic marker in early breast cancer: a meta-analysis of published studies involving 12,155 patients. Br J Cancer. 2007, 96: 1504-1513. 10.1038/sj.bjc.6603756.
Stuart-Harris R, Caldas C, Pinder SE, Pharoah P: Proliferation markers and survival in early breast cancer: a systematic review and meta-analysis of 85 studies in 32,825 patients. Breast. 2008, 17: 323-334. 10.1016/j.breast.2008.02.002.
Chang JC, Makris A, Gutierrez MC, Hilsenbeck SG, Hackett JR, Jeong J, Liu ML, Baker J, Clark-Langone K, Baehner FL, Sexton K, Mohsin S, Gray T, Alvarez L, Chamness GC, Osborne CK, Shak S: Gene expression patterns in formalin-fixed, paraffin-embedded core biopsies predict docetaxel chemosensitivity in breast cancer patients. Breast Cancer Res Treat. 2008, 108: 233-240. 10.1007/s10549-007-9590-z.
Darb-Esfahani S, Loibl S, Muller BM, Roller M, Denkert C, Komor M, Schluns K, Blohmer JU, Budczies J, Gerber B, Noske A, du Bois A, Weichert W, Jackisch C, Dietel M, Richter K, Kaufmann M, von Minckwitz G: Identification of biology-based breast cancer types with distinct predictive and prognostic features: role of steroid hormone and HER2 receptor expression in patients treated with neoadjuvant anthracycline/taxane-based chemotherapy. Breast Cancer Res. 2009, 11: R69-10.1186/bcr2363.
Estevez LG, Cuevas JM, Anton A, Florian J, Lopez-Vega JM, Velasco A, Lobo F, Herrero A, Fortes J: Weekly docetaxel as neoadjuvant chemotherapy for stage II and III breast cancer: efficacy and correlation with biological markers in a phase II, multicenter study. Clin Cancer Res. 2003, 9: 686-692.
Sjostrom J, Blomqvist C, Heikkila P, Boguslawski KV, Raisanen-Sokolowski A, Bengtsson NO, Mjaaland I, Malmstrom P, Ostenstadt B, Bergh J, Wist E, Valvere V, Saksela E: Predictive value of p53, mdm-2, p21, and mib-1 for chemotherapy response in advanced breast cancer. Clin Cancer Res. 2000, 6: 3103-3110.
Paik S, Tang G, Shak S, Kim C, Baker J, Kim W, Cronin M, Baehner FL, Watson D, Bryant J, Costantino JP, Geyer CE, Wickerham DL, Wolmark N: Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. J Clin Oncol. 2006, 24: 3726-3734. 10.1200/JCO.2005.04.7985.
Viale G, Regan MM, Mastropasqua MG, Maffini F, Maiorano E, Colleoni M, Price KN, Golouh R, Perin T, Brown RW, Kovacs A, Pillay K, Ohlschlegel C, Gusterson BA, Castiglione-Gertsch M, Gelber RD, Goldhirsch A, Coates AS: Predictive value of tumor Ki-67 expression in two randomized trials of adjuvant chemoendocrine therapy for node-negative breast cancer. J Natl Cancer Inst. 2008, 100: 207-212. 10.1093/jnci/djm289.
Bartlett JM, Munro AF, Dunn JA, McConkey C, Jordan S, Twelves CJ, Cameron DA, Thomas J, Campbell FM, Rea DW, Provenzano E, Caldas C, Pharoah P, Hiller L, Earl H, Poole CJ: Predictive markers of anthracycline benefit: a prospectively planned analysis of the UK National Epirubicin Adjuvant Trial (NEAT/BR9601). Lancet Oncol. 2010, 11: 266-274. 10.1016/S1470-2045(10)70006-1.
Mamounas EP, Bryant J, Lembersky B, Fehrenbacher L, Sedlacek SM, Fisher B, Wickerham DL, Yothers G, Soran A, Wolmark N: Paclitaxel after doxorubicin plus cyclophosphamide as adjuvant chemotherapy for node-positive breast cancer: results from NSABP B-28. J Clin Oncol. 2005, 23: 3686-3696. 10.1200/JCO.2005.10.517.
Berry DA, Cirrincione C, Henderson IC, Citron ML, Budman DR, Goldstein LJ, Martino S, Perez EA, Muss HB, Norton L, Hudis C, Winer EP: Estrogen-receptor status and outcomes of modern chemotherapy for patients with node-positive breast cancer. JAMA. 2006, 295: 1658-1667. 10.1001/jama.295.14.1658.
Kostopoulos I, Arapantoni-Dadioti P, Gogas H, Papadopoulos S, Malamou-Mitsi V, Scopa CD, Markaki S, Karagianni E, Kyriakou V, Margariti A, Kyrkou E, Pavlakis K, Zaramboukas T, Skordalaki A, Bourli A, Markopoulos C, Pectasides D, Dimopoulos MA, Skarlos D, Fountzilas G: Evaluation of the prognostic value of HER-2 and VEGF in breast cancer patients participating in a randomized study with dose-dense sequential adjuvant chemotherapy. Breast Cancer Res Treat. 2006, 96: 251-261. 10.1007/s10549-005-9062-2.
Hayes DF: Is there a standard type and duration of adjuvant chemotherapy for early stage breast cancer?. Breast. 2009, 18: S131-134.
Andre F, Hatzis C, Anderson K, Sotiriou C, Mazouni C, Mejia J, Wang B, Hortobagyi GN, Symmans WF, Pusztai L: Microtubule-associated protein-tau is a bifunctional predictor of endocrine sensitivity and chemotherapy resistance in estrogen receptor-positive breast cancer. Clin Cancer Res. 2007, 13: 2061-2067. 10.1158/1078-0432.CCR-06-2078.
Noguchi S: Predictive factors for response to docetaxel in human breast cancers. Cancer Sci. 2006, 97: 813-820. 10.1111/j.1349-7006.2006.00265.x.
Aas T, Borresen AL, Geisler S, Smith-Sorensen B, Johnsen H, Varhaug JE, Akslen LA, Lonning PE: Specific P53 mutations are associated with de novo resistance to doxorubicin in breast cancer patients. Nat Med. 1996, 2: 811-814. 10.1038/nm0796-811.
Wahl AF, Donaldson KL, Fairchild C, Lee FY, Foster SA, Demers GW, Galloway DA: Loss of normal p53 function confers sensitization to Taxol by increasing G2/M arrest and apoptosis. Nat Med. 1996, 2: 72-79. 10.1038/nm0196-72.
Pusztai L: Gene expression profiling of breast cancer. Breast Cancer Res. 2009, 11: S11-
Goldhirsch A, Wood WC, Coates AS, Gelber RD, Thurlimann B, Senn HJ: Strategies for subtypes--dealing with the diversity of breast cancer: highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann Oncol. 2011, 22: 1736-1747. 10.1093/annonc/mdr304.
Cheang MC, Chia SK, Voduc D, Gao D, Leung S, Snider J, Watson M, Davies S, Bernard PS, Parker JS, Perou CM, Ellis MJ, Nielsen TO: Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst. 2009, 101: 736-750. 10.1093/jnci/djp082.
Urruticoechea A, Smith IE, Dowsett M: Proliferation marker Ki-67 in early breast cancer. J Clin Oncol. 2005, 23: 7212-7220. 10.1200/JCO.2005.07.501.
Viale G, Giobbie-Hurder A, Regan MM, Coates AS, Mastropasqua MG, Dell'Orto P, Maiorano E, MacGrogan G, Braye SG, Ohlschlegel C, Neven P, Orosz Z, Olszewski WP, Knox F, Thurlimann B, Price KN, Castiglione-Gertsch M, Gelber RD, Gusterson BA, Goldhirsch A: Prognostic and predictive value of centrally reviewed Ki-67 labeling index in postmenopausal women with endocrine-responsive breast cancer: results from Breast International Group Trial 1-98 comparing adjuvant tamoxifen with letrozole. J Clin Oncol. 2008, 26: 5569-5575. 10.1200/JCO.2008.17.0829.
Yerushalmi R, Woods R, Ravdin PM, Hayes MM, Gelmon KA: Ki67 in breast cancer: prognostic and predictive potential. Lancet Oncol. 2010, 11: 174-183. 10.1016/S1470-2045(09)70262-1.
Bonetti M, Gelber RD: A graphical method to assess treatment-covariate interactions using the Cox model on subsets of the data. Stat Med. 2000, 19: 2595-2609. 10.1002/1097-0258(20001015)19:19<2595::AID-SIM562>3.0.CO;2-M.
Acknowledgements
We thank the French and Belgium pathologists involved in the PACS01 trial for kindly providing the tumor blocks. This work was supported in part by Inserm, Institut Paoli-Calmettes, and grants from Ligue Nationale Contre le Cancer (label DB), Fédération Nationale des Centres de Lutte Contre le Cancer.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
JJ and FB designed the concept of the study. HR, DS, PK, FA, MC, PV and FB contributed to the accrual of patients in the trial. JJ, ECJ and FPL participated in the central pathological reading of samples, and carried out IHC analyses. JMB, BE and PF performed the statistical analyses. ALM collected histo-clinical data and samples. JJ, DB and FB were responsible for interpretation of results. JJ and FB wrote the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
13058_2011_2848_MOESM1_ESM.DOC
Additional file 1: Table S1 (WORD file). Characteristics of patients in the PACS01 trial and in this sub-study. (DOC 59 KB)
13058_2011_2848_MOESM3_ESM.DOC
Additional file 3: Table S3 (WORD file). Multivariate analysis of DFS according to histo-clinical variables. (DOC 44 KB)
13058_2011_2848_MOESM4_ESM.PPT
Additional file 4: Figure S1 (POWER POINT file). Adjusted hazard ratios associated with docetaxel addition (forest plots) according to histo-clinical variables. (PPT 908 KB)
13058_2011_2848_MOESM5_ESM.DOC
Additional file 5: Table S4 (WORD file). Univariate and multivariate analyses of 34 antibodies for interaction with chemotherapy arm. (DOC 288 KB)
13058_2011_2848_MOESM6_ESM.DOC
Additional file 6: Table S5 (WORD file). Molecular subtypes and correlations with histo-clinical features and IHC results. (DOC 286 KB)
13058_2011_2848_MOESM7_ESM.PPT
Additional file 7: Figure S2 (POWER POINT file). Disease-free survival according to molecular subtypes and docetaxel. Kaplan-Meier DFS curves in patients with luminal A (A), luminal B (B), HER2-overexpressing (C) and triple-negative (D) tumor according to docetaxel addition. (PPT 72 KB)
13058_2011_2848_MOESM8_ESM.DOC
Additional file 8: Table S6 (WORD file). Univariate and multivariate analyses of molecular subtypes for interaction with chemotherapy arm. (DOC 46 KB)
13058_2011_2848_MOESM9_ESM.DOC
Additional file 9: Table S7 (WORD file). Univariate and multivariate analyses of molecular subtypes for interaction with chemotherapy arm, using another definition for luminal B and HER2-overexpressing subtypes. (DOC 46 KB)
13058_2011_2848_MOESM11_ESM.DOC
Additional file 11: Table S9 (WORD file). Univariate and multivariate analyses of molecular subtypes for interaction with chemotherapy arm, using 15% cut-off for Ki67 for the definition of luminal subtypes. (DOC 51 KB)
13058_2011_2848_MOESM14_ESM.DOC
Additional file 14: Table S12 (WORD file). Univariate and multivariate analyses "per quartiles" for interaction with chemotherapy arm. (DOC 482 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Jacquemier, J., Boher, JM., Roche, H. et al. Protein expression, survival and docetaxel benefit in node-positive breast cancer treated with adjuvant chemotherapy in the FNCLCC - PACS 01 randomized trial. Breast Cancer Res 13, R109 (2011). https://doi.org/10.1186/bcr3051
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/bcr3051